

ABSTRACT
Background: Carbon monoxide (CO) was previously only considered as a highly toxic pollutant since it binds to hemoglobin with high affinity. Recently, however, it has been recognized as a signaling molecule with regulatory roles in many physiological and pathophysiological processes within the cardiovascular system. The aim of this study was to clarify the behavior of CO in patients with acute coronary syndrome (ACS).
Methods: We assessed 235 patients with suspected ACS, 98 smokers (88 male, 62 ± 14 years) and 137 nonsmokers (77 male, 72 ± 13 years), who had undergone emergent cardiac catheterization and blood sampling for calculation of carboxyhemoglobin (COHb). Patients were categorized into 4 groups: smoking patients with ACS (n=77), smoking patients without ACS (n=21), non-smoking patients with ACS (n=97), and non-smoker patients without ACS (n=40). We investigated whether biomarkers were related to COHb levels.
Results: LogCOHb was significantly higher in the smoking patients compared to non-smoking patients (0.30 ± 0.12 vs. 0.45 ± 0.18, P < 0.01). Interestingly, among the non-smoking patients, COHb was increased in the ACS patients compared to the non ACS patients (0.31 ± 0.12 vs. 0.25 ± 0.12 P < 0.01). In contrast, among the smoking patients, there was no difference in COHb between the ACS and non-ACS patients (0.45 ± 0.18 vs. 0.44 ± 0.18, n.s.). There were no correlations between COHb and any of the biomarkers.
Conclusions: These results suggest that endogenous CO may be useful to assess the risk of cardiovascular stress.
Keywords: carbon monoxide; acute coronary syndrome; nonsmoker, biomarker
ABBREVIATIONS
CO, carbon monoxide; COHb, carboxyhemoglobin; ACS, acute coronary syndrome; HO, heme oxygenase
INTRODUCTION
Carbon monoxide (CO), which was previously only considered as a highly toxic pollutant due to its high-affinity binding to hemoglobin1), has recently been recognized as a signaling molecule with regulatory roles in many physiological and pathophysiological processes within the cardiovascular, immune, and nervous systems2-5). The physiological signaling effects of CO involve modulation of soluble guanylate cyclase and subsequent upregulation of cyclic guanosine monophosphate (cGMP) production similar to those of nitric oxide (NO)6). CO is endogenously produced during the processes of heme catabolism via heme oxygenase (HO)7). Since protective roles of HO under various pathophysiological processes have been reported8,9), the roles of CO under these processes have been currently concerned10,11). Moreover, the vasodilatation properties of CO, which also has anti-apoptotic and anti-inflammatory potentials2,3), have been investigated in the cardiovascular system12). We previously reported that synergetic antioxidant and vasodilatory action of carbon monoxide attenuated angiotensin II-induced cardiac hypertrophic change13). Although the cytoprotective roles of CO has been investigated by in vitro studies using CO releasing molecules (11) and in vivo studies using high exogenous CO exposure14), the behavior of CO under pathophysiological conditions is still unknown. It has been widely reported that healthy smokers have high concentration levels of CO, which is measured by carboxyhemoglobin (COHb) (15), but the endogenous CO concentration levels of those with various pathophysiological conditions are unclear. In the present study, we clarified the behavior of CO in patients with acute coronary syndrome (ACS), and investigated whether CO could be a novel biomarker of ACS.
METHODS
Subject and study protocol
This study enrolled 235 consecutive patients who had visited the emergency room because of chest pain and underwent emergency cardiac catheterization at Fukushima Medical University Hospital between 2011 and 2013. COHb levels were measured by blood gas analysis (ABL-800 FLEX, Radiometer, Copenhagen, Denmark) which was using aortic blood sampling obtained from all participants. Patients with shock vital, performance of cardiopulmonary resuscitation, and performance of intubation were excluded. The presence of ACS was defined as coronary total occlusion or advanced coronary stenosis by emergency cardiac catheterization, and patients who were not recognized as having significant coronary stenosis were defined as non-ACS patients. We examined several biomarkers, such as creatinine kinase (CK), troponin I, brain natriuretic peptide (BNP), which were samples collected at the same time as blood gas collection. In addition, we investigated the coronary risk factors hypertension, diabetes mellitus, and dyslipidemia.
Hypertension was defined as the recent use of antihypertensive drugs, or the recent diagnosis of hypertension with a systolic pressure of ≥140 mmHg and/or diastolic blood pressure of ≥90 mmHg. Diabetes mellitus was defined as the recent use of insulin or antidiabetic drugs, the recent diagnosis of diabetes mellitus with a fasting blood glucose value of ≥126 mg/dl, and/or a hemoglobin A1c value (NGSP) of ≥6.5%. Dyslipidemia was defined as the recent use of cholesterol-lowering drugs, a recent diagnosis of dyslipidemia with a triglyceride value of ≥150 mg/dl, a low-density lipoprotein cholesterol value of ≥140 mg/dl, and/or a high-density lipoprotein cholesterol value of <40 mg/dl. A smoker was defined as a person who had smoked any number of cigarettes until onset of symptoms. A nonsmoker was defined as a person who had never smoked before, or had discontinued smoking since before visiting our hospital. Since smoking affected endogenous CO levels, we compared COHb between ACS patients and non-ACS patients within a nonsmoker population. Furthermore, we investigated whether biomarkers such as CK, troponin I, and BNP were associated with COHb levels. The protocol was approved by the Ethical Committee of Fukushima Medical University.
Statistical analysis
Data are expressed as mean±SD or number (%). Because COHb levels were not normally distributed, we made log transformation for COHb. Continuous variables were compared using an unpaired Student’s t-test as appropriate, and categorical data were analyzed with the chi-squared test. We compared carbon monoxide concentrations between ACS and non-ACS using multivariable-adjusted model in non-smokers and smokers, respectively. Spearman’s correlation coefficient was used to assess the relationship between COHb and biomarkers. The optimal cut off value of COHb for the diagnosis of ACS was assessed using the receiver operating characteristics (ROC) curve analysis. All statistical analyses were done using SPSS version 21 (SPSS, Chicago, IL, USA). A P value of <0.05 was considered statistically significant.
RESULTS
In patients who had been admitted with chest pain, the prevalence of ACS was 74.0% (174/235). Among these, 77.0% (134/174) were male, as shown in Table 1. In addition, the percentage of smokers among the ACS and non-ACS patients were 44.2% (77/174) , and 34.4% (21/61), respectively. Among both the ACS and non-ACS patients, the smokers were significantly younger than the nonsmokers and had a larger percentage of males. Furthermore, ACS patients who were smokers tended to have a higher body mass index than those who did not smoke. As for coronary risk factors, no significant differences were observed regarding the prevalence of hypertension and dyslipidemia between each group. The ACS patients had a high prevalence of diabetes mellitus compared with the non-ACS patients in both smokers and nonsmokers (Table 1).
Table 1.
Comparisons of clinical features among all patients.
| ACS | Non-ACS | P-value | |||
| Nonsmoker (n=97) |
Smoker (n=77) |
Nonsmoker (n=40) |
Smoker (n=21) |
||
| Age (years) | 72 ± 12†† | 62 ± 14** | 71 ± 16†† | 62 ± 17** | <0.001 |
| Male gender (n, %) | 61 (63) | 73 (95) | 16 (40) | 15 (71) | <0.001 |
| Body mass index (kg/cm2) | 23.6 ± 2.9†† | 25.6 ± 3.9** | 22.5 ± 3.1†† | 24.9 ± 15.6 | <0.001 |
| Co-morbidity | |||||
| Hypertension (n, %) | 62 (64) | 43 (56) | 30 (75) | 12 (57) | 0.215 |
| Diabetes (n, %) | 35 (36) | 30 (39) | 6 (15) | 2 (10) | 0.005 |
| Dyslipidemia (n, %) | 49 (51) | 38 (49) | 12 (30) | 6 (29) | 0.052 |
Data given as mean ± SD or n (%).
ACS, acute coronary syndrome.
*P<0.05 and **P<0.01 vs. nonsmoker ACS patients, †P<0.05 and ††P<0.01 vs. smoker ACS patients.
As shown in Figure 1, COHb was significantly higher in the smokers compared to nonsmokers (0.30 ± 0.12 vs. 0.45 ± 0.18, P < 0.01) in all groups. Figure 2 shows comparisons of COHb between ACS and non-ACS patients in the nonsmoker population. COHb was significantly higher in the ACS patients compared to non-ACS patient in nonsmoker population (0.31 ± 0.12 vs. 0.25 ± 0.12 P < 0.01). However, no difference was observed in COHb between the ACS and non-ACS patients in the smoker population (0.45 ± 0.18 vs. 0.44 ± 0.18, n.s.).
Fig. 1.
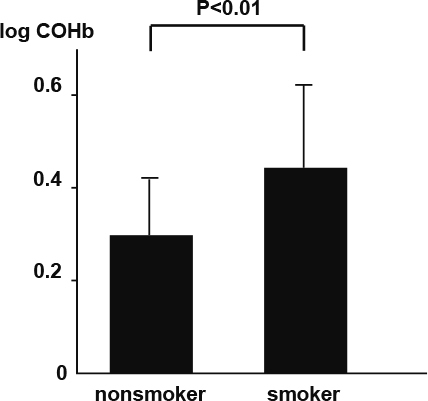
Comparison of COHb between smokers and nonsmokers in all subjects.
Fig. 2.
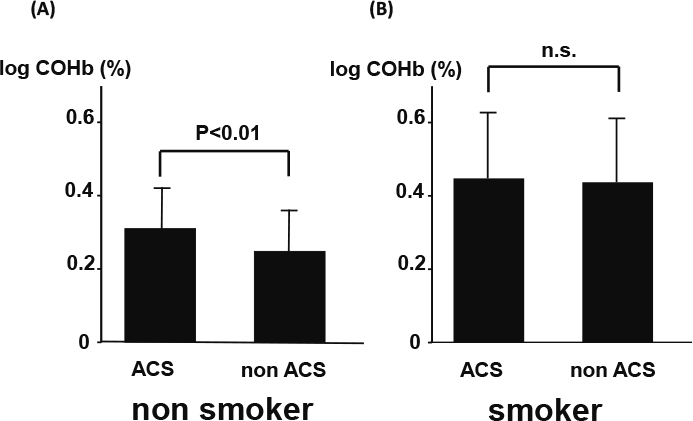
Comparison of COHb between ACS and non-ACS patients (A) nonsmoker and (B) smoker populations. There data were adjusted for gender, age and BMI.
The relationships between COHb and the biomarkers (troponin I, CK, BNP) are shown in Figure 3. There were no correlations between COHb and any of the biomarkers.
Fig. 3.
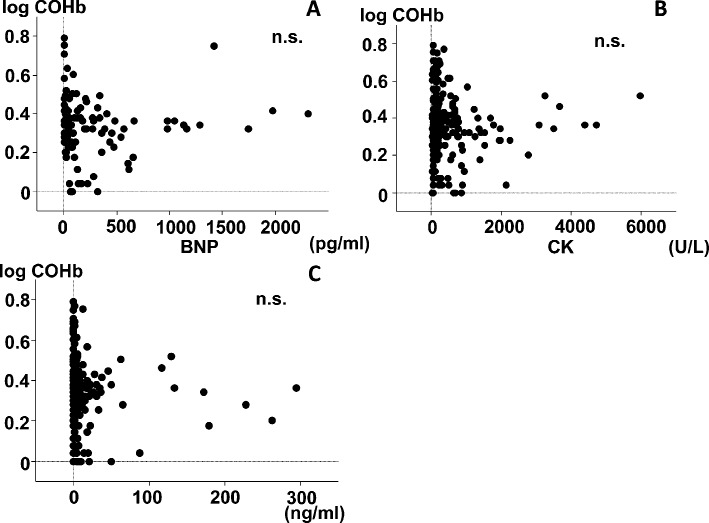
Relations between COHb and biomarkers in the nonsmoker population. (A) BNP, brain natriuretic peptide; (B) CK; creatinine kinase; (C) Troponin I
Figure 4 shows the ROC analysis for ACS diagnosis by COHb in nonsmokers, with an optimal cut off value of COHb at 0.95%. The area under the curve was 0.60, and the sensitivity and specificity were 69.1% and 55.0%, respectively.
Fig. 4.
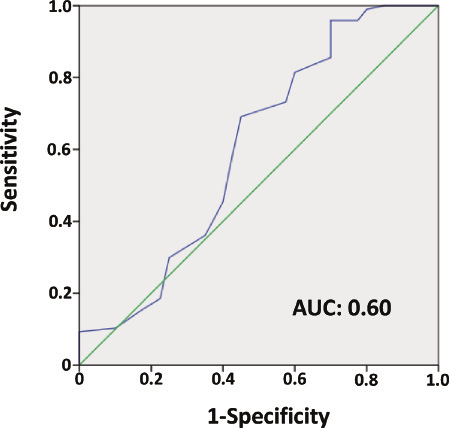
Receiver operating characteristic (ROC) curve analysis of COHb for diagnosis of acute coronary syndrome.
DISCUSSION
We have shown here for the first time that COHb, assessed using blood gas analysis, is significantly elevated in a nonsmoker population with ACS. CO is usually regarded as a toxic species that disrupts cellular respiration because CO binds to hemoglobin with high affinity (1). However, CO is produced endogenously in the process of heme catabolism by only HO, which is a heme-catabolizing enzyme that converts heme into biliverdin, iron, and CO. HO has two isoforms: HO-1, an inducible form, and HO-2, a constitutive form5). HO-1 expression is induced by various pathophysiological conditions16-18). Biliverdin, a byproduct of heme catabolism, is endogenously converted bilirubin, and protects tissues by antioxidative behavior19). CO, another product of heme catabolism, has profound signaling effects on intracellular signaling processes. The vasodilating properties of CO have been investigated in the cardiovascular system12,20). CO also has antiapoptotic2) and anti-inflammatory3) potentials. In addition, we showed previously that CO also had antioxidative behavior13).
Tobacco smoking is an important source of CO as a pollutant15). COHb commonly reaches a level of two or more percent in smokers and may even exceed 10 percent, compared to around one percent in nonsmokers15). In this study, COHb was increased in the smoker patients who were influenced strongly by exogenous CO, compared to nonsmoker patients (P < 0.01). Among the smoker patients, there was no difference in COHb between the ACS and non-ACS patients. In contrast, among the nonsmoker patients, COHb was increased in the ACS patients compared to the non-ACS patients (P < 0.05). Of the nonsmokers, COHb concentrations were more increased in the ACS patients than those in the non-ACS patients. Because CO is heavily influenced by tobacco smoking, the endogenous CO was masked with exogenous CO in the smoker patients.
A number of recent studies, both in vivo and in vitro, have reported that HO-1 and its product CO have a cytoprotective role against ischemic/reperfusion injury. Stein et al. reported that pre-treatment with CO-releasing molecules prior to coronary occlusion in mice markedly reduced infarct size10). The CO-releasing molecules triggered a cardioprotective signaling cascade that recruited the transcription factors NF-kB, STAT1/3, and Nrf2 with a subsequent increase in cardioprotective and anti-apoptotic molecules in the myocardium leading to a late preconditioning-mimetic infarct-sparing effect11). It has been reported that after exposure to CO had reached toxic levels, several cardiac manifestations, including arrhythmias and electrocardiographic alterations21), acute myocardial infarction22), takotsubo cardiomyopathy23), and cardiogenic shock24), occur. There are only a few studies that have investigated the relationship between cardiovascular disease and CO at levels non-toxic to the human body. Cheng et al. reported that high exhaled CO levels predicted the development of metabolic syndrome and future cardiovascular disease events25). Moreover, they suggested that elevated CO is a marker of greater subclinical cardiovascular disease burden and a potential key component in the progression from subclinical to clinical cardiovascular disease26). Both studies indicated that chronic sustentation of high CO levels was reflected in cardiovascular disease progression. In the present study, there are two reasons why CO was elevated at ACS onset. The first is that ACS patients have chronic elevation of endogenous CO concentration, which result in high COHb in the emergency room. The other reason is that acute reaction during myocardial ischemia elevates endogenous CO concentration, also resulting in high COHb at emergency room. Because we did not obtain blood gas analysis before ACS onset, the reason for elevated CO concentrations at onset was not clear.
Although the detailed mechanisms of association between the elevation of endogenous CO and the onset of ACS remain unclear, our study suggests that CO during the acute phase might provide important clues to the pathogenesis of myocardial salvage following ischemia. The mechanisms of how to elevate COHb concentrations and how to produce the cytoprotective effects in ACS patients are not revealed. Further investigation of oxidative stress, apoptosis and inflammation markers at the same time when blood gas analysis is obtained is considered necessary to reveal this mechanism. Therefore, the cut-off values required for CO to function as a cellular defense mechanism was not determinate in the present study. Generally, Trop I and CK are markers of myocardial damage used for the diagnosis of acute myocardial infarction and the estimate of infarction size27,28). Moreover, BNP is a marker of heart failure used to confirm diagnosis, measure the severity of left ventricular compromise, quantify the functional class, estimate the prognosis, predict future cardiac events29-32), and evaluate the efficacy of therapy. In this study no correlation between COHb and biomarkers troponin I, CK and BNP, was observed. Because the ROC curve used to identify ACS by COHb indicated that the sensitivity was 69.1% and specificity was 55.0%, we believe that COHb is not an effective biomarker for diagnosing ACS. While it has been clearly reported that troponin I is a main maker of acute myocardial infarction, there are quite a few cases where troponin I does not increase, such as in unstable angina pectoris, in which the myocardium does not come into necrosis, as well as in the super early phase of acute myocardial infarction. Therefore, in the present study we investigated ACS patients who were not only affected by acute myocardial infarction but also unstable angina pectoris. Some ACS patients with high COHb had low levels of troponin I, as shown in Fig. 3. Similar results were observed regarding CK and BNP levels. Assuming that CO was induced by HO-1 at the exact time of ACS onset, we believe that the heart is the first organ in human body in which CO concentration is elevated. Because we investigated by blood gas analysis collected from arterial blood in this study, it is considered that COHb measured by blood gas analysis was systemic CO concentration. Therefore, we suggest that systemic COHb is less accurate than regional COHb in cardiomyocytes and coronary arteries. The sensitivity and specificity of COHb by itself are less likely to make a diagnosis of ACS. However, it is suggested that COHb is treated as a cardiovascular stress marker because CO is induced by HO-1 under various cardiovascular stresses.
STUDY LIMITATIONS
The present study was a cross-section analysis of a single institution. The number of subjects was relatively small. Hence, prospective studies with a larger subject group are needed.
CONCLUSIONS
Our results suggest that endogenous CO may be useful to assess the risk against cardiovascular stress.
DISCLOSURES
Conflict of Interest: None declared
REFERENCES
- 1.Ernst A, Zibrak JD. Carbon monoxide poisoning. N Engl J Med, 339: 1603-1608, 1998. [DOI] [PubMed] [Google Scholar]
- 2.Brouard S, Otterbein LE, Anrather J, et al. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med, 192: 1015-1026, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Otterbein LE, Zuckerbraun BS, Haga M, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nature Med, 9: 183-190, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov, 9: 728-743, 2010. [DOI] [PubMed] [Google Scholar]
- 5.Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev, 57: 585-630, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Furchgott RF, Jothianandan D. Endothelium-dependent and -independent vasodilation involving cyclic GMP: relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessels, 28: 52-61, 1991. [DOI] [PubMed] [Google Scholar]
- 7.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide:from basic science to therapeutic applications. Physiol Rev, 86: 583-650, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Poss KD, Tonga S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natal Aced Sic USA, 94: 10925-10930, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yachie A, Niida Y, Wada T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest, 103: 129-135, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stein AB, Guo Y, Tan W, et al. Administration of a CO-releasing molecule induces late preconditioning against myocardial infarction. J Mol Cell Cardiol, 38: 127-134, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stein AB, Bolli R, Dawn B, et al. Carbon monoxide induces a late preconditioning-mimetic cardioprotective and antiapoptotic milieu in the myocardium. J Mol Cell Cardiol, 52: 228-236, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide:from basic science to therapeutic applications. Physiol Rev, 86: 583-650, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi A, Ishikawa K, Matsumoto H, Kimura S, Kamiyama Y, Maruyama Y. Synergetic antioxidant and vasodilatory action of carbon monoxide in angiotensin II - induced cardiac hypertrophy. Hypertension, 50: 1040-1048, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Fujimoto H, Ohno M, Ayabe S, et al. Carbon monoxide protects against cardiac ischemia-reperfusion injury in vivo via MAPK and Akt-eNOS pathways. Arterioscler Thromb Vasc Biol, 24: 1848-1853, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Hee J, Callais F, Momas I, et al. Smokers' behaviour and exposure according to cigarette yield and smoking experience. Pharmacol Biochem Behav, 52(1): 195-203, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Foresti R, Clark JE, Green CJ, Motterlini R.Thiol compounds interact with nitric oxide in regulating heme oxygenase-1 induction in endothelial cells. Involvement of superoxide and peroxynitrite anions. J Biol Chem, 272(29): 18411-18417, 1997. [DOI] [PubMed] [Google Scholar]
- 17.Ishikawa K, Navab M, Leitinger N, Fogelman AM, Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J Clin Invest, 100(5): 1209-1216, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vile GF, Tyrrell RM. Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. J Biol Chem, 268(20): 14678-14681, 1993. [PubMed] [Google Scholar]
- 19.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science, 27; 235(4792): 1043- 6, 1987. [DOI] [PubMed] [Google Scholar]
- 20.Johnson RA, Lavesa M, DeSeyn K, Scholer MJ, Nasjletti A. Heme oxygenase substrates acutely lower blood pressure in hypertensive rats. Am J Physiol, 271: H1132-8, 1996. [DOI] [PubMed] [Google Scholar]
- 21.Gandini C, Castoldi AF, Candura SM, et al. Carbon monoxide cardiotoxicity. J Toxicol Clin Toxicol, 39: 35-44, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Tucciarone M, Dileo PA, Castro ER, Guerrero M. Myocardial infarction secondary to carbon monoxide poisoning: an uncommon presentation of a common condition. Case report and review of the literature. Am J Ther, 16: 462-465, 2009. [DOI] [PubMed] [Google Scholar]
- 23.Jung YS, Lee JS, Min YG, et al. Carbon monoxide-induced cardiomyopathy. Circ J, 78: 1437-1444, 2014. [DOI] [PubMed] [Google Scholar]
- 24.McMeekin JD, Finegan BA. Reversible myocardial dysfunction following carbon monoxide poisoning. Can J Cardiol, 3: 118-121, 1987. [PubMed] [Google Scholar]
- 25.Cheng S, Lyass A, Massaro JM, O’Connor GT, Keaney JF Jr, Vasan RS. Exhaled carbon monoxide and risk of metabolic syndrome and cardiovascular disease in the community. Circulation, 122: 1470-1477, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng S, Enserro D, Xanthakis V, et al. Association of exhaled carbon monoxide with subclinical cardiovascular disease and their conjoint impact on the incidence of cardiovascular outcomes. Eur Heart J, 35(42): 2980-2987, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Circulation, 126: 2020-2035, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Wright RS, Anderson JL, Adams CD, et al. 2011 ACCF/AHA focused update of the Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction (updating the 2007 guideline):a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in collaboration with the American College of Emergency Physicians, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol, 57(19): 1920-1959, 2011. [DOI] [PubMed] [Google Scholar]
- 29.Yasue H, Yoshimura M, Sumida H, et al. Localization of mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation, 90: 195-203, 1994. [DOI] [PubMed] [Google Scholar]
- 30.McDonagh TA, Robb SD, Murdoch DR, et al. Biochemical detection of left-ventricular systolic dysfunction. Lancet, 351: 9-13, 1998. [DOI] [PubMed] [Google Scholar]
- 31.Maisel AS, Krishnaswamy P, Nowak R, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med, 347: 161-167, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Ogawa K, Oida A, Sugimura H, et al. Clinical significance of blood brain natriurtic peptide concentration measurement in the detection of heart disease in untreated outpatients: Comparison of electrocardiography, chest radiography and echocardiography. Circ J, 66: 122-126, 2002. [DOI] [PubMed] [Google Scholar]