

Abstract
The simultaneous and spatially controlled display of different proteins on nanocarriers is a desirable property not often achieved in practice. Here, we report the use of clathrin triskelions as a versatile platform for functional protein display. We hypothesized that site-specific molecular epitope recognition would allow for effective and ordered protein attachment to clathrin triskelions. Clathrin binding peptides (CBPs) were genetically fused to mCherry and green fluorescent protein (GFP), expressed, and loaded onto clathrin triskelions by site-specific binding. Attachment was confirmed by surface plasmon resonance. mCherry fusion proteins modified with various CBPs displayed binding affinities between 470 nM and 287 μM for the clathrin triskelions. Simultaneous attachment of GFP-Wbox and mCherry-Cbox fusion constructs to the clathrin terminal domain was verified by Förster resonance energy transfer. The circulating half-lives, area under the curve, and the terminal half-lives of GFP and mCherry were significantly increased when attached to clathrin triskelions. Clathrin triskelion technology is useful for the development of versatile and multifunctional carriers for spatially controlled protein or peptide display with tremendous potential in nanotechnology, drug delivery, vaccine development, and targeted therapeutic applications.
Keywords: clathrin triskelions, protein display, molecular epitope recognition, spatial control, protein based carriers
Graphical abstract
Introduction
The simultaneous and spatially controlled display of multiple proteins would be valuable for the development of multifunctional biomaterials for drug and gene delivery, vaccination, and other applications. Precise control of spatial distribution - including the orientation, proximity, and stoichiometry of multiple ligands - is challenging and has yet to be achieved in common polymeric and lipidic nanocarriers. Many conjugation approaches, especially via amine chemistry, produce a heterogeneous mixture of poorly defined nanocarriers, making it difficult to study the spatial and stoichiometric effects of targeting ligands on cellular binding [1].
The unique properties, highly defined structures, and biocompatibility of natural proteins have made them materials of choice for nanomedicine, bionanotechnology, and materials sciences. Commonly used protein-based carriers include virus-like particles, hepatitis B virus capsids, ferritin nanocages, and bacterial S-layers [2]. The protein subunits of these carriers self-assemble into highly ordered symmetrical cages and capsids. Genetic modifications and functionalization of these carriers allow site-specific control of ligand display [3, 4].
Clathrin triskelions are a unique class of protein-based carriers. Their three legs that radiate symmetrically from a central C-terminal hub [5] allow for the versatile and simultaneous display of proteins or peptides. Each leg possesses an N-terminal domain comprising multiple distinct molecular epitopes for specific binding to conserved peptide sequences. This allows for site-specific attachment of three unique decorations to each leg for the customizable delivery of proteins, drugs, and therapeutic cargoes. Clathrin triskelion monomers can exist in isolation but are also able to assemble into larger protein structures. Depending on the conditions used (e.g. buffer composition and pH), triskelions can form spherical cages of different sizes (~40–150 nm), making them highly versatile carriers for protein display [6, 7].
Clathrin is ubiquitously expressed in all eukaryotic cells where it coordinates internalization via clathrin-mediated endocytosis [8]. It is a 180 kDa protein that arranges its flexible legs in an antiparallel direction to self-assemble into polyhedral cage structures [9] that facilitate the uptake and shuttling of cargo inside cells [10]. Various accessory proteins containing binding domains that interact with molecular epitopes found on the clathrin triskelions orchestrate the formation and collapse of clathrin-coated vesicles (CCVs) during membrane destabilization and clathrin assembly [11]. We hypothesize that a fusion protein containing a binding domain specific for these clathrin epitopes could be used to specifically attach any peptide or protein for ordered display via molecular epitope recognition.
Due to the multiple molecular epitopes found on the N-terminal hub of the clathrin triskelions, proteins can be attached simultaneously to produce a multi-display delivery system. These epitopes provide flexibility for site-specific attachment of ligands, proteins, or therapeutic cargoes to the clathrin triskelions. Very few studies have exploited clathrin’s specific binding motifs for downstream applications. For instance, triskelions forming clathrin cages have been used (i) as nucleation points for metallic nanoparticle synthesis [12, 13] and (ii) as potential biosensors by exploiting their assembling and disassembling properties [14]. However, it is currently unknown whether the clathrin triskelions can be used for the simultaneous binding of two macromolecules at two very close binding sites (within 25 angstroms). Moreover, the stability of clathrin/protein interactions under physiological conditions has yet to be characterized.
Binding via molecular epitope recognition motifs not only allows the precise control of stoichiometry and spatial distance between attached proteins but also the precise regulation of protein orientation [4]. This is especially important if a displayed protein’s activity is dependent on epitope accessibility. For example, amine coupling often leads to non-specific reactions of exposed lysines and possibly inactivation of the reactive site of the protein [15, 16]. The clathrin delivery platform overcomes these obstacles.
Here, we demonstrate the use of clathrin triskelions for the simultaneous display of multiple proteins. We designed green fluorescent protein (GFP) and mCherry fusion proteins containing peptides recognizing two distinct epitopes located at the N-terminal domain of the clathrin triskelions. GFP and mCherry were selected as proof-of-concept candidate fluorophores because their fluorescence at different wavelengths allows for the accurate determination of simultaneous binding. Surface plasmon resonance (SPR) and Förster resonance energy transfer (FRET) were used to confirm binding of GFP and mCherry fusion proteins to clathrin triskelions. Further, clathrin-protein complex stability under physiological conditions was assessed in in vivo pharmacokinetic (PK) studies. Clathrin triskelions represent a useful technology for a range of therapeutic applications such as protein therapeutics, cancer therapy, and vaccination.
2. Materials and methods
2.1 Materials
All chemicals and solvents were purchased from Fisher Scientific (Hampton, NH), unless otherwise stated. Coomassie blue stain and polyvinylidene fluoride (PVDF) membrane were purchased from BioRad (Hercules, CA). Protease inhibitor tablets, lysozyme, 2xYT, enhanced chemiluminescence (ECL) western substrate, DyLight 594 and 650 NHS ester, Pierce 20K and 150K molecular weight cut-off (MWCO) concentrating tubes, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), and N-hydroxysuccinimide (NHS) were obtained from Thermo Scientific (Waltham, MA). Isopropyl thiogalactosidase (IPTG) and anti-clathrin heavy chain mouse mAb X22 were purchased from EMD Millipore (Billerica, MA). Ficoll 400 was purchased from Alfa Aesar. Restriction enzymes and ligases were obtained from New England BioLabs (Ipswich, MA). Sepharose CL-6B and Tween 20 were obtained from Sigma-Aldrich (St. Louis, MO). NuPage LDS sample buffer (4X) and DH5α electrocompetent cells were purchased from Life Technologies (Grand Island, NY). Wizard PCR and Gel Purification Kit and (2S,3S)-1,4-bis(sulfanyl)butane-2,3-diol (DTT) were purchased from Promega (Madison, WI). Bio-Tek E.Z.N.A. Plasmid Mini Kit was obtained from OMEGA (Norcross, GA). E. cloni BL21 (DE3) electrocompetent cells were obtained from Lucigen (Middleton, WI). All primers and oligonucleotide sequences for cloning were from Integrated DNA Technologies (Coralville, IA).
2.2 Recombinant DNA constructs
An mCherry cloning vector (#29722, Addgene, Cambridge, MA) was used for the mCherry gene amplification. The PCR amplified mCherry product containing 5′-NheI and 3′-HindIII restriction sites was purified and subcloned into the bacterial expression vector pET-21a (Novagen, Merck Millipore, Billerica, MA). Codon-optimized oligonucleotide sequences for expression in E. coli were used for the clathrin binding peptide (CBP) domains. Overlapping oligonucleotides coding for the CBPs with 5′-BsrG1 and 3′-HindIII restriction sites were cloned into the pET-21a-mcherry vector. The pET-21a-mCherry-CBP vector encodes for mCherry-CBP containing a C-terminal poly-histidine tag for purification by immobilized metal affinity chromatography (IMAC), proceeded by a thrombin cleavage sequence. All plasmid sequences were confirmed by Sanger sequencing.
2.3 Protein expression and purification
mCherry, mCherry-CBP fusion proteins, GFP, and GFP-CBP fusion proteins were expressed in BL21 (DE3). A 1L 2xYT culture was inoculated with 1% of the overnight culture and grown to an OD600 of ~0.8. Protein expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and the culture was incubated overnight at 24°C. The cells were harvested 18 h post-induction by centrifugation. The cells were resuspended in phosphate buffered saline (PBS) containing protease inhibitors and lysozyme (1mg/ml) followed by lysis via sonication. The cellular debris was pelleted by a 45 min centrifugation step at 17,000g at 4°C. Ni2+-affinity chromatography was used to isolate and purify the proteins in the supernatant. Following mCherry isolation, the C-terminal His-tag was cleaved with thrombin (GE Healthcare, Buckinhamshire, UK). The thrombin was removed by size exclusion chromatography using a Sepharose CL-6B column (Sigma-Aldrich, St. Louis, MO). Fractions were collected and analyzed by SDS-PAGE and mass spectrometry (Q Exactive Plus mass spectrometer) to verify the protein purity and molecular mass.
2.4 Western blotting
The protein concentration in the clathrin sample was quantified with the Bradford Protein assay (BioRad). After denaturing the protein samples with 4x LDS sample buffer and 50 mM DTT at 65°C for 5 min, the triskelion sample was separated on a 9% SDS-PAGE gel (Natural Diagnostics, Atlanta, GA). Electrophoresis was performed in SDS running buffer for 1.5 h at 150 V. Protein was then wet-transferred to a PVDF membrane in Tris-glycine transfer buffer (20% methanol) for 2 h at 100 V. The membrane was washed with Tris-buffered saline containing 0.5% tween 20 (TBST), followed by blocking with 5% (w/v) dried milk in TBST for 1 h. The PVDF membrane was incubated with an anti-clathrin heavy chain mouse mAb X22 (#CP45-100UL, EMD Millipore) at a 1:500 dilution in TBST/BSA buffer overnight at 4°C. After incubation with rabbit anti-mouse HRP secondary antibody (#7076S, Cell Signaling Technology, Danvers, MA) at 1:10,000 dilution for 1h at RT and thorough washing, ECL substrate was added. Images were acquired using a BioRad ChemiDoc imager.
2.5 Triskelion isolation
Triskelion monomers were isolated as previously described [12]. Briefly, bovine brains were flash frozen at −80°C cut into small cubes, and homogenized with a blender in HKM (25 mM HEPES, 125 mM potassium sulfate, 5 mM magnesium acetate, 1mM DTT) buffer. Following homogenization, the sample was spun at 6,000× g for 20 min. The supernatant was collected and separated from the pellet by gauze filtration and then spun in an Optima XE-90 Ultracentrifuge (Beckman Coulter) at 43,000× g for 40 min at 4°C. The supernatant was removed and the resulting pellet resuspended in HKM buffer. After resuspension, an equal volume of 12.5% Ficoll and 12.5% sucrose in HKM buffer were added to the solution before sonication for 1 min. The solution was spun down at 25,000× g for 20 min at 4°C before collecting and pooling the supernatant. After mixing the supernatant with 3 volumes of HKM buffer, the diluted supernatant was spun at 34,000g for 60 min at 4°C to pellet CCVs, which were then suspended in dissociation buffer (1 M Tris, 1 mM EDTA) for a final spin at 25,000× g for 30 min at 4°C to remove partial cages. The solution containing the triskelions was further concentrated using Pierce protein concentrators with a 150K molecular weight cut-off (MWCO) at 3,000 × g for 45 min at 4°C.
2.6 Clathrin triskelion imaging with transmission electron microscopy
Samples of clathrin triskelions were prepared at ~1 mg/mL. For negative staining, a 1% sample of uranyl acetate was prepared. Type A carbon TEM grids (Ted Pella, Redding, CA) were placed on Whatman filter paper (GE Healthcare) and 20 μL of the sample was dropped onto the grid. The grid was left to air-dry for 5 min. Immediately after, 20 μL of the 1% uranyl acetate was added to the grid. After a 3-min incubation period the remaining liquid was wicked away and the grids were allowed to air dry. The grid was analyzed by a FEI Techni F20 G2 transmission electron microscope (FEI, Hillsboro, Oregon) at an accelerating voltage of 200 kV.
2.7 Surface plasmon resonance (SPR)
Binding analysis was performed on a SR7000DC SPR instrument (Reichert, Buffalo, NY). The clathrin triskelions were immobilized on the surface of a carboxymethyl dextran hydrogel gold chip by standard amine coupling chemistry. Briefly, a 300 μL solution of 50 μg/mL clathrin suspended in immobilization buffer (10 mM sodium acetate, pH 5) was injected over the left channel at 15 μL/min for 6 min. A ligand density of ~500–1000 resonance units (μRU) of clathrin was immobilized as per the manufacturer’s recommendations. The same ligand density was used throughout all the experiments. Following immobilization, 1M ethanolamine pH 8.5 was injected over both channels for 10 min at 25 μL/min to block any activated groups on the gold chip. A channel without clathrin immobilization was used as a reference. All experiments were performed at 25°C at a flow rate of 35 μL/min. PBST (pH 7.4) was used as a running buffer. mCherry-CBP fusion proteins were buffer exchanged into running buffer using 20K MWCO Pierce concentrators and serially diluted to obtain final concentrations. All samples were run in duplicate and repeated twice. Data are represented as mean ± sd (n=3). The gold chip was regenerated using a 50 μL injection of 10 mM glycine, pH 2 to remove bound analyte. Binding data were analyzed in Scrubber (BioLogic Software). A 1:1 binding ratio Hill model was used to fit the binding interaction data.
2.8 Förster Resonance Energy Transfer (FRET) assay
mCherry-Cbox and GFP-Wbox were pre-associated with clathrin proteins and serially diluted in mouse serum (50%, 40%, 33%, 25%, 10%, and 5%). The samples were incubated at 37°C to mimic physiological conditions and analyzed in triplicate at each serum concentration. GFP was excited at 488 nm, and the fluorescence was measured at 610 nm (the emission of mCherry). The measurements were performed on a FilterMax F5 multi-mode microplate reader by molecular devices (Sunnyvale, CA).
2.9 In vitro loading assay
The theoretical loading efficiency was calculated using the number of molecules present in the incubation reaction. A known amount of clathrin was mixed simultaneously with varying amounts of molar excess of mCherry-Cbox and GFP-Wbox. The reaction mixture was incubated at room temperature with rocking for 48 h. The unbound fluorescent protein was removed by a concentrating tube equipped with a 150 kDa molecular weight cut-off membrane. The concentration of the fluorescent protein was quantified using a standard curve and the equation y=mx+b where y is the fluorescent intensity and x is the concentration of the protein. The following equation was then used to calculate the loading efficiency for mCherry and GFP to clathrin triskelions:
Where MWC and MWFP are the molecular weight for clathrin and the fluorescent proteins, respectively, CC and CFP are the concentrations of clathrin and fluorescent protein, respectively, and nC and nFP are the number of binding sites for clathrin and fluorescent protein, respectively (i.e. 3 for clathrin and 1 for the fluorescent protein).
2.10 In vivo pharmacokinetics analysis
Male CD-1 mice (6–8 weeks) were obtained from Charles River Laboratories (Wilmington, MA) and dosed with 15 mg/kg mCherry or GFP by tail-vein injections. 50 ul blood samples were taken at 5, 10, 15, 30, 45, 60 min, 2, 3, 4, 6, 8, 12, and 24 h by submandibular bleeding. A serial sampling strategy was implemented. Blood samples were spun down at 3,000 rpm for 10 min and the serum was collected. mCherry, GFP, and Dylight 594 were quantified by fluorescence measurements with a FilterMax F5 microplate reader (Molecular Devices). All animal experiments were approved by the Institutional Animal Care and Use Committee of the University at Buffalo.
2.11 Data analysis and statistics
Parameter estimates of a 2-compartment pharmacokinetic model were analyzed using the Adapt 5 (BMSR, Los Angeles, CA) maximum likelihood algorithm. Non-compartmental analysis (NCA) was performed in Phoenix WinNonlin (Certara, Princeton, NJ). AUC calculations were normalized to the injected dose. Comparisons between groups were analyzed using unpaired t-test. All statistical analyses were performed in Prism 5 (GraphPad Software, La Jolla, CA).
3. Results
3.1. Triskelion isolation and characterization
Clathrin and accessory proteins were isolated from bovine brain using differential centrifugation [8, 17]. A single band corresponding to 180 kDa clathrin triskelions was obtained, as shown by the SDS-PAGE analysis (Figure 1A). The identity of the clathrin triskelions was confirmed by western blotting using anti-clathrin heavy chain antibody (Figure 1B). The successful isolation of the clathrin triskelions was further confirmed by transmission electron microscopy (TEM). The TEM image shows the typical three-legged structure of the clathrin triskelions [7]. Each leg is ~45–50 nm in length (Figure 1C).
Figure 1.
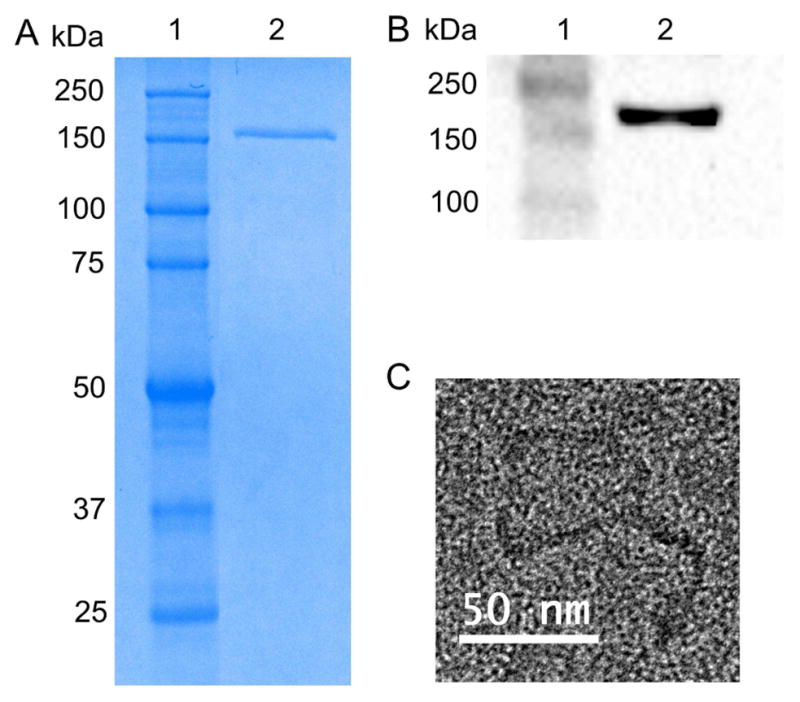
Clathrin isolation from bovine brains. (A) SDS-PAGE of the triskelion sample (MW ~180 kDa) on a 9% gel: ladder (lane 1), clathrin triskelion (lane 2). (B) A western blot analysis confirms the presence of clathrin in the sample. An anti-clathrin heavy chain antibody was used to detect the clathrin triskelion: ladder (lane 1), clathrin triskelion (lane 2). (C) TEM image of a clathrin triskelion.
3.2. Rational design and expression of mCherry-CBP and GFP-CBP fusion proteins
The N-terminal structure, commonly referred to as the clathrin terminal domain (CTD), has a seven-blade β-propeller conformation (Figure 2A) [18] with three unique binding sites. This includes the well-characterized clathrin-binding domain (C-box: located between blades I and II), additional sites that have been identified at distinct CTD locations (Figure 2B) [19]. To identify the most suitable CBD for dual display of proteins on clathrin triskelions, the binding affinity of three different peptides derived from accessory protein sequences shown to bind to clathrin triskelions were assessed [20–23]: (i) the LLDLD-motif, also termed C-box, derived from amphiphysin and known to bind between blades I and II of the β-propeller; (ii) the PWDLW motif (W-box) derived from SNX9 and amphiphysin that binds to the top of the β-propeller; and (iii) the LLGDL motif (R-box) found in β-arrestin that binds to the pocket present between blades IV and V (Figure 2A&B). These peptide binding motifs were linked to mCherry and GFP to obtain the mCherry-Cbox, mCherry-TriCBP, and the GFP-Wbox fusion proteins (Figure 2C).
Figure 2.

(A) A schematic representation of the secondary structure of a clathrin terminal domain. Roman numerals correspond to the blade number of the β-propeller (the PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC) and the PDB code 2XZG [24]. (B) Known binding sites on the clathrin terminal domain. (C) Schematic representation of the mCherry-Cbox, GFP-Wbox, and the mCherry-Tri-CBP. TCS represents the thrombin cleavage site and H6 stands for the hexa-histidine tag. The linker region is composed of glycine and serine repeats (SGGSGG)n. Detailed nucleotide and amino acid sequences for the constructs used in the study are shown in the supporting information, Table S1 and Table S2.
Computer-aided protein-protein docking simulations (SYBYL) were used to rationally design a Tri-CBP to interact with the three binding sites of the clathrin triskelion terminal domain. The linker lengths [SGGSGG]n were optimized to provide sufficient flexibility between the peptide-binding domains. The protein-docking algorithm Surflex-Dock in SYBYL was used to visualize binding interactions between the mCherry-CBP fusions to the CTD at the C-box and W-box domains (Figure 3).
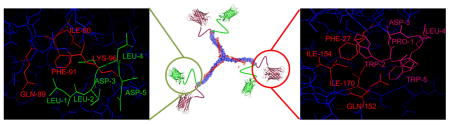
Figure 3.

(A&B) Surface and stick presentation of the C-box CBP (green) binding to the clathrin terminal domain (blue). The images were generated from the crystal structure RCSB PDB ID: 2XZG. (C&D) Surface and stick representations depict the binding of the W-box (pink; interacting amino acids shown in red) domain to the clathrin terminal domain (blue). Speckled light blue is the corresponding binding region. Docking and imaging rendering were performed in SYBYL.
Ile80, Gln89, Phe91, and Lys96 contribute to binding of the C-Box (LLDLD) via hydrophobic interactions but also hydrogen bonding and electrostatic interactions; Phe27, Gln152, Ile154, and Ile170 contribute at the W-box domain (PWDLW) via predominantly hydrophobic interactions but also through hydrogen bonding and electrostatic interactions; Arg188 (R), and Gln 192 contribute to binding of the R-box domain [25].
The purified fusion proteins were analyzed by SDS-PAGE (Figure 4A&B) and mass spectrometry to confirm their molecular weights (Figure S1&4C). The molecular weights of the fusion proteins were in good agreement with the expected molecular weights. The expected mCherry-Cbox molecular weight was 29.8 kDa and the molecular weight determined by mass spectrometry was 29.6 kDa, which also agreed well with SDS-PAGE. Although the mCherry- TriCBP protein ran higher than expected on the SDS-PAGE, mass spectrometry results show major peaks at 35.2 kDa that match well with the expected molecular weight. The SDS-PAGE result is most likely due to amino acid composition of the protein [26]. The mCherry-TriCBP and the GFP-Wbox also exhibited comparable theoretical and experimentally determined molecular weights. Genetic modifications and attachment to the clathrin triskelions did not affect the fluorescence activity of the fluorescent proteins. The emission spectra of the mCherry, mCherry-Cbox, and mCherry triskelions clearly overlay, indicating no change in the activity of the fluorescent proteins. The same results were obtained with GFP, GFP-Wbox, and GFP-Wbox attached to the clathrin triskelions (supporting information Figure S2).
Figure 4.

(A) SDS-PAGE of the mCherry-CBP fusion construct loaded on a 12% gel: ladder (lane 1), unmodified mCherry (lane 2), mCherry-Cbox (lane 3), mCherry-Tri-CBP (lane 4) (B) SDS-PAGE of the GFP-Wbox fusion constructs loaded on a 12% gel: ladder (lane 1), GFP-Wbox (lane 2), unmodified GFP (lane 3). (C) The molecular weights of mCherry, mCherry-Cbox, mCherry-TriCBP, GFP, and GFP-Wbox constructs as determined by mass spectrometry (supporting information, Figure S1).
3.3. Determining binding affinity via surface plasmon resonance
The binding affinities of the mCherry-CBP fusion proteins were determined by SPR. A range of CBP concentrations were assayed to determine binding interactions with the clathrin triskelions (Figure 5A–D). Binding interactions were fitted using a Hill equation or modified Hill equation (for the Tri-CBP). No binding was observed when unmodified mCherry and GFP were assayed with only a negligible response signal detected (data not shown). The mCherry-CBP fusion protein interacted with the clathrin triskelions by molecular epitope recognition. The modified Hill equation described two affinity protein-protein interaction characteristics of the Tri-CBP (equation shown in the supporting information, Figure S3) with a first KD1 of ~470 nM and a second KD2 of ~274 μM for the R-box domain (Figure 5C), confirmed by SPR data for the single R-box domain (287 μM) (Figure 5D). The estimated binding affinity of the C-box domain was 40 μM, consistent with previous reports (~22 μM) [27]. Binding was increased ~85-fold with the rationally designed Tri-CBP. The GFP fusion protein targeting the W-box was also generated and was found to bind to clathrin with a KD of 3.9 μM (Figure 5A, Figure S4).
Figure 5.

Binding affinity of the CBPs to the clathrin triskelions: (A) GFP-Wbox. (B) mCherry-Cbox (C) mCherry-TriCBP (D) mCherry-Rbox. Data are represented as mean ± sd (n=3).
3.4. Attachment of mCherry-Cbox and GFP-Wbox fusion protein to clathrin triskelions
FRET was used to assess overall loading efficiency of mCherry-Cbox and GFP-Wbox fusion proteins to the clathrin triskelions (Figure 6A). Control experiments showed that no FRET signal was obtained when clathrin triskelions were incubated with mCherry and GFP without any binding peptides. When ~90% of the sites were occupied, the FRET signal was 5.5E+07. The FRET signal decreased by half when only 50% of the sites were occupied (Figure 6B). Loading capacity was controlled by modulating the amount of mCherry-Cbox and GFP-Wbox added to the clathrin triskelions. Loading efficiency varied between 10.7% ± 2.7% and 82.9 ± 11.8% (Figure 6C). Together with the SPR analysis, these results demonstrate that both the mCherry-Cbox and the GFP-Wbox bound to the clathrin N-terminus in a site-specific manner. Moreover, controlling ligand molarity modulated the loading efficiency.
Figure 6.

(A) mCherry and GFP fluorescent protein FRET pair spectral profiles. (B) FRET analysis of ~100% attachment of mCherry-Cbox/GFP-Wbox, ~50% attachment of mCherry-Cbox/GFP-Wbox, and 0% attachment of unmodified mCherry and GFP (C) Loading efficiency of mCherry-Cbox and GFP-Wbox simultaneously attached to clathrin triskelions. (D) Stability of GFP-Wbox and mCherry-Cbox simultaneously attached to clathrin triskelions in the presence of serum. (C&D) Data are represented as mean ± sd (n=3).
3.5. Stability in presence of serum
FRET was used to assess the stability of the clathrin bound mCherry-Cbox and GFP-Wbox under physiological conditions. Clathrin triskelions containing bound mCherry-Cbox and GFP-Wbox were incubated at 37°C with increasing serum concentrations to evaluate the binding stability (Figure 6D). mCherry-Cbox and GFP-Wbox attached to clathrin triskelions displayed high stability when incubated with increasing concentrations of serum. After 24h of incubation at 37°C in the presence of 50% serum, only ~20% of the mCherry-Cbox and GFP-Wbox were competed off the clathrin triskelions.
3.6. In vivo PK studies
To assess the stability of the clathrin bound mCherry-Cbox and GFP-Wbox in vivo, the circulating half-lives of the unmodified fluorescent proteins and fusion proteins were determined in male CD-1 mice. Serum data was fit to a two-compartment PK model (Figure 7). The half-lives of unmodified and fusion fluorescent proteins were consistent with the circulating half-life of mKate a fluorescent protein of similar molecular weight [28]. The area under the curve (AUC) was calculated using the linear trapezoidal method for all samples, terminal half-lives were calculated using non-compartmental analysis, and the circulating half-lives were determined using the two-compartment PK model (Table 1). Attachment of fluorescent proteins to clathrin significantly increased exposure compared to the unmodified protein by at least two-fold.
Figure 7.

CD-1 mice (n=3) were dosed by tail-vein injections with 15 mg/kg of recombinant fluorescent proteins attached to clathrin. (A) Serum time course of unmodified mCherry, mCherry-Cbox, and mCherry-Cbox attached to clathrin. (B) Serum time course of unmodified GFP, GFP-Wbox, and GFP-Wbox attached to clathrin. Data are presented as mean ± sd.
Table 1.
Circulating half-lives, dose normalized AUC, and terminal half-lives of unmodified fluorescent proteins, CBP fluorescent proteins, and the fluorescent protein-clathrin conjugates. Statistical analysis was performed by means of an unpaired two-tailed t-test with *p < 0.05, **p < 0.01, and ***p < 0.001. In all statistical analyses the CBP attached to clathrin was compared to the corresponding CBPs.
| Protein Construct | Half-Life ± SE (min) | Dose Normalized AUC ± SE (min/mL) | Terminal Half-Life ± SE (min) |
|---|---|---|---|
| Clathrin triskelions | 53.5 ± 12.2 | 21.5 ± 1.4 | 597 ± 87.4 |
| Unmodified mCherry | 10.1 ± 1.8 | 4.9 ± 0.2 | 82.4 ± 10.2 |
| Unmodified GFP | 5.8 ± 2.3 | 4.5 ± 0.2 | 15.8 ± 1.0 |
| GFP-Wbox | 6.4 ± 0.4 | 4.6 ± 0.2 | 51.6 ± 5.0 |
| mCherry-Cbox | 4.3 ± 0.2 | 6.0 ± 0.6 | 102 ± 17.5 |
| mCherry-Cbox Clathrin | 17.1 ± 0.6*** | 11.4 ± 0.7*** | 1,130 ± 401** |
| GFP-Wbox Clathrin | 28.1 ± 0.4*** | 11.4 ± 0.8*** | 733 ± 108 ** |
The model-estimated total circulating half-life increased by ~4-fold when GFP-Wbox or mCherry-Cbox was attached to the clathrin triskelions. Exposure increased 2-fold for the GFP-Wbox and mCherry-Cbox attached to clathrin and the terminal half-life was significantly increased at least 12-fold compared to the fusion proteins alone (Figure 7 & Table 1). The concentration time courses of GFP and mCherry were qualitatively similar with comparable AUCs, half-lives, and terminal half-lives (Figure 7A&B, and Table 1). When mCherry-Cbox and GFP-Wbox were simultaneously attached to the clathrin triskelions (Figure 8A&B), both displayed similar stability in circulation. After an initial dissociation from the clathrin triskelions of about ~70–75%, the fraction of mCherry-Cbox and GFP-Wbox associated with the triskelions remained stable for at least 12h. Both mCherry-Cbox and GFP-Wbox remained associated to the clathrin triskelions at comparable percentages (Figure 8B). No statistical differences between mCherry-Cbox and GFP-Wbox were observed (two-tailed unpaired T-test).
Figure 8.

(A) Serum time course of mCherry-Cbox and GFP-Wbox associated with clathrin triskelions. Free clathrin was labeled with DyLight 594 for quantification. (B) Percentage of mCherry-Cbox and GFP-Wbox simultaneously associated with clathrin triskelions as a function of time. No statistical differences between mCherry-Cbox and GFP-Wbox were observed (two-tailed unpaired T-test, p=0.33).
4. Discussion
Clathrin triskelions represent a versatile scaffold for simultaneous protein attachment and display. Here, we demonstrate the successful use of clathrin triskelions for the dual display of two proof-of-concept proteins. Via molecular epitope recognition we were able to control the spatial and stoichiometric attachment of GFP and mCherry to clathrin triskelions.
We rationally designed and developed peptide sequences that allowed simultaneous attachment of recombinant proteins or peptides to the clathrin triskelions by molecular epitope recognition. In contrast to coupling strategies that covalently attach proteins via amine groups such as lysine conjugation, binding by molecular epitope recognition allows the precise control of protein orientation and prevents inadvertent cross-linking and the disruption of protein bioactivity sometimes caused by coupling to multiple lysines [15, 16].
Peptide sequences were selected to bind to the C-box and W-box of the clathrin triskelions for dual display of mCherry and GFP. Although, these sites are in close proximity (only 25 Å apart), 80–90% of all available sites were occupied when loaded with mCherry-Cbox and GFP-Wbox. Independent loading of the mCherry-Cbox and the GFP-Wbox shows that the loading efficiency is comparable to the loading efficiency when both the GFP-Wbox and mCherry-Cbox were co-incubated (supporting information Table S3). This indicates that both sites are suitable for the simultaneous attachment of proteins and that steric hindrance is not a major factor. The binding affinities of the peptide sequences tested were in the low micromolar range (4 μM and 40 μM for W-box and C-box, respectively). As shown by SPR, these unique binding domains had distinct binding affinities that ranged from 4 μM for the W-box to 287 μM for the R-box. Using a rational design approach, we engineered a Tri-CBP that could bind to three distinct clathrin triskelion sites simultaneously: the C-box, the W-box, and the R-box. The crystal structure of clathrin was used to optimize the linker lengths connecting the three peptide thereby allowing sufficient flexibility. For our Tri-CBP fusion protein, which encompassed three distinct binding motifs on the clathrin N-terminus, at least two distinct binding interactions occurred, most likely because all three sites interacted synergistically to produce a KD1 of 470 nM for the de novo designed Tri-CBP. This represents an 85-fold increase in binding affinity compared to the single C-box domain. Miele et al. reported a peptide containing both the C-box and W-box bound with an affinity of about 5 μM [27]. Interestingly, our data suggest that a second distinct binding interaction occurs, which is most likely mediated by the R-box (calculated KD2 = 274 μM). An mCherry fusion construct containing solely the R-box was assayed by SPR to establish whether this second interaction site was indeed characteristic of the R-box domain. Consistent with our hypothesis, the R-box domain bound to clathrin with an estimated binding affinity of about 287 μM. Although, we could increase the binding affinity to clathrin triskelions, this did not further improve the circulating half-life of mCherry bound to clathrin trikelions (supporting information Figure S6). The R-box was not selected for further validation and characterization due to its relatively poor binding affinity (KD = 287 μM) to the clathrin triskelion. Based on the fundamental understanding of binding phenomena, a binding affinity in the high micromolar range is likely to provide minimal interactions at physiological protein concentrations in vivo [29]. Thus, we chose the C-box and W-box for simultaneous display of the GFP and mCherry.
Loading experiments demonstrated that adjusting the amount of proteins incubated with the clathrin triskelions controlled the loading efficiency. When the clathrin triskelions were incubated with a 6 molar excess of mCherry and GFP, 83–95% of all available sites were occupied. 20–30% site occupancy was obtained with a 2-molar excess of the mCherry-Cbox or GFP-Wbox, and a 10–20% occupancy was obtained when a 1:1 molar ratio was used.
Our PK analysis showed that our approach represents an effective site-specific binding strategy that does not require chemical modification and that also effectively increases the circulating half-life. Furthermore, clathrin triskelions mediated a significant increase in exposure of the attached protein cargo, calculated as the AUC. There was an almost 2-fold increase in the mCherry-Cbox/clathrin triskelion AUC compared to the unattached mCherry-Cbox, an 11-fold increase in terminal half-life (1,130 min vs. 102 min; p<0.01), and almost a 4–fold increase in total circulating half-life (17.1 min vs. 4.29 min; p<0.001). A similar trend was observed for the GFP-Wbox attached to the clathrin triskelions. A 4-fold increase in total circulating half-life (28.1 min vs. 6.42 min; p<0.05), a 2.5-fold increase in AUC, and a 14-fold increase in terminal half-life (733 min vs. 51.6 min; p<0.01) were observed. Both mCherry and GFP have a molecular weight of ~28kDa and are thus liable to rapid renal elimination after systemic administration. Here, the clathrin triskelions increase the overall molecular weight of mCherry and GFP to above the renal elimination threshold (30–50 kDa) [30, 31]. This is likely to have protected the fluorescent proteins from rapid renal elimination and increased the overall circulating half-life of the fluorescent protein (Figure 7 & supporting information Figure S5).
Further, molecular epitope recognition allows the simultaneous display of two proteins precisely positioned at the N-termini of the clathrin triskelions. Clathrin associated mCherry-Cbox and GFP-Wbox circulated longer than unattached proteins: after 3h ~28% of the mCherry-Cbox and GFP-Wbox remained associated with the clathrin triskelions. After 12h in circulation, ~10% of the mCherry-Cbox and GFP-Wbox remained attached. Both mCherry-Cbox and GFP-Wbox remained associated with the clathrin triskelions at the same percentages. Taken together, these data strongly indicate that these binding sites are suitable for the simultaneous co-display of proteins. While in vitro serum stability data showed that 80% of the proteins remained associated with the clathrin triskelions even after 24h of incubation, the dissociation of the proteins from the triskelions occurred faster in vivo. The dissociation could be caused by the presence of naturally occurring proteins with clathrin triskelions binding sites, such as amphiphysin and SNX9, both of which are present on plasma membranes [32, 33]. Longer retention of the mCherry-Cbox and the GFP-Wbox may be necessary in some applications, which may be achievable by developing peptide motifs that show a higher affinity for the clathrin triskelions, possibly obtained through phage display.
Site-specific attachment of clathrin triskelions via molecular epitope recognition has many implications for protein display: 1) dual targeting of surface receptors, 2) avidity interactions, 3) cancer vaccination (by displaying either two tumor antigens or a tumor antigen and an immune-adjuvant), and 4) induction of receptor di-, tri- or multimerization. The ability of single triskelions to form clathrin cages of different sizes and shapes further extends the versatility of this approach as a drug delivery platform. The symmetry of the assembled cages allows for spatially controlled protein, peptide, and ligand attachment in an organized display. Viruses and virus-like particles (VLPs) have shown that highly ordered protein display in the form of antigens is vital for stimulating an immune response. Ordered display by clathrin cage technology, therefore, holds promise for inducing immune responses for vaccination purposes.
5. Conclusion
Here we demonstrated the suitability of clathrin triskelions as a versatile platform for simultaneous display and in vivo delivery of different proteins. Although GFP and mCherry were used as proof-of-concept, the platform is generalizable for the delivery of other therapeutic proteins, targeting ligands, small molecules, and macromolecules. The fluorescent proteins were simultaneously attached to clathrin by molecular epitope recognition and displayed enhanced in vivo characteristics compared to free proteins. The clathrin triskelion represents a biomimetic nanocarrier that exhibits organization, easy attachment, and multiple molecular epitopes capable of attaching distinct proteins. This highly adaptable technology shows enormous potential for site-specifically controlling the orientation of an attached protein to a clathrin scaffold via molecular epitope recognition. This could have important applications in protein delivery and cancer vaccination where a controlled, multivalent, and oriented protein display is critical.
Supplementary Material
Acknowledgments
We thank Dr. Brian Kay (University of Illinois at Chicago) and his lab for providing training in cloning and protein expression techniques. We would like to thank Dr. Joseph Balthasar (SUNY Buffalo) for generously sharing his Reichert SPR instrument with us. We thank Patrick Glassman (SUNY Buffalo) for technical assistance with the SPR measurements and Kevin Welle (University of Rochester) for assisting us with mass spectrometry measurements. JN acknowledges support by the NIH through awards 1R21EB021454-01 and 1R21HL126082-01A1. SF is supported by the Allan Barnett Fellowship.
Footnotes
Author contributions
MBD and JN conceived and designed the experiments. MBD, SWF, ML, and DCP performed the experiments. MBD, SWF, and JN analyzed the data. MBD, SPK, and JN wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aubin-Tam ME, Hamad-Schifferli K. Structure and function of nanoparticle-protein conjugates. Biomed Mater. 2008;3:034001. doi: 10.1088/1748-6041/3/3/034001. [DOI] [PubMed] [Google Scholar]
- 2.Uchida M, Klem MT, Allen M, Suci P, Flenniken M, Gillitzer E, et al. Biological containers: Protein cages as multifunctional nanoplatforms. Adv Mater. 2007;19:1025–42. [Google Scholar]
- 3.Lucon J, Qazi S, Uchida M, Bedwell GJ, LaFrance B, Prevelige PE, et al. Use of the interior cavity of the P22 capsid for site-specific initiation of atom-transfer radical polymerization with high-density cargo loading. Nat Chem. 2012;4:781–8. doi: 10.1038/nchem.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venter PA, Dirksen A, Thomas D, Manchester M, Dawson PE, Schneemann A. Multivalent Display of Proteins on Viral Nanoparticles Using Molecular Recognition and Chemical Ligation Strategies. Biomacromolecules. 2011;12:2293–301. doi: 10.1021/bm200369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ungewickell E, Branton D. Assembly Units of Clathrin Coats. Nature. 1981;289:420–2. doi: 10.1038/289420a0. [DOI] [PubMed] [Google Scholar]
- 6.Fotin A, Cheng Y, Sliz P, Grigorieff N, Harrison SC, Kirchhausen T, et al. Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature. 2004;432:573–9. doi: 10.1038/nature03079. [DOI] [PubMed] [Google Scholar]
- 7.Heuser J, Kirchhausen T. Deep-Etch Views of Clathrin Assemblies. Journal of Ultrastructure Research. 1985;92:1–27. doi: 10.1016/0889-1605(85)90123-5. [DOI] [PubMed] [Google Scholar]
- 8.Vigers GPA, Crowther RA, Pearse BMF. 3-Dimensional Structure of Clathrin Cages in Ice. Embo Journal. 1986;5:529–34. doi: 10.1002/j.1460-2075.1986.tb04242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirchhausen T. Clathrin. Annual Review of Biochemistry. 2000;69:699–727. doi: 10.1146/annurev.biochem.69.1.699. [DOI] [PubMed] [Google Scholar]
- 10.Sorkin A. Cargo recognition during clathrin-mediated endocytosis: a team effort. Current Opinion in Cell Biology. 2004;16:392–9. doi: 10.1016/j.ceb.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Jin AJ, Nossal R. Rigidity of triskelion arms and clathrin nets. Biophysical Journal. 2000;78:1183–94. doi: 10.1016/S0006-3495(00)76676-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schoen AP, Schoen DT, Huggins KNL, Arunagirinathan MA, Heilshorn SC. Template Engineering Through Epitope Recognition: A Modular, Biomimetic Strategy for Inorganic Nanomaterial Synthesis. Journal of the American Chemical Society. 2011;133:18202–7. doi: 10.1021/ja204732n. [DOI] [PubMed] [Google Scholar]
- 13.Huggins KNL, Schoen AP, Arunagirinathan MA, Heilshorn SC. Multi-Site Functionalization of Protein Scaffolds for Bimetallic Nanoparticle Templating. Advanced Functional Materials. 2014;24:7737–44. [Google Scholar]
- 14.Dannhauser PN, Platen M, Boning H, Schaap IA. Durable protein lattices of clathrin that can be functionalized with nanoparticles and active biomolecules. Nat Nanotechnol. 2015;10(11):954–957. doi: 10.1038/nnano.2015.206. [DOI] [PubMed] [Google Scholar]
- 15.Kalkhof S, Sinz A. Chances and pitfalls of chemical cross-linking with amine-reactive N-hydroxysuccinimide esters. Anal Bioanal Chem. 2008;392:305–12. doi: 10.1007/s00216-008-2231-5. [DOI] [PubMed] [Google Scholar]
- 16.Salvucci ME. Covalent modification of a highly reactive and essential lysine residue of ribulose-1,5-bisphosphate carboxylase/oxygenase activase. Plant Physiol. 1993;103:501–8. doi: 10.1104/pp.103.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crowther RA, Pearse BMF. Assembly and Packing of Clathrin into Coats. Journal of Cell Biology. 1981;91:790–7. doi: 10.1083/jcb.91.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ter Haar E, Musacchio A, Harrison SC, Kirchhausen T. Atomic structure of clathrin: A beta propeller terminal domain joins an alpha zigzag linker. Cell. 1998;95:563–73. doi: 10.1016/s0092-8674(00)81623-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collette JR, Chi RJ, Boettner DR, Fernandez-Golbano IM, Plemel R, Merz AJ, Geli MI, Traub LM, Lemmon SK. Clathrin Functions in the Absence of the Terminal Domain Binding Site for Adaptor-associated Clathrin-Box Motifs. Mol Biol Cell. 2009;20:3401–3413. doi: 10.1091/mbc.E08-10-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dell’Angelica EC, Klumperman J, Stoorvogel W, Bonifacino JS. Association of the AP-3 adaptor complex with clathrin. Science. 1998;280:431–4. doi: 10.1126/science.280.5362.431. [DOI] [PubMed] [Google Scholar]
- 21.Ramjaun AR, McPherson PS. Multiple amphiphysin II splice variants display differential clathrin binding: Identification of two distinct clathrin-binding sites. Journal of Neurochemistry. 1998;70:2369–76. doi: 10.1046/j.1471-4159.1998.70062369.x. [DOI] [PubMed] [Google Scholar]
- 22.Kang DS, Kern RC, Puthenveedu MA, von Zastrow M, Williams JC, Benovic JL. Structure of an Arrestin2-Clathrin Complex Reveals a Novel Clathrin Binding Domain That Modulates Receptor Trafficking. Journal of Biological Chemistry. 2009;284:29860–72. doi: 10.1074/jbc.M109.023366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willox AK, Royle SJ. Functional Analysis of Interaction Sites on the N-Terminal Domain of Clathrin Heavy Chain. Traffic. 2012;13:70–81. doi: 10.1111/j.1600-0854.2011.01289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, et al. Role of the Clathrin Terminal Domain in Regulating Coated Pit Dynamics Revealed by Small Molecule Inhibition (vol 146, pg 471, 2011) Cell. 2011;146:841. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 25.Lemmon SK, Traub LM. Getting in Touch with the Clathrin Terminal Domain. Traffic. 2012:13. doi: 10.1111/j.1600-0854.2011.01321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guan Y, Zhu Q, Huang D, Zhao S, Jan Lo L, Peng J. An equation to estimate the difference between theoretically predicted and SDS PAGE-displayed molecular weights for an acidic peptide. Scientific Reports. 2015;5:13370. doi: 10.1038/srep13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adriana E, Miele PJW, Evans Philip R, Traub Linton M, Owen David J. Two distinct interaction motifs in amphiphysin bind two independent sites on the clathrin terminal domain β-propeller. Nature Structural and Molecular Biology. 2004;11:242–8. doi: 10.1038/nsmb736. [DOI] [PubMed] [Google Scholar]
- 28.Sockolosky JT, Kivimae S, Szoka FC. Fusion of a short peptide that binds immunoglobulin G to a recombinant protein substantially increases its plasma half-life in mice. PLoS One. 2014;9:e102566. doi: 10.1371/journal.pone.0102566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gemmecker G. NMR Spectroscopy in Drug Development and Analysis. Wiley-VCH Verlag GmbH; 2007. NMR as a Tool in Drug Research; pp. 135–54. [Google Scholar]
- 30.Fox ME, Szoka FC, Frechet JM. Soluble polymer carriers for the treatment of cancer: the importance of molecular architecture. Acc Chem Res. 2009;42:1141–51. doi: 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Venkatachalam MA, Rennke HG. The structural and molecular basis of glomerular filtration. Circ Res. 1978;43:337–47. doi: 10.1161/01.res.43.3.337. [DOI] [PubMed] [Google Scholar]
- 32.Shin N, Ahn N, Chang-Ileto B, Park J, Takei K, Ahn SG, et al. SNX9 regulates tubular invagination of the plasma membrane through interaction with actin cytoskeleton and dynamin 2. J Cell Sci. 2008;121:1252–63. doi: 10.1242/jcs.016709. [DOI] [PubMed] [Google Scholar]
- 33.Zanner R, Gratzl M, Prinz C. Expression of the endocytic proteins dynamin and amphiphysin in rat gastric enterochromaffin-like cells. J Cell Sci. 2004;117:2369–76. doi: 10.1242/jcs.01091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.