

Abstract
The main characteristic of alcohol use disorder is the consumption of large quantities of alcohol despite the negative consequences. The transition from the moderate use of alcohol to excessive, uncontrolled alcohol consumption results from neuroadaptations that cause aberrant motivational learning and memory processes. Here, we examine studies that have combined molecular and behavioural approaches in rodents to elucidate the molecular mechanisms that keep the social intake of alcohol in check, which we term ‘stop pathways’, and the neuroadaptations that underlie the transition from moderate to uncontrolled, excessive alcohol intake, which we term ‘go pathways’. We also discuss post-transcriptional, genetic and epigenetic alterations that underlie both types of pathways.
Keywords: Addiction, Epigenetics in the nervous system, Kinases Molecular neuroscience, Neural circuits, Neurotrophic factors
Introduction
According to the World Health Organization, 10–16% of individuals who consume alcohol and are aged 15 years or older engage in repeated, excessive episodic drinking (WHO, 2014) and are considered to be ‘problem drinkers’ (Enoch et al., 2002). Many of these individuals have a mild to moderate form of alcohol use disorder (AUD) and thus are maladaptively preoccupied with alcohol craving, seeking and consumption, despite the negative consequences of these activities (American Psychiatric Publishing, 2013). A subset of problem drinkers has a severe form of AUD, which is characterized by a dependence on alcohol and is commonly referred to as ‘alcoholism’. These individuals typically exhibit compulsive alcohol use and a loss of behavioural control, as well as alcohol tolerance and withdrawal symptoms, which may include anxiety, depressive episodes, social withdrawal, insomnia, nausea and seizures, which can be lethal (WHO, 2014) (Enoch et al., 2002) (American Psychiatric Publishing, 2013).
Studies using animal models of alcohol consumption provided convincing data suggesting that alcohol, like other drugs of abuse, activates molecular cascades within the mesocorticolimbic system that ultimately encode drug reward and reinforcement (Koob et al., 2010). Over time, and in part owing to the aberrant activation of mesocorticolimbic and nigrostriatal pathways, a transition from moderate to excessive use of alcohol may occur (Koob et al., 2010). This transition is often associated with dysphoria and negative reinforcement mechanisms, leading to persistent cycles of excessive drug taking and withdrawal (Koob et al., 2013) (Wise & Koob, 2014), which are thought to result from long-lasting molecular neuroadaptations (Hyman et al., 2006).
In this Review, we examine the molecular signalling mechanisms that prompt or prevent alcohol use and abuse by focusing on studies that have used rodent paradigms to model human patterns of drinking: that is, moderate alcohol intake, excessive consumption, dependence, craving and relapse (Boxes 1,2). We term the signalling pathways that contribute to the transition from moderate to uncontrolled excessive alcohol intake and to alcohol dependence as ‘go pathways’ (Fig. 1). As addiction is thought to be a maladaptive form of learning and memory (Hyman et al., 2006) (Torregrossa et al., 2011), we focus mainly on go-pathway molecules that have been linked to synaptic plasticity, learning and memory. We term the endogenous pathways that work in the opposite direction to the go pathways, and thus promote resilience against the development of AUD and keep alcohol intake in moderation, the ‘stop pathways’ (Fig. 2). Note that, in this article, we do not discuss the interaction between alcohol and G protein-coupled receptors or ion channels, and we do not address the mechanisms underlying processes such as tolerance, sensitization, intoxication and neuroinflammation (Crews et al, 2014) (Ron & Messing, 2013) (Ahmadiantehrani et al. 2014) (Rothenfluh et al., 2014).
Box 1. Behavioural models of alcohol consumption.
Models of moderate alcohol consumption
Continuous access to alcohol in a two-bottle choice (10%CA2BC)
A home cage-based voluntary drinking procedure in which rodents are given continuous access to one bottle of water and one bottle of alcohol in tap water (typically 10% (vol/vol); the alcohol concentration may vary (but remains mostly under 15%)). This training protocol typically results in alcohol consumption levels of 2–5 g per kg per 24 h in mice and <1 g per kg per 24 h in rats; the animals show no escalation in alcohol consumption across the weeks and a blood alcohol concentration (BAC) of 50 mg per dl (mouse). Thus, this protocol is used as a model of moderate drinking. Variants of this protocol use exposure to a gradually increasing concentration of alcohol (for example, from 4% to 16%) or the addition of saccharin (0.2%) to the alcohol solution.
Operant self-administration of 10% alcohol (10%OSA)
In this paradigm, animals typically press a lever to receive a contingent oral reward of alcohol (the premise of this procedure is that drugs of abuse control behaviour by functioning as positive reinforcers196). To obtain lever pressing for a moderate amount of alcohol, the rats are pre-trained on a continuous access to one bottle of water and one bottle of alcohol (typically 10% alcohol in tap water) protocol or undergo a sucrose fading protocol197. Indeed, this approach typically leads to a moderate consumption level of 0.2–0.5 g per kg per 1 h, which generates a BAC of 5–20 mg per dl (Ref. 197).
Oral alcohol and sucrose operant self-administration (AS-OSA)
This protocol is similar to that described above, except that lever presses deliver a solution containing alcohol (typically 8–12%) and sucrose (~2%).
Models of excessive drinking
Intermittent access to 20% alcohol in a two-bottle choice (20%IA2BC)
This is a voluntary drinking procedure that is applied in the animal’s home cage. Rodents are given 24 hours of concurrent access to one bottle containing typically 20% alcohol in tap water and one bottle of water every other day, followed by 24 hours of alcohol deprivation. Rodents typically show an escalation of alcohol intake and preference across the weeks, eventually reaching alcohol consumption levels of 15–20 g per kg per 24 h in C57BL/6 mice and 5–6 g per kg per 24 h in rats, inducing BACs that correspond to binge drinking levels (~80 mg per dl per 0.5 h for rats and 97.9–179.4 mg per dl per 2 h for mice)197, 198, 199.
Continuous access to escalating concentrations of alcohol in a two-bottle choice (ESC-CA2BC)
This is a variant of the continuous access protocol, which is used mostly in mice; it models the transition from moderate to excessive alcohol intake by gradually increasing the concentration of alcohol (for example, from 4% to 20% over the course of a few days). Alcohol intake levels start at 1.5 g per kg (at 3% alcohol) and escalate to 15 g per kg (at 20% alcohol)113.
Limited access to alcohol (20%LA)
Models such as the ‘drinking in the dark’ procedure in mice promote high, binge-like drinking by introducing one bottle containing 20% alcohol each day, typically for 2–4 hours, beginning 3 hours into the dark cycle. This procedure results in an alcohol consumption level of 8 g per kg per 4 h of alcohol and a BAC of 160 mg per dl (Ref. 200).
Operant self-administration of 20% alcohol (20%OSA)
To model excessive drinking, animals undergo intermittent access to 20% alcohol in a two-bottle choice (as described above) and, subsequently, operant self-administration of alcohol, with rats pressing the lever to obtain a 20% solution of alcohol197. Operant responding and alcohol intake are high, and rats typically consume 0.4–1.0 g per kg per 0.5 h, which generates a BAC of 30–90 mg per dl (Ref. 197).
Alcohol dependence paradigm (ADEP)
To model alcohol dependence, animals are chronically pre-exposed to alcohol in a non-contingent manner by using vapour inhalation or ingestion of a liquid diet144, 199, 201, 202. Pre-exposure of the animals to high alcohol concentrations by vapour inhalation leads to high levels of alcohol self-administration and can produce BACs of 150–250 mg per dl in rats and 175 mg per dl in mice203. In these animals, alcohol withdrawal produces physical and motivational symptoms of alcohol dependence201, 202. This model provides the advantage of experimentally controlling the BAC levels.
Intermittent access to alcohol and quinine adulteration (IA2BC-QN)
This protocol is used to model compulsive-like alcohol intake, despite the unpleasant consequences (that is, the bitter taste of quinine). Mice are trained in an intermittent access to alcohol in a two-bottle choice procedure. After achieving a stable consumption, quinine (0.1 g per l) is added to the alcohol solution83.
Box 2. Behavioural models of alcohol relapse.
Alcohol deprivation effect
In this model, animals are given voluntary access to alcohol for several days, weeks or months, are then deprived of alcohol for a certain period of time, and subsequently presented again with alcohol. This protocol leads to a robust, albeit temporary, enhancement in alcohol consumption after the period of deprivation, which is considered to model relapse204. A variant of this protocol uses long-term continuous access to alcohol in a four-bottle choice, in which animals have continuous access to four bottles, each containing water, 5% alcohol, 10% alcohol or 20% alcohol (vol/vol)115. The alcohol intake is then measured after the alcohol deprivation period204.
Reinstatement
This protocol models relapse to drug or alcohol seeking; animals are trained to respond for alcohol reinforcement, typically by pressing a lever. Then, after extinction of the responding by no reward delivery, the non-reinforced pressing on the alcohol-associated lever is significantly enhanced by alcohol priming injections, alcohol-related cues (for example, a light, a tone, or the odour and taste of alcohol) or stressors205.
Reacquisition
This is an operant animal model of relapse to drug or alcohol consumption; the training is similar to that of the reinstatement procedure, except that a non-contingent alcohol primer, which is delivered in the reward port at the beginning of the test session, triggers a rapid reacquisition of operant responding for alcohol205.
Figure 1. Signalling pathways underlying the go pathways.
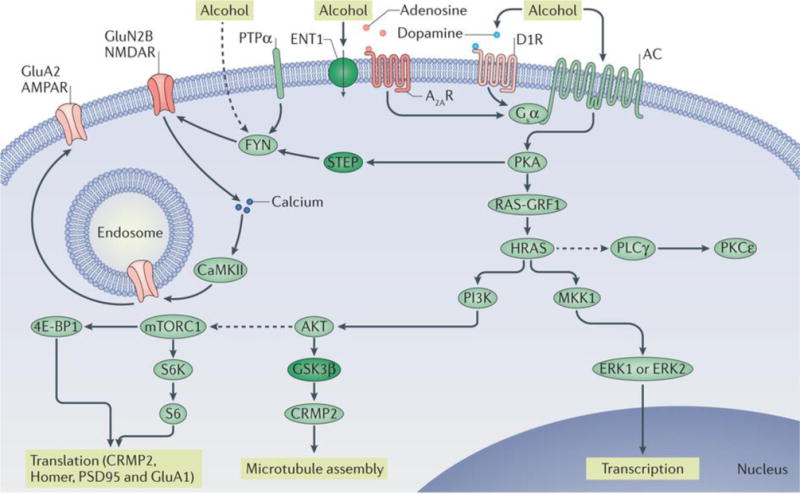
Repeated cycles of excessive alcohol exposure and withdrawal lead to aberrant activation of specific intracellular signalling cascades (the go pathways), the nature of which varies depending on the brain region. Collectively, these alcohol-related changes in intracellular signalling cascades drive long-lasting, detrimental behavioural phenotypes associated with alcohol abuse, such as excessive consumption, alcohol seeking, craving and relapse. Protein kinase A (PKA) has a central role in the go pathways. PKA is activated by adenylyl cyclase (AC), which hydrolyses ATP to cyclic AMP and is activated by alcohol through several mechanisms, including the inhibition of equilibrative nucleoside transporter 1 (ENT1) (inactivated proteins are shown in dark green) and subsequent activation of the Gsα-coupled adenosine A2A receptors (A2ARs) and/or the activation of the Gsα-coupled dopamine D1 receptor (D1R). cAMP binds to the regulatory subunit of PKA, thus freeing the catalytic subunit of the kinase to phosphorylate its substrates, which include striatum-enriched protein-tyrosine phosphatase (STEP). Activation of FYN requires the recruitment of protein-tyrosine phosphatase-α (PTPα), which dephosphorylates an inhibitory phosphorylation site. STEP is inactivated by PKA phosphorylation, which enables the sustained activation of FYN. Alcohol might also activate FYN by additional mechanisms (as indicated by the dashed line). When FYN is activated, it phosphorylates the GluN2B subunit of NMDA-type glutamate receptors (NMDARs), resulting in enhancement of NMDAR activity. Calcium entry via the NMDARs activates calcium/calmodulin-dependent protein kinase type II (CaMKII), resulting in autophosphorylation of the kinase. CaMKII phosphorylates the AMPA-type glutamate receptor (AMPAR) subunit GluA2, resulting in forward trafficking of these receptors to the synaptic membrane. Another target of PKA is RAS-specific guanine nucleotide-releasing factor 1 (RAS-GRF1), which, when activated, promotes the transition of the small GTP-binding proteins HRAS and KRAS from inactive GDP-bound forms to active GTP-bound forms. Activation of HRAS leads to the activation of phosphoinositide 3-kinase (PI3K). PI3K, in turn, activates AKT, which phosphorylates glycogen synthase kinase 3β (GSK3β), thus inhibiting its activity. When GSK3β activity is reduced, collapsin response mediator protein 2 (CRMP2) binds to tubulin, enabling microtubule assembly. AKT, through intermediate proteins, also activates mechanistic target of rapamycin complex 1 (mTORC1) (as indicated by the dashed line). mTORC1 phosphorylates its substrates eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) and p70 S6 kinase (S6K), which then phosphorylates its substrate S6. 4E-BP1, S6K and S6 are part of the ribosomal translational machinery, and activation of mTORC1 initiates the translation of postsynaptic density protein 95 (PSD95), Homer, CRMP2 and GluA1. HRAS also activates mitogen-activated protein kinase kinase 1 (MKK1), which in turn activates extracellular signal-regulated kinases 1 and 2 (ERK1/2), inducing gene transcription. Moreover, HRAS indirectly activates (as indicated by the dashed line) phospholipase Cγ (PLCγ), which in turn activates protein kinase Cε (PKCε).
Figure 2. Signalling pathways underlying the stop pathways.
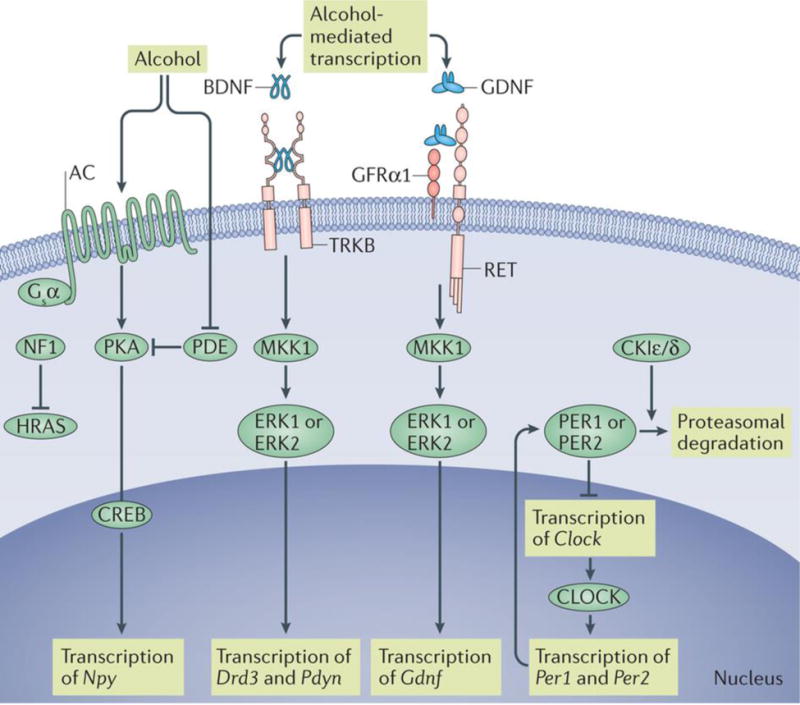
Moderate, but not excessive, alcohol consumption causes activation of intracellular signalling cascades that lead to the expression of genes encoding growth factors or neuropeptides. These signalling molecules have a protective role by promoting downstream cascades that prevent the dominance of the go pathway and the establishment of detrimental behavioural phenotypes. Moderate intake of alcohol increases the levels of brain-derived neurotrophic factor (Bdnf) mRNA. Binding of BDNF to tropomyosin-related kinase B (TRKB) activates extracellular signal-regulated kinase 1 and 2 (ERK1/2) signalling, thus promoting the expression of Drd3 (which encodes the dopamine D3 receptor) and Pdyn (which encodes preprodynorphin). Similarly, binding of glial cell line-derived neurotrophic factor (GDNF) to its receptors RET and GDNF family receptor α1 (GFRα1) results in the activation of ERK1/2 and the induction of the transcription of Gdnf. Activated protein kinase A (PKA) phosphorylates the transcription factor cyclic AMP response element (CRE)-binding protein (CREB). PKA-mediated phosphorylation of CREB results in the activation of the transcription machinery and in the induction of expression of genes, including neuropeptide Y (Npy). Activity of PKA is terminated by 3′,5′-cyclic nucleotide phosphodiesterase (PDE), which hydrolyses cAMP to 5′ AMP. Alcohol-induced inhibition of PDE activity enables the sustained activation of PKA. Activity of HRAS and KRAS is terminated by GTPase-activating proteins, including neurofibromin (NF1). Circadian locomoter output cycles protein kaput (CLOCK) drives the transcription of Per1 and Per2, which encode the period proteins. PER1 and PER2 suppress the transcription of Clock. The casein-kinases Iε and Iδ (CKIε/δ) phosphorylate PER1 and PER2, leading to the proteasomal degradation of these proteins. AC, adenylyl cyclase; MKK1, mitogen-activated protein kinase kinase 1.
Go pathways promote excessive drinking
Below, we describe examples of how voluntary alcohol intake initiates intracellular signalling cascades within the mesocorticolimbic and nigrostriatal regions to produce adaptations that ultimately drive alcohol-drinking behaviours (Fig. 1).
PKA
Cyclic AMP-dependent protein kinase A (PKA) is a serine/threonine kinase that has a central role in learning and memory13, 14 and in behavioural responses to drugs of abuse15; indeed, PKA is a key initiator of many actions of alcohol. PKA is activated by adenylyl cyclase (AC), which itself is activated by Gsα protein-coupled receptor stimulation (Figs 1,2). Global inhibition of PKA activity, either by a reduction in Gsα expression16 or through the deletion of the genes encoding AC1 and AC8, reduces alcohol intake in a mouse model of moderate alcohol consumption (the continuous access to alcohol in a two-bottle choice (10%CA2BC) paradigm17; Box 1). Furthermore, infusion of a Gβγ inhibitory peptide within the nucleus accumbens (NAc) prevents alcohol-induced nuclear translocation of PKA and PKA-stimulated gene expression, and decreases alcohol consumption in rats in the 10%CA2BC paradigm18. Together, these studies suggest that PKA has an important role in mechanisms that promote alcohol intake. Alcohol is thought to activate PKA via, in part, the inhibition of equilibrative nucleoside transporter 1 (ENT1), which controls the extracellular levels of adenosine19. In fact, Ent1-knockout mice consume more alcohol than wild-type littermates in both the 10%CA2BC paradigm and a model of excessive alcohol consumption (the limited access to alcohol (20%LA) procedure20; Box 1). In addition, these mice exhibit faster acquisition of alcohol self-administration phenotypes in moderate alcohol-consumption models (10%CA2BC and the operant self-administration of 10% alcohol (10%OSA) paradigms; Box 1), which may owe to reduced activity of the adenosine A1 receptors (A1Rs) in the NAc21. Adenosine also binds to A2ARs, which are coupled to Gsα and PKA (Fig. 1), and the inhibition of A2ARs reduces alcohol consumption in rats in a moderate alcohol consumption model (10%OSA paradigm)22, 23. Alcohol-induced activation of PKA leads to the phosphorylation of numerous PKA substrates, including the FYN kinase inhibitor, striatum-enriched protein-tyrosine phosphatase (STEP; also known as PTPN5)24 and RAS-specific guanine nucleotide-releasing factor 1 (RAS-GRF1)25. The consequences of these phosphorylation events are outlined below.
FYN signalling pathway
The tyrosine-protein kinase FYN has an important role in synaptic plasticity, learning and memory26, 27. The activity of FYN is negatively regulated by STEP, which dephosphorylates and thereby inhibits the kinase28. The activity of STEP is negatively regulated by PKA, which phosphorylates the phosphatase and thereby inhibits its catalytic activity26 (Fig. 1). In mice, excessive alcohol consumption (for example, repeated cycles of binge drinking and alcohol withdrawal in the intermittent access to 20% alcohol in a two-bottle choice (20%IA2BC) paradigm; Box 1) results in PKA-mediated phosphorylation of STEP (and thus inhibition of the phosphatase) in the dorsomedial striatum (DMS)24. As alcohol inhibits STEP activity, a robust and sustained activation of FYN is detected in the DMS of rodents consuming high levels of alcohol (20%IA2BC paradigm)24, 29, 30. In fact, the requirement of PKA for alcohol-mediated activation of FYN was first reported in hippocampal slices31. Alcohol-mediated inhibition of STEP also enables the activation of protein-tyrosine phosphatase-α (PTPα)32, which is required for the activation of FYN33. Indeed, excessive alcohol intake (20%IA2BC model) increases the interaction between FYN and PTPα, which in turn contributes to the sustained activation of the kinase30. Interestingly, alcohol-induced inhibition of STEP, as well as activation of FYN and PTPα, can be detected in the DMS but not in other striatal regions (that is, the dorsolateral striatum (DLS) and the NAc)24, 29, 30, 32, demonstrating that the molecular changes induced by alcohol intake are highly selective.
The consequence of alcohol-mediated activation of FYN is the phosphorylation of the GluN2B subunit of NMDA-type glutamate receptors (NMDARs)29, which in turn produces a robust and long-lasting activation of NMDARs in the DMS29. Calcium entry through the NMDARs activates calcium/calmodulin-dependent protein kinase type II (CaMKII)34. Although the direct role in alcohol consumption of CaMKII in the DMS has not been determined, moderate alcohol consumption in mice (10%CA2BC paradigm) increases CaMKII activity in the central amygdala (CeA), which, in turn, contributes to alcohol reward35. Moreover, transgenic mice expressing an inactive mutant form of CaMKII exhibit a delay in the onset of moderate alcohol intake in an excessive-alcohol-consumption model (the continuous access to escalating concentrations of alcohol in a two-bottle choice (ESC-CA2BC) paradigm36; Box 1). Long-term potentiation, a cellular signature of synaptic plasticity, depends on NMDARs and on CaMKII-dependent trafficking of AMPA-type glutamate receptors (AMPARs) to the synaptic membrane37. GluN2B-dependent long-term potentiation, as well as the trafficking of AMPAR subunits, is detected in the DMS of rats consuming high levels of alcohol (20%IA2BC paradigm)38. Furthermore, repeated cycles of binge drinking and alcohol withdrawal in mice (20%IA2BC model) increase AMPAR activity and produce structural alterations in DMS neurons expressing the dopamine D1 receptor (D1R)39. Finally, phosphorylation of AMPARs and an increase in the AMPAR to NMDAR ratio are also detected in the CeA of mice self-administering a moderate level of a sweetened alcohol solution (the oral alcohol and sucrose operant self-administration (AS-OSA) protocol35; Box 1).
Importantly, in support of the possibility that the FYN signalling pathway is indeed a mechanism underlying excessive alcohol drinking, the pharmacological inhibition of FYN, GluN2B or AMPARs in the DMS of rats or the knockdown of PTPα in the DMS of rodents reduces excessive alcohol intake and the reinstatement of operant responding (20%IA2BC and 20%LA paradigms)29, 38, 40 (Box 2). Similarly, the inhibition of CaMKII or AMPARs in the CeA reduces moderate alcohol intake35. Conversely, the global or DMS-specific knockdown of STEP increases alcohol intake in mice in moderate and excessive alcohol consumption models (10%CA2BC, 20%IA2BC and ESC-CA2BC paradigms24, 41; Box 1).
HRAS signalling pathway
Another important contributor to synaptic plasticity and memory processes is HRAS, a small GTP-binding protein42. Rats with a history of excessive alcohol intake and abstinence show activation of HRAS in the NAc25, and downregulation of HRAS expression or inhibition of its activity in the NAc attenuates the self-administration of alcohol in models of excessive alcohol intake in mice (20%IA2BC paradigm) and rats (20%IA2BC and 20%LA models)25. In line with these findings, escalation of alcohol drinking (20%LA procedure) is not detected in mice with heterozygous knockout of the gene encoding another member of the RAS GTPases, KRAS43.
HRAS activity in the CNS is positively regulated by RAS-GRF1 and RAS-GRF2 (Ref. 44) (Fig. 1). PKA-mediated phosphorylation enhances the activity of RAS-GRF1, thus increasing the activity of HRAS45, and an increase in the level of phosphorylated RAS-GFR1 is detected in the NAc of rats self-administering high levels of alcohol25 (20%IA2BC paradigm followed by the operant 20% alcohol self-administration (20%OSA) protocol; Box 1). The levels of Rasgrf2 and Hras transcripts are higher in the brains of mice that are bred to consume high levels of alcohol than in the brains of mice that do not consume high levels of alcohol46, and global deletion of Rasgrf2 increases the moderate intake of alcohol in mice (ESC-CA2BC paradigm)47.
Downstream of HRAS is phosphoinositide 3-kinase (PI3K), which activates AKT (also known as PKB); AKT, in turn, phosphorylates and inactivates glycogen synthase kinase 3β (GSK3β)48 (Fig. 1). In parallel to activating HRAS, excessive alcohol intake in rats (20%OSA paradigm) and mice (20%LA model) activates PI3K and AKT and inhibits GSK3β in the NAc49, 50, 51. Furthermore, alcohol-induced inactivation of GSK3β in the NAc prevents the phosphorylation of the GSK3β substrate collapsin response mediator protein 2 (CRMP2; also known as DRP2), enabling CRMP2 to bind to tubulin and hence to promote microtubule assembly51. Importantly, focal inhibition of PI3K or AKT, or downregulation of Crmp2 mRNA levels, in the NAc robustly reduces excessive alcohol drinking in rodents (shown in the 20%IA2BC and 20%LA models for mice and in the 20%IA2BC and 10%OSA paradigms for rats)49, 50, 51.
Through intermediate steps, AKT also activates mechanistic target of rapamycin complex 1 (mTORC1) (Fig. 1), a kinase that is responsible for the initiation of dendritic protein translation, synaptic plasticity, memory processes52, 53 and addiction54. mTORC1 is robustly activated in the NAc of rodents consuming high levels of alcohol (20%IA2BC paradigm), as indicated by the increased phosphorylation of its targets, p70 S6 kinase (S6K) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1)55 (Fig. 1). Interestingly, in mice, a single session of excessive alcohol intake (20%IA2BC model) is sufficient to activate mTORC1 signalling in D1R-expressing but not D2R-expressing neurons in the NAc shell56. These findings suggest that alcohol exerts its actions on signalling in a cell type-specific manner, although further investigation is required to determine whether this phenomenon is more generalized.
Alcohol-induced initiation of the mTORC1-dependent translational machinery leads to increased levels of the synaptic proteins Homer, postsynaptic density protein 95 (PSD95; also known as DLG4), the GluA1 subunit of AMPARs and CRMP2 in the NAc49, 51, 55, 56 (Fig. 1). The cellular consequences of alcohol-dependent mTORC1-mediated translation of synaptic proteins are the enhancement of synaptic plasticity56 and, potentially, an increase in structural plasticity51. Importantly, systemic, as well as intra-NAc, administration of the mTORC1 inhibitor rapamycin attenuates alcohol seeking and drinking in excessive-alcohol-consumption models (shown in the 20%IA2BC model for mice and in the 20%IA2BC and 20%OSA paradigms for rats)55, 56.
Finally, retrieval of alcohol-associated memories in rats with a history of excessive alcohol intake (20%OSA paradigm) activates mTORC1 and leads to increased synthesis of synaptic proteins in the rat CeA, medial prefrontal cortex (mPFC) and orbitofrontal cortex57, suggesting that in these regions mTORC1 has a role in the reconsolidation of alcohol-associated memories. Importantly, systemic or intra-CeA administration of rapamycin immediately after memory retrieval disrupts the reconsolidation of alcohol-associated memories and produces a long-lasting suppression of relapse57, as measured in a retention test after abstinence and then reacquisition of 20%OSA protocol (Box 2).
Taken together, these findings indicate that the PI3K–AKT–mTORC1 axis has a central role in the neuroadaptations that underlie go-pathway phenotypes such as alcohol seeking, drinking and relapse.
PKCε signalling
HRAS is also upstream of phospholipase Cγ (PLCγ), which can activate protein kinase C (PKC) isoforms, including PKCε (encoded by PRKCE58 (Fig. 1). Although a link between alcohol-mediated activation of HRAS and PLCγ–PKC signalling has yet to be determined, PKCε has a well-documented and important role in go pathways. Specifically, Prkce-knockout mice consume less alcohol than do wild-type littermates and exhibit a decrease in preference for alcohol over water (ESC-CA2BC and 10%OSA models)59, 60. Conditional overexpression of the kinase in the forebrain of mice with Prkce deletion rescued the alcohol-consumption phenotypes61, suggesting that the kinase contributes to alcohol-consumption behaviours. Studies in mice further revealed that the loci of PKCε action are the NAc62 and the amygdala63 (20%LA procedure). The amygdala is a focal region of circuitries underlying stress and negative reinforcement64, and studies have shown that amygdalar PKCε has a role in the mechanisms that underlie anxiety-like behaviour in mice65. Specifically, in the CeA of Prkce-knockout mice, there is reduced expression of corticotropin-releasing factor (CRF)66, a neuropeptide implicated in mechanisms underlying stress and anxiety67. In agreement, a reduction in CRF-induced enhancement of type A GABA receptor-dependent inhibitory postsynaptic potentials was detected in the CeA of Prkce-knockout mice compared with wild-type littermates68. Finally, global knockout or a CeA-specific knockdown of PKCε attenuated anxiety-like behaviours in mice65, 66. Together, these PKCε-related results suggest that this kinase is a mediator of the interplay between stress and heightened alcohol intake.
ERK1/2
Another kinase downstream of HRAS is mitogen-activated protein kinase kinase 1 (MKK1; also known as MEK1), which activates extracellular signal-regulated kinase 1 (ERK1; also known as MAPK3) and ERK2 (also known as MAPK1)69. ERK1 and ERK2 (from here on referred to as ERK1/2) have well-established roles in learning, memory and addiction70 (Fig. 1). Transcripts of genes in the ERK1/2 pathway are enriched in the brains of mice that are genetically selected to consume high levels of alcohol46, and cue-induced reinstatement of alcohol seeking (Box 2) is associated with activation of ERK1/2 in the NAc shell and in the basolateral amygdala (BLA) of alcohol-preferring rats71. Curiously, however, ERK1/2 signalling has mainly been associated with stop pathways (see below).
Stop pathways gate drinking
Here, we describe examples of molecular adaptations that gate the level of alcohol intake, keeping alcohol consumption to moderate levels, and thus oppose the go pathways (Fig. 2). The stop pathways also provide clues as to why some individuals become problem drinkers and exhibit AUD phenotypes, whereas the majority of people do not.
BDNF and GDNF
Increasing evidence suggests that brain-derived neurotrophic factor (BDNF), which has roles in CNS development, plasticity, learning and memory72, and glial cell line-derived neurotrophic factor (GDNF), which is important for the function of dopaminergic neurons73, are components of the stop pathways. Both Bdnf and Gdnf are alcohol-responsive genes; moderate (10%CA2BC paradigm) but not high (20%IA2BC model) levels of alcohol intake increase the level of Bdnf transcripts in the DLS of rodents74, 75, and Gdnf mRNA levels are elevated in the ventral tegmental area (VTA) of rats consuming alcohol for a short, but not prolonged, period of time (1 week versus 7 weeks of the 20%IA2BC paradigm)76. In a moderate alcohol-consumption model (10%CA2BC paradigm), Bdnf-heterozygous mice and conditional Bdnf-knockout mice consume more alcohol than wild-type littermates74, 77, 78, whereas global increases in BDNF levels in mice reduce alcohol intake74. Similarly, Gdnf-heterozygous mice consume more alcohol than wild-type mice after a period of deprivation (20%OSA paradigm)79.
In support of the possibility that BDNF gates the levels of alcohol intake, knockdown of BDNF in the DLS increases, whereas intra-DLS infusion of BDNF attenuates, the moderate intake of alcohol in rodents (shown in the 10%CA2BC model for mice and in the 10%OSA paradigm for rats)78, 80, 81. Furthermore, overexpression of BDNF in the mPFC, or systemic administration of an agonist of the BDNF receptor tropomyosin-related kinase B (TRKB), normalizes the level of alcohol intake in mice from excessive and compulsive to moderate (shown in the models 10%CA2BC and 20%IA2BC or in the intermittent access to alcohol and quinine adulteration (IA2BC-QN) paradigm82, 83; Box 1). Similarly, BDNF in the CeA and medial amygdala (MeA) of rats controls moderate alcohol intake (10%CA2BC model) as well as withdrawal-induced anxiety-like behaviours84, 85. The role of GDNF in alcohol consumption is comparable to that of BDNF. Short hairpin RNA-mediated knockdown of GDNF in the VTA or NAc of rats accelerates the escalation of excessive alcohol intake (20%IA2BC paradigm) and increases the intensity of relapse (that is, the amount of alcohol consumed) after abstinence (10%IA2BC and 10%OSA paradigms, alcohol deprivation effect, reinstatement and reacquisition tests76, 86; Box 2). Furthermore, infusion of GDNF into the VTA of rats reduces excessive alcohol intake (20%IA2BC model)87, and overexpression of this growth factor in the mesolimbic system prevents the transition from moderate to excessive alcohol consumption (20%IA2BC and 10%OSA paradigms)86. Finally, GDNF administration into the VTA of rats blocks relapse to alcohol drinking87. Together, these data suggest that alcohol induces upregulation of both BDNF and GDNF and that these neurotrophic factors act to keep alcohol intake in check.
Binding of BDNF to TRKB and binding of GDNF to the receptor tyrosine kinase RET and its co-receptor GDNF family receptor α1 (GFRα1) activate ERK1/2, PLCγ and PI3K signalling pathways72, 73 (Fig. 2). BDNF and GDNF gate alcohol drinking via the activation of ERK1/2 in the DLS and VTA, respectively81, 87. BDNF controls alcohol consumption, at least in part, through ERK1/2-induced expression of preprodynorphin (also known as proenkephalin B) and D3R in the dorsal striatum88, 89, and via increased expression of activity-regulated cytoskeleton-associated protein (ARC) in the CeA and MeA90, 91, 92. Interestingly, moderate consumption of alcohol (10%CA2BC paradigm) increases neurogenesis in the hippocampal dentate gyrus of mice by a TRKB-dependent mechanism93. GDNF reduces drinking by normalizing the deficient firing of dopaminergic neurons in the VTA and the low dopamine levels in the NAc that occur during withdrawal from excessive alcohol intake86, 94 and by upregulating its own expression95.
If the stop-pathway molecules counteract the motivation for alcohol, then the transition from moderate to excessive, uncontrolled consumption should stem, at least in part, from a breakdown of these protective pathways. In line with this possibility, long-term consumption of high (but not moderate) amounts of alcohol (20%IA2BC paradigm) reduces Bdnf expression in the mPFC of mice that are not dependent on alcohol75, 82 and alcohol-dependent rats (shown in the alcohol-dependence paradigm (ADEP)96; Box 1). Similarly, long-term, excessive alcohol drinking produces a reduction in the levels of GDNF in the VTA of rats (20%IA2BC model)76. Interestingly, these data are in line with human studies, which found that both BDNF and GDNF levels are reduced in blood serum samples from alcohol-dependent humans97, 98.
The discovery of these types of protective homeostatic pathways also raises the question as to why some individuals become problem drinkers and exhibit AUD phenotypes, whereas most people do not. One possibility is that innate differences in the level of specific growth factors may account for different alcohol-drinking levels; indeed, Bdnf levels in the CeA and MeA are much lower in an alcohol-preferring line of rats (P rats) than in a line of alcohol-non-preferring rats (NP rats)99, and Gdnf levels in the VTA of rats are negatively correlated with the amount of alcohol consumed76. Another possibility is that loss-of-function mutations within stop-pathway-related genes increase the susceptibility and/or the intensity of AUD. In fact, loss-of-function mutations in BDNF may confer a genetic risk for the development of excessive alcohol drinking. Specifically, a well-described loss-of-function mutation in BDNF (G196A; also known as polymorphism rs6265) produces an amino acid substitution (Val66Met) that leads to reduced activity-dependent release of BDNF100, 101. We recently found that this polymorphism promotes compulsive-like alcohol consumption in a transgenic mouse model (IA2BC-QN paradigm)83, and human studies suggest that the Val66Met polymorphism is associated with earlier onset of alcoholism102, higher risk of relapse103 and slower recovery of the grey matter after a period of abstinence104.
ERK1/2
As stated above, BDNF and GDNF in the DLS and VTA, respectively, transduce their signal to gate the level of alcohol drinking through ERK1/2 (Refs 78,87). In line with the possible role of ERK1/2 as stop-pathway molecules, systemic inhibition of MKK, the upstream activator of ERK1/2 (Fig. 2), increases moderate and excessive alcohol intake in mice (AS-OSA and 20%LA models)105, 106, 107, suggesting that alcohol-induced activation of ERK1/2 is required for keeping consumption under control.
Circadian rhythm genes
Circadian rhythm clock genes, which allow the precise adaptation of organisms to the external and internal environment, have been implicated in psychiatric disorders108. Interestingly, clock genes are also emerging as potential players in alcohol-mediated actions109, specifically in the stop pathways (Fig. 2). Mice expressing a mutation in the clock gene Per1 (which encodes PERIOD 1) show increased alcohol intake following social defeat110. Furthermore, mice expressing a mutant variant of Per2 that encodes a non-functional protein exhibit high levels of alcohol intake and an increase in the motivation to consume alcohol111. In humans, single point mutations in PER1 and PER2 are associated with heavy alcohol drinking and an increased sensitivity to stress in young adults110, 112. Further evidence supporting a role for circadian genes in stop pathways stems from the findings that mice with a dominant negative mutation in circadian locomoter output cycles protein kaput (Clock) show increases in the intake of and preference for high amounts of alcohol113, and that virus-mediated knockdown of CLOCK in the VTA of wild-type mice facilitates the escalation from moderate to excessive alcohol intake (ESC-CA2BC paradigm)113. Finally, systemic administration of an inhibitor of casein-kinases Iε and Iδ, which are responsible for PER2 proteasomal degradation114, decreases alcohol consumption after deprivation of alcohol in rats115 (Box 2).
PKA and CREB signalling
As described above, PKA is a central player in the go pathways; however, intriguingly, PKA signalling also has a major role in stop pathways. For example, genetic knockout of Adcy5 (which encodes AC5) increases alcohol intake in rodents116, and inhibition of PKA activity through a global deletion of the gene encoding the PKA RII regulatory subunit increases alcohol consumption in mice117. One well-described substrate of PKA is the transcription factor cAMP response element (CRE)-binding protein (CREB)14. PKA-mediated phosphorylation of CREB initiates CRE-dependent gene transcription14, and a large body of work has identified a unique role for the PKA–CREB axis in the amygdala as a ‘gatekeeper’ of heightened anxiety and alcohol intake118 (Fig. 2). Specifically, Creb-heterozygous mice consume more alcohol and have a greater preference for alcohol over water than their wild-type littermates (10%CA2BC paradigm)84. Further studies using this paradigm revealed that the inhibition of PKA activity in the CeA of rats decreased the level of phosphorylated CREB, and this effect was associated with increased levels of anxiety-like behaviour and alcohol intake119, 120. By contrast, activation of PKA in the CeA produced the opposite effects (10%CA2BC model)119, 120. A subsequent study showed that the downstream target of CREB in the amygdala is neuropeptide Y (NPY)121. NPY is abundantly expressed in the brain and is anxiolytic when administered into the CNS122. Knockout of Npy in mice increases alcohol consumption, whereas transgenic overexpression of NPY reduces it123. Interestingly, P rats, which drink alcohol excessively and show greater anxiety-like behaviour, have lower levels of CREB phosphorylation and NPY in the amygdala than NP rats120, suggesting that an increased susceptibility to developing anxiety and AUD may owe to innate differences in the PKA–CREB–NPY axis. However, this possibility needs further investigation in human studies.
The activity of PKA is terminated by 3′,5′-cyclic nucleotide phosphodiesterases (PDEs), a family of enzymes that hydrolyse cAMP and thus terminate PKA activity (Fig. 2). In rodents, inhibition of PDE activity leads to sustained activation of PKA and to a reduction in alcohol intake in several alcohol-consumption paradigms124, 125, 126. Moreover, TP-10, a specific inhibitor of the PDE10A isoform, reduces relapse-like alcohol self-administration in rats127, and PDE10A levels correlate with the levels of alcohol consumption in rats during stress-induced relapse128, 129. Together, these data suggest that, specifically in the amygdala, PKA activity is required to produce neuroadaptations that gate alcohol-drinking behaviours.
Other potential stop-pathway molecules
Protein kinase Mζ (PKMζ), anaplastic lymphoma kinase (ALK), LIM domain only protein 3 (LMO3) and neurofibromin 1 (NF1) may also contribute to the stop pathways. The expression of PKMζ, which is an isoform of PKC, is increased in the NAc of mice in response to consumption of alcohol and a period of 24 hours of withdrawal, and global knockout of Prkcz (which encodes PKMζ) in mice increases alcohol intake in models of excessive alcohol intake (20%IA2BC and 20%LA paradigms)130. The biological function of PKMζ in the CNS is a matter of controversy131, 132, and the possibility that this kinase contributes to stop mechanisms should be further explored. The striatal expression levels of Alk or of the transcription regulator Lmo3 negatively correlate with the amount of alcohol consumed in a mouse model of excessive alcohol intake, and the genetic deletion of Alk or a genetic reduction in Lmo3 levels increases excessive alcohol intake in this paradigm133, 134, 135. Finally, the activity of HRAS and KRAS is terminated by NF1 (Ref. 136); a recent study reported that Nf1-heterozygous mice consume less alcohol than wild-type littermates (shown in the ADEP followed by 20%LA procedure) and that this reduced consumption of alcohol may be associated with increased baseline GABA release in the CeA137. These findings are rather surprising given the central role of RAS signalling in the go pathways; however, this may be another example of how signalling molecules can have opposite roles depending on the brain region they function through.
Epigenetic modifications
The majority of studies described above focus on a single gene or a signalling cascade; however, it is highly plausible that alcohol exerts its pleiotropic actions by affecting central molecular hubs, which in turn initiate the transcription or translation of a diverse group of genes. Non-coding RNAs, such as microRNAs (miRNAs), as well as epigenetic mechanisms that control the structure of chromatin, such as DNA methylation and histone acetylation, have emerged in this past decade as an exciting field to explore in addiction research138, 139, 140, which could fulfil the requirement of such hubs that transduce both the go and stop pathways.
Histone acetylation
Mice with a history of excessive alcohol intake show reduced levels of histone H4 acetylation in the NAc (20%LA procedure)141, and alcohol withdrawal-induced anxiety-like behaviour is associated with decreased levels of histone H3 and H4 acetylation in the CeA and MeA of rats and a corresponding increase in histone deacetylase (HDAC) activity142. Furthermore, P rats exhibit a higher basal level of anxiety-like behaviour and lower levels of histone H3 acetylation than NP rats143. Higher levels of amygdalar HDAC2 protein are also detected in P rats than in NP rats, and knockdown of HDAC2 in the CeA of P rats reverses excessive alcohol drinking and anxiety-like phenotypes143. Together, these results indicate that high levels of HDAC activity, chromatin condensation and, consequently, low expression levels of specific genes may have a role in the comorbidity of anxiety, stress and excessive alcohol drinking. In line with this possibility, systemic administration of HDAC inhibitors141 or knockdown of HDAC2 in the CeA143 causes marked reductions in excessive alcohol seeking and drinking in rodents. By contrast, the levels of HDACs are reduced and histone H3 methylation is reported to be increased in hippocampal regions in response to moderate consumption of alcohol (10%CA2BC paradigm)93.
DNA methylation
A recent elegant study established a direct link between DNA methylation and alcohol dependence144. Specifically, it showed that the levels of DNA methyltransferase 1 (DNMT1) are increased in the mPFC of rats with a history of alcohol dependence, and that this increase leads to the hypermethylation of DNA and to the downregulation of the expression of a group of synaptic genes144. Moreover, inhibition of DNMT1 activity in the mPFC restored the expression levels of the selected synaptic genes and prevented escalation of alcohol intake in alcohol-dependent animals144. In line with these findings, DNMT1 levels are markedly higher in the NAc of mice with a history of excessive alcohol intake than in alcohol-naive controls, and the systemic administration of DNMT1 inhibitors reduces alcohol consumption in these mice (20%IA2BC paradigm)141. In line with the studies in rodents, a genome-wide epigenomic approach identified profound disturbances in the DNA methylation status of numerous genes in peripheral samples from alcohol-dependent people145 and in the post-mortem brains of individuals with alcoholism146. Furthermore, a recent study in humans reported that hypermethylation of the gene encoding protein phosphatase 1G (PPM1G) is associated with early escalation of alcohol use147. PPM1G is a serine/threonine phosphatase, and thus it is plausible that a reduction in the levels of PPM1G in the brain is associated with hyperphosphorylation of various substrates. Finally, DNA methylation of the genes encoding nerve growth factor (NGF) and BDNF was increased and the expression level of these neurotrophic factors was decreased, in the serum of people with alcoholism148, 149. NGF signalling is an important contributor to normal CNS function150, and, as described above, BDNF has a central role in the stop pathways. Thus, DNA methylation-dependent reduction of the expression of these genes could contribute to the mechanisms that manifest in alcohol addiction.
miRNAs
The role of miRNAs in drug addiction139 and, more specifically, in AUD151, 152, 153 is starting to emerge. A pioneering study showed that exposure to alcohol upregulates the levels of miR-9 and, in doing so, downregulates the levels of large-conductance calcium- and voltage-gated potassium channel (BK channel; encoded by KCNMA1), which in turn contribute to alcohol tolerance154. Although interesting, these results were obtained in a cell culture model and, therefore, need to be further explored in vivo. A direct link between alcohol drinking and miRNAs stems from two recent studies that found that protracted withdrawal from alcohol in alcohol-dependent rats (ADEP)96 or excessive alcohol intake in non-dependent mice (20%IA2BC model)82 increases the expression of miR-206 (Ref. 96) and miR-30a-5p82, respectively, in the mPFC. Both miRNAs target Bdnf, and an inverse correlation between the levels of these miRNAs and BDNF levels was observed in both paradigms82, 96. Importantly, overexpression of miR-206 (Ref. 96) and miR-30a-5p82 produced an increase in alcohol self-administration in non-dependent rats and mice, respectively (20%IA2BC model and ADEP). Furthermore, inhibition of miR-30a-5p function in the mPFC of mice restored the levels of Bdnf mRNA and, consequently, reverted alcohol intake from excessive to moderate levels82. Overexpression of miR-382 in the NAc of mice attenuated excessive alcohol consumption and preference (20%IA2BC paradigm)155; however, more studies are necessary to establish a direct link between endogenous miR-382 and alcohol intake. Furthermore, the levels of miR-10a and miR-21 were elevated in the blood of humans who consumed excessive amounts of alcohol and were exposed to a stressful cue156; it would be of interest to test the role of these specific miRNAs in animal studies.
The results described above show that alcohol exposure modifies the expression of miRNAs. Notably, changes in miRNA profiling were detected in the mPFC of alcohol-dependent rats157, and the levels of miRNAs were elevated in the PFC in post-mortem tissue from humans with a severe AUD158, 159. Interestingly, although alcohol intake produces changes in the levels of numerous miRNAs, the animal studies discussed above indicate that manipulation of the function of a single miRNA is sufficient to produce robust behavioural changes. This suggests that each miRNA is sufficient, but not necessary, to drive the alcohol-drinking behaviour.
Together, these data support the notion that epigenetic modifications, which alter gene expression and/or mRNA degradation, have the potential of serving as network hubs that change the molecular landscape in response to alcohol exposure. It is noteworthy that the upstream initiators of HDACs, the activity of DNMT1 or the expression of miRNAs in response to alcohol exposure are unknown and should be further explored. Another interesting question is whether any of these epigenetic modifications are directly linked to alterations in synaptic transmission and synaptic plasticity.
From signalling to circuitries
AUD is a complex disorder, with various clusters of behavioural phenotypes characterizing different stages of the condition (for examples, binge drinking and intoxication, withdrawal and negative affect, preoccupation and anticipation, craving and relapse)4. These abnormal behavioural phenotypes are thought to result from malfunctioning of several brain circuitries4, 160. Thus, intracellular signalling cascades affected by alcohol drinking must be taken in the context of their effects on brain circuits. Below, we provide some selected examples to illustrate the potential interplay between molecular signalling cascades and circuits (Fig. 3).
Figure 3. Signalling, neural circuits and alcohol.
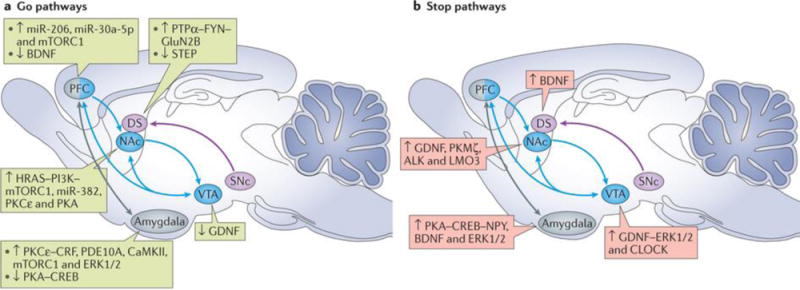
The go (part a) and stop (part b) pathways affect the function of several neural circuits. In the schematics, up arrows depict activation or increased expression of a molecule, whereas down arrows depict decreases in expression and/or activity of a molecule. The mesocorticolimbic circuitry (blue) mediates reward processing. Glial cell line-derived neurotrophic factor (GDNF), signalling via extracellular signal-regulated kinases 1 and 2 (ERK1/2) and circadian locomoter output cycles protein kaput (CLOCK), acts as a stop-pathway molecule in the ventral tegmental area (VTA) and dampens alcohol drinking, whereas HRAS–phosphoinositide 3-kinase (PI3K)–mechanistic target of rapamycin complex 1 (mTORC1), calcium/calmodulin-dependent protein kinase type II (CaMKII), protein kinase Cε (PKCε) and microRNA-382 (miR-382) act as go-pathway molecules in the nucleus accumbens (NAc) and promote alcohol drinking. In the prefrontal cortex (PFC), mTORC1 (go pathway) is activated in a reconsolidation of an alcohol-seeking session, and brain-derived neurotrophic factor (BDNF) levels are reduced after excessive drinking in response to increases in miR-206 and miR-30a-5p levels (go-pathway molecules). The nigrostriatal circuitry (purple) has a role in goal-directed behaviours (dorsomedial striatum (DMS)), habitual learning and compulsive behaviour (dorsolateral striatum (DLS)). The tyrosine-protein kinase FYN–protein-tyrosine phosphatase-α (PTPα)–GluN2B signalling pathway is centred in the DMS (go pathway), and BDNF–ERK1/2 signalling mainly occurs in the DLS (stop pathway). The reduction in activity of striatum-enriched protein-tyrosine phosphatase (STEP) in the DMS also contributes to the go pathway. The extended amygdala (grey) is implicated in the negative emotional state that characterizes alcohol withdrawal and relapse. The go-pathway actions of PKCε–corticotropin-releasing factor (CRF) and 3′,5′-cyclic nucleotide phosphodiesterase 10A (PDE10A) are centred in the central amygdala (CeA) and basolateral amygdala, respectively; CaMKII and ERK1/2 also have a role in go-pathway signalling in the CeA. These signalling molecules promote anxiety-like and alcohol-drinking behaviours. By contrast, the BDNF–ERK1/2 (stop) pathway in the CeA and medial amygdala (MeA) dampens these phenotypes. In addition, malfunctioning of cyclic AMP-dependent protein kinase A (PKA)–cAMP response element (CRE)-binding protein (CREB) signalling in the CeA and MeA also contributes to the interplay between stress and heightened drinking. Finally, mTORC1 (go pathway) in the CeA mediates alcohol-associated memory reconsolidation. ALK, anaplastic lymphoma kinase; DS, dorsal striatum; LMO3, LIM domain only protein 3; NPY, neuropeptide Y; PKMζ, protein kinase Mζ; SNc, substantia nigra pars compacta.
The mesolimbic system
The best-characterized brain circuit involved in addiction is the mesolimbic system, which consists of projections from the VTA to limbic regions such as the NAc, hippocampus and amygdala (Fig. 3). The mesolimbic system has a major role in learning, memory, motivation and reward161, and drugs of abuse are considered to ‘hijack’ this circuit162. During episodes of alcohol intake, the activity of the mesolimbic system increases, resulting in enhanced release of dopamine in the NAc163, which correlates with reinforcement of drug taking164. By contrast, the transition from drug use to abuse is typically associated with a decrease in mesolimbic activity, and a reduction in dopamine levels in the NAc is associated with abstinence6, 165. This dopaminergic deficiency is thought to have a key role in the allostatic mechanisms that cause a progressive reduction in the hedonic set point, resulting in increased alcohol seeking and intake6, 165.
Several of the aforementioned go- and stop-pathway molecules act within the mesolimbic system, and it is highly likely that imbalanced signalling within neurons projecting from the VTA to the limbic regions contributes to the transition from initial, moderate consumption of alcohol to alcohol abuse (Fig. 3). For example, the first drink of alcohol activates mTORC1 in D1R-expressing neurons in the NAc to stimulate synaptic plasticity and promote alcohol consumption56. In parallel, short-term consumption of alcohol leads to an increase in the expression of GDNF in the VTA76, enabling the activation of the ERK1/2 pathway to dampen consumption86, 87. Activation of mTORC1 in the NAc is maintained in response to long-term excessive drinking55, which is accompanied by the activation of PKCε62. However, long-term excessive drinking of alcohol also dysregulates GDNF signalling in the VTA76, which is correlated with mesolimbic dopamine deficiency86, 94. Thus, it is plausible that an orchestrated balance between go and stop processes, mediated by the mesolimbic dopaminergic signalling cascades, prevents or promotes escalation to pathological alcohol-drinking behaviours.
The nigrostriatal system
It is increasingly accepted that the dopaminergic nigrostriatal system, which projects from the substantia nigra to the dorsal striatum (Fig. 3), has a crucial role in the habitual and compulsive nature of alcohol addiction166, 167. Alcohol consumption is initially maintained by goal-directed behaviours, which are controlled by the NAc and the DMS; however, chronic drug and alcohol intake attenuates cortical control, and the subcortical dominance shifts from the DMS to the DLS, a brain region that drives habit learning167, 168, thus leading to impulsive and compulsive behaviours associated with addiction.
The FYN signalling cascade in the DMS promotes synaptic and structural plasticity, which, in turn, induce and maintain excessive alcohol consumption29, 40. By contrast, BDNF signalling in the DLS keeps alcohol intake in moderation78, 80, 81, possibly by preventing habit learning and/or compulsive behaviours. Thus, it is plausible that BDNF in the DLS maintains the drinking behaviour in a controlled, goal-directed manner. However, excessive drinking over a prolonged period leads to the inhibition of the actions of BDNF in the corticostriatal circuitry78, enabling the FYN pathway in the DMS to take over, followed by DLS-dependent habit formation via a yet-to-be-identified signalling pathway.
The extended amygdala
In addition to these mesencephalic–striatal circuits, the negative emotional state that often accompanies alcohol withdrawal is thought to engage the extended amygdala, an area composed of several basal forebrain regions, including the bed nucleus of the stria terminalis, the BLA, the CeA and the posterior shell of the NAc4, 165. These structures receive afferent connections from cortical and subcortical limbic regions, the midbrain and the lateral hypothalamus, and in turn project to the ventral pallidum, the VTA and the lateral hypothalamus64, 169, 170. The extended amygdala includes major components of the stress regulatory system of the brain, which also have a major role in negative reinforcement, in which alcohol intake serves to suppress negative states, to control behaviour4, 64. Activation of PKCε and the inhibition of PKA in the CeA, MeA or BLA of rodents drive anxiety-like behaviour during alcohol withdrawal and promote alcohol consumption65, 66, 119, 120, 127, 171. Thus, the balance between the activation state of PKA and PKCε in the extended amygdala seems to have a crucial role in the transition from positive to negative reinforcement mechanisms that govern the escalation and maintenance of alcohol intake after repeated alcohol intoxication.
Cortico-amygdalar pathways
The cortico-amygdalar networks mediate the retrieval and maintenance of long-term fear memories172, as well as fear extinction173. Retrieval of alcohol-associated memories activates mTORC1 in the mPFC, orbitofrontal cortex and CeA57; thus, this kinase is implicated in synaptic plasticity changes in the cortico-amygdalar circuitries that drive the retrieval and reconsolidation of these persistent memories, leading to relapse to alcohol seeking and drinking. Interestingly, cortico-amygdalar projections provide a link from mesocorticolimbic and corticostriatal neurons to the extended amygdala, as well as direct mesolimbic connections.
Together, the data discussed above indicate that alcohol can cause a multi-target imbalance in various signalling pathways within several brain circuitries (Fig. 3), which drive and maintain excessive, compulsive alcohol-drinking behaviours. A major challenge is identifying a direct link between the molecular cascades and the brain circuitries. The emergence of new state-of-the-art techniques that enable optical or chemical stimulation of intracellular signalling174, 175, 176 will allow the examination of signalling in the context of circuitries and behaviour. Indeed, using optogenetics, a recent study explored the circuit-specific role of ΔFOSB, a truncated product of the Fosb gene, in alcohol-drinking behaviours177, and showed that alcohol consumption induces ΔFOSB expression selectively in D1R-expressing neurons across all striatal regions. The authors used optogenetic tools to enhance neuronal activity in limbic regions that send synaptic inputs to the NAc, showing distinct patterns of ΔFOSB induction in medium spiny neuron subtypes in the NAc core and shell177.
Conclusions and open questions
Several interesting themes have emerged from the studies described above. First, alcohol use leads to brain region-specific and, potentially, neuron- and circuit-specific neuroadaptations that prevent or promote the transition from moderate to excessive alcohol use (the stop pathways and go pathways, respectively) (Figs 1,2,3). Second, alcohol-induced activation or inhibition of intracellular signalling seems to be determined by the amount of alcohol consumed and by the duration of exposure. For example, the levels of BDNF are increased in the DLS only in response to moderate alcohol intake, and mTORC1 signalling is activated only in response to high intake. Third, the same signalling molecules (for example, PKA and ERK1/2) contribute to both the go and stop pathways, making it likely that the opposing actions of these molecules are determined by the locus of activity.
Although alcohol seems to alter molecular cascades in a spatiotemporal-dependent manner, it is unclear how a seemingly nonspecific agent such as alcohol can produce such restricted signalling events. Indeed, this point leads to the fundamental question: how does alcohol exactly work? One possibility is that the primary effects of alcohol, like of other drugs of abuse, occur through a well-defined, limited number of binding partners, which then initiate the intracellular events. Several channels have been identified that directly interact with alcohol178, 179. However, these interactions cannot explain all, or even most, of the actions of alcohol on signalling.
Another intriguing possibility is that alcohol alters the architecture of membranes, especially the composition of lipid rafts. Specifically, participants in the go and stop pathways, such as HRAS, PLCγ, RET, TRKB and AC, reside in lipid rafts180, 181. By virtue of its structure, alcohol can easily penetrate membrane lipids, and pioneering studies in the 1970s discovered that alcohol can alter membrane fluidity and cholesterol content182, 183. Cholesterol content varies across brain regions, and thus alcohol may regulate a broad range of cell-surface receptor-induced signalling in a brain region-specific manner by changing the structure, fluidity and composition of membrane lipids, including cholesterol-enriched rafts. Several studies have started to address this possibility in cells184, 185 and in vivo30, 186, 187. However, data from animal models of alcohol consumption are sparse30, in part owing to technical limitations.
A related question is: how does alcohol simultaneously change the expression level and/or the activity of so many genes? Many of the signalling cascades described herein can potentially activate the transcription of immediate-early genes, including the FOS family of transcription factors. In fact, FOS immunoreactivity is increased in numerous brain regions in response to alcohol intake or withdrawal in rodents (for examples, see Refs188,189,190,191). FOS labelling is commonly used as a marker of neuronal activation; however, surprisingly little is known about the downstream consequences of FOS activation. This interesting line of investigation is now possible with the use of Fos–lacZ transgenic rats in combination with fluorescence-activated cell sorting (FACS), which allows the sorting of FOS-positive neurons192. Another intriguing possibility is that epigenetic modifications are upstream initiators of some, or most, of the molecular changes that occur in response to alcohol consumption and act as molecular network hubs that change the intracellular molecular landscape. Although, as described above, large-scale human genomic and epigenomic studies have been conducted in recent years, further studies are required to determine whether all, or a subset, of these genes contribute to (or prevent) the development of AUD and/or are related to pathological by-products of the disorder. Finally, it is noteworthy that epigenetic changes in the offspring of animals that have consumed alcohol have been recently reported193, and this exciting line of research merits further investigation.
It is important to note that the signalling cascades described in this Review have been directly linked to alcohol intake. However, the potential contribution of other alcohol-sensitive signalling molecules (Box 3) requires further investigation. Finally, the identification of genetic loci associated with AUD has been challenging, most likely because of the complexity of the disorder. Recent elegant investigations that combined animal and human studies have pushed the field forward, leading to the identification of mutations within novel signalling genes that may contribute to the innate differences in the susceptibility to develop these disorders (Box 4).
Interestingly, the go and stop pathways consist of different signalling and regional pathways that are not necessarily dependent on each other. For example, in the go pathways, blocking the alcohol-induced activation of the PI3K–AKT–mTORC1 pathway in the NAc prevents excessive alcohol drinking independently of the activation of the FYN–GluN2B–AMPAR pathway in the DMS, and vice versa. Thus, the normal function of each of the different components in the go pathways is sufficient, but not necessary, for the establishment and/or maintenance of alcohol-drinking behaviours. Furthermore, the same signalling pathways may have different roles in different phases and phenotypes of AUD. This interesting conclusion provides new promising opportunities for developing treatments for AUD, as targeting a single molecule may have sufficient efficacy to reduce alcohol-drinking behaviours. Several drugs that act on signalling have been approved by the US Food and Drug Administration (FDA) for other indications and have the potential to be used for the treatment of AUD. For example, in rodents, the FDA-approved drug cabergoline suppresses excessive alcohol drinking and relapse by upregulating GDNF in the VTA194, and the FDA-approved drug rapamycin decreases alcohol-drinking behaviours or prevents relapse55, 57. Rapamycin is an immunosuppressant, and it is therefore unlikely that it can be used to treat AUD; however, the rapid development of second-generation mTORC1 inhibitors195 provides an exciting new therapeutic opportunity. Finally, targeting enzymes that control epigenetic modifications may be another promising pharmacotherapeutic strategy, as it may be sufficient to inhibit a hub to produce desirable changes in behaviour. For example, the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA), an FDA-approved drug, is an effective inhibitor of alcohol-drinking behaviours in rodent models141. Thus, elucidating the molecular mechanisms underlying alcohol-drinking behaviours should allow us to move forward to translational paths for the treatment of AUD.
Supplementary Material
Acknowledgments
This Review is supported by the US National Institute on Alcohol Abuse and Alcoholism (NIAAA) of the National Institutes of Health (NIH-NIAAA RO1 AA016848, NIAAA R37 AA016848, NIH-NIAAAP50 AA017072, R01AA014366 and U01AA023489) to D.R. and by the Israel Science Foundation (ISF 968–13 and 1916–13), the Brain & Behavior Research Foundation (NARSAD 19114), the German Israel Foundation (GIF I-2348-105.4/2014) and the National Institute of Psychobiology in Israel (NIPI 110-14-15) to S.B.
References
- 1.World Health Organization. Global status report on alcohol and health 2014. WHO; 2014. [Google Scholar]
- 2.Enoch MA, Goldman D. Problem drinking and alcoholism: diagnosis and treatment. Am Fam Physician. 2002;65:441–448. [PubMed] [Google Scholar]
- 3.American Psychiatric Association. The Diagnostic and Statistical Manual of Mental Disorders DSM-5. 5th. American Psychiatric Publishing; 2013. [Google Scholar]
- 4.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koob GF. In: Behavioral Neurobiology of Alcohol Addiction. Sommer WH, Spanagel R, editors. Springer; 2013. pp. 3–30. [DOI] [PubMed] [Google Scholar]
- 6.Wise RA, Koob GF. The development and maintenance of drug addiction. Neuropsychopharmacology. 2014;39:254–262. doi: 10.1038/npp.2013.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- 8.Torregrossa MM, Corlett PR, Taylor JR. Aberrant learning and memory in addiction. Neurobiol Learn Mem. 2011;96:609–623. doi: 10.1016/j.nlm.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crews FT, Vetreno RP. Neuroimmune basis of alcoholic brain damage. Int Rev Neurobiol. 2014;118:315–357. doi: 10.1016/B978-0-12-801284-0.00010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ron D, Messing RO. In: Behavioral Neurobiology of Alcohol Addiction. Sommer WH, Spanagel R, editors. Springer; 2013. pp. 87–126. [DOI] [PubMed] [Google Scholar]
- 11.Ahmadiantehrani S, Warnault V, Legastelois R, Ron D. In: Neurobiology of Alcohol Dependence. Nohrona A, Cui C, Harris R, Crabbe J, editors. Elsevier; 2014. pp. 155–171. [Google Scholar]
- 12.Rothenfluh A, Troutwine B, Ghezzi A, Atkinson NS. In: Neurobiology of Alcohol Dependence. Nohrona A, Cui C, Harris R, Crabbe J, editors. Elsevier; 2014. pp. 467–494. [Google Scholar]
- 13.Abel T, Nguyen PV. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res. 2008;169:97–115. doi: 10.1016/S0079-6123(07)00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. doi: 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee AM, Messing RO. Protein kinases and addiction. Ann NY Acad Sci. 2008;1141:22–57. doi: 10.1196/annals.1441.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wand G, Levine M, Zweifel L, Schwindinger W, Abel T. The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J Neurosci. 2001;21:5297–5303. doi: 10.1523/JNEUROSCI.21-14-05297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maas JW, Jr, et al. Calcium-stimulated adenylyl cyclases are critical modulators of neuronal ethanol sensitivity. J Neurosci. 2005;25:4118–4126. doi: 10.1523/JNEUROSCI.4273-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao L, et al. βγ dimers mediate synergy of dopamine D2 and adenosine A2 receptor-stimulated PKA signaling and regulate ethanol consumption. Cell. 2002;109:733–743. doi: 10.1016/s0092-8674(02)00763-8. [DOI] [PubMed] [Google Scholar]
- 19.Mailliard WS, Diamond I. Recent advances in the neurobiology of alcoholism: the role of adenosine. Pharmacol Ther. 2004;101:39–46. doi: 10.1016/j.pharmthera.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Choi DS, et al. The type 1 equilibrative nucleoside transporter regulates ethanol intoxication and preference. Nat Neurosci. 2004;7:855–861. doi: 10.1038/nn1288. [DOI] [PubMed] [Google Scholar]
- 21.Nam HW, et al. Adenosine transporter ENT1 regulates the acquisition of goal-directed behavior and ethanol drinking through A2A receptor in the dorsomedial striatum. J Neurosci. 2013;33:4329–4338. doi: 10.1523/JNEUROSCI.3094-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arolfo MP, Yao L, Gordon AS, Diamond I, Janak PH. Ethanol operant self-administration in rats is regulated by adenosine A2 receptors. Alcohol Clin Exp Res. 2004;28:1308–1316. doi: 10.1097/01.alc.0000139821.38167.20. [DOI] [PubMed] [Google Scholar]
- 23.Thorsell A, Johnson J, Heilig M. Effect of the adenosine A2a receptor antagonist 3,7-dimethyl-propargylxanthine on anxiety-like and depression-like behavior and alcohol consumption in Wistar rats. Alcohol Clin Exp Res. 2007;31:1302–1307. doi: 10.1111/j.1530-0277.2007.00425.x. [DOI] [PubMed] [Google Scholar]
- 24.Darcq E, et al. Inhibition of striatal-enriched tyrosine phosphatase 61 in the dorsomedial striatum is sufficient to increased ethanol consumption. J Neurochem. 2014;129:1024–1034. doi: 10.1111/jnc.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben Hamida S, et al. The small G protein H-Ras in the mesolimbic system is a molecular gateway to alcohol-seeking and excessive drinking behaviors. J Neurosci. 2012;32:15849–15858. doi: 10.1523/JNEUROSCI.2846-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohnishi H, Murata Y, Okazawa H, Matozaki T. Src family kinases: modulators of neurotransmitter receptor function and behavior. Trends Neurosci. 2011;34:629–637. doi: 10.1016/j.tins.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Trepanier CH, Jackson MF, MacDonald JF. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2012;279:12–19. doi: 10.1111/j.1742-4658.2011.08391.x. [DOI] [PubMed] [Google Scholar]
- 28.Goebel-Goody SM, et al. Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacol Rev. 2012;64:65–87. doi: 10.1124/pr.110.003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, et al. Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J Neurosci. 2010;30:10187–10198. doi: 10.1523/JNEUROSCI.2268-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibb SL, Hamida SB, Lanfranco MF, Ron D. Ethanol-induced increase in Fyn kinase activity in the dorsomedial striatum is associated with subcellular redistribution of protein tyrosine phosphatase α. J Neurochem. 2011;119:879–889. doi: 10.1111/j.1471-4159.2011.07485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yaka R, Phamluong K, Ron D. Scaffolding of Fyn kinase to the NMDA receptor determines brain region sensitivity to ethanol. J Neurosci. 2003;23:3623–3632. doi: 10.1523/JNEUROSCI.23-09-03623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J, Kurup P, Foscue E, Lombroso PJ. Striatal-enriched protein tyrosine phosphatase regulates the PTPα/Fyn signaling pathway. J Neurochem. 2015;134:629–641. doi: 10.1111/jnc.13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhandari V, Lim KL, Pallen CJ. Physical and functional interactions between receptor-like protein-tyrosine phosphatase α and p59fyn. J Biol Chem. 1998;273:8691–8698. doi: 10.1074/jbc.273.15.8691. [DOI] [PubMed] [Google Scholar]
- 34.Coultrap SJ, Bayer KU. CaMKII regulation in information processing and storage. Trends Neurosci. 2012;35:607–618. doi: 10.1016/j.tins.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salling MC, et al. Moderate alcohol drinking and the amygdala proteome: identification and validation of calcium/calmodulin dependent kinase II and AMPA receptor activity as novel molecular mechanisms of the positive reinforcing effects of alcohol. Biol Psychiatry. 2014;79:430–442. doi: 10.1016/j.biopsych.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Easton AC, et al. αCaMKII autophosphorylation controls the establishment of alcohol drinking behavior. Neuropsychopharmacology. 2013;38:1636–1647. doi: 10.1038/npp.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, et al. Ethanol-mediated facilitation of AMPA receptor function in the dorsomedial striatum: implications for alcohol drinking behavior. J Neurosci. 2012;32:15124–15132. doi: 10.1523/JNEUROSCI.2783-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, et al. Alcohol elicits functional and structural plasticity selectively in dopamine D1 receptor-expressing neurons of the dorsomedial striatum. J Neurosci. 2015;35:11634–11643. doi: 10.1523/JNEUROSCI.0003-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben Hamida S, et al. Protein tyrosine phosphatase α in the dorsomedial striatum promotes excessive ethanol-drinking behaviors. J Neurosci. 2013;33:14369–14378. doi: 10.1523/JNEUROSCI.1954-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Legastelois R, Darcq E, Wegner SA, Lombroso PJ, Ron D. Striatal-enriched protein tyrosine phosphatase controls responses to aversive stimuli: implication for ethanol drinking. PLoS ONE. 2015;10:e0127408. doi: 10.1371/journal.pone.0127408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye X, Carew TJ. Small G protein signaling in neuronal plasticity and memory formation: the specific role of Ras family proteins. Neuron. 2010;68:340–361. doi: 10.1016/j.neuron.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Repunte-Canonigo V, et al. Genome-wide gene expression analysis identifies K-ras as a regulator of alcohol intake. Brain Res. 2010;1339:1–10. doi: 10.1016/j.brainres.2010.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feig LA. Regulation of neuronal function by Ras-GRF exchange factors. Genes Cancer. 2011;2:306–319. doi: 10.1177/1947601911408077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baouz S, et al. Sites of phosphorylation by protein kinase A in CDC25Mm/GRF1, a guanine nucleotide exchange factor for Ras. J Biol Chem. 2001;276:1742–1749. doi: 10.1074/jbc.M005770200. [DOI] [PubMed] [Google Scholar]
- 46.Mulligan MK, et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci USA. 2006;103:6368–6373. doi: 10.1073/pnas.0510188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stacey D, et al. RASGRF2 regulates alcohol-induced reinforcement by influencing mesolimbic dopamine neuron activity and dopamine release. Proc Natl Acad Sci USA. 2012;109:21128–21133. doi: 10.1073/pnas.1211844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cozzoli DK, et al. Binge drinking upregulates accumbens mGluR5–Homer2–PI3K signaling: functional implications for alcoholism. J Neurosci. 2009;29:8655–8668. doi: 10.1523/JNEUROSCI.5900-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neasta J, Ben Hamida S, Yowell QV, Carnicella S, Ron D. AKT signaling pathway in the nucleus accumbens mediates excessive alcohol drinking behaviors. Biol Psychiatry. 2011;70:575–582. doi: 10.1016/j.biopsych.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu F, et al. mTORC1-dependent translation of collapsin response mediator protein-2 drives neuroadaptations underlying excessive alcohol drinking behaviors. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.12. http://dx.doi.org/10.1038/mp.2016.12. [DOI] [PMC free article] [PubMed]
- 52.Buffington SA, Huang W, Costa-Mattioli M. Translational control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci. 2014;37:17–38. doi: 10.1146/annurev-neuro-071013-014100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neasta J, Barak S, Hamida SB, Ron D. mTOR complex 1: a key player in neuroadaptations induced by drugs of abuse. J Neurochem. 2014;130:172–184. doi: 10.1111/jnc.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neasta J, Ben Hamida S, Yowell Q, Carnicella S, Ron D. Role for mammalian target of rapamycin complex 1 signaling in neuroadaptations underlying alcohol-related disorders. Proc Natl Acad Sci USA. 2010;107:20093–20098. doi: 10.1073/pnas.1005554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beckley JT, Laguesse S, Phamluong K, Wegner SA, Ron D. The first alcohol drink triggers mTORC1-dependent synaptic plasticity in nucleus accumbens dopamine D1 receptor neurons. J Neurosci. 2016;36:701–13. doi: 10.1523/JNEUROSCI.2254-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barak S, et al. Disruption of alcohol-related memories by mTORC1 inhibition prevents relapse. Nat Neurosci. 2013;16:1111–1117. doi: 10.1038/nn.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Azzi A, Boscoboinik D, Hensey C. The protein kinase C family. Eur J Biochem. 1992;208:547–557. doi: 10.1111/j.1432-1033.1992.tb17219.x. [DOI] [PubMed] [Google Scholar]
- 59.Hodge CW, et al. Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCε. Nat Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- 60.Olive MF, Mehmert KK, Messing RO, Hodge CW. Reduced operant ethanol self-administration and in vivo mesolimbic dopamine responses to ethanol in PKCε-deficient mice. Eur J Neurosci. 2000;12:4131–4140. doi: 10.1046/j.1460-9568.2000.00297.x. [DOI] [PubMed] [Google Scholar]
- 61.Choi DS, Wang D, Dadgar J, Chang WS, Messing RO. Conditional rescue of protein kinase C ε regulates ethanol preference and hypnotic sensitivity in adult mice. J Neurosci. 2002;22:9905–9911. doi: 10.1523/JNEUROSCI.22-22-09905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cozzoli DK, et al. Protein kinase C epsilon activity in the nucleus accumbens and central nucleus of the amygdala mediates binge alcohol consumption. Biol Psychiatry. 2015;79:443–451. doi: 10.1016/j.biopsych.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lesscher HM, et al. Amygdala protein kinase C epsilon controls alcohol consumption. Genes Brain Behav. 2009;8:493–499. doi: 10.1111/j.1601-183X.2009.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janak PH, Tye KM. From circuits to behaviour in the amygdala. Nature. 2015;517:284–292. doi: 10.1038/nature14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hodge CW, et al. Decreased anxiety-like behavior, reduced stress hormones, and neurosteroid supersensitivity in mice lacking protein kinase Cε. J Clin Invest. 2002;110:1003–1010. doi: 10.1172/JCI15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lesscher HM, et al. Amygdala protein kinase C epsilon regulates corticotropin-releasing factor and anxiety-like behavior. Genes Brain Behav. 2008;7:323–333. doi: 10.1111/j.1601-183X.2007.00356.x. [DOI] [PubMed] [Google Scholar]
- 67.Zorrilla EP, Logrip ML, Koob GF. Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol. 2014;35:234–244. doi: 10.1016/j.yfrne.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bajo M, Cruz MT, Siggins GR, Messing R, Roberto M. Protein kinase C epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proc Natl Acad Sci USA. 2008;105:8410–8415. doi: 10.1073/pnas.0802302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 70.Pascoli V, Cahill E, Bellivier F, Caboche J, Vanhoutte P. Extracellular signal-regulated protein kinases 1 and 2 activation by addictive drugs: a signal toward pathological adaptation. Biol Psychiatry. 2014;76:917–926. doi: 10.1016/j.biopsych.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 71.Schroeder JP, et al. Cue-induced reinstatement of alcohol-seeking behavior is associated with increased ERK1/2 phosphorylation in specific limbic brain regions: blockade by the mGluR5 antagonist MPEP. Neuropharmacology. 2008;55:546–554. doi: 10.1016/j.neuropharm.2008.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshii A, Constantine-Paton M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol. 2010;70:304–322. doi: 10.1002/dneu.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- 74.McGough NN, et al. RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci. 2004;24:10542–10552. doi: 10.1523/JNEUROSCI.3714-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Logrip ML, Janak PH, Ron D. Escalating ethanol intake is associated with altered corticostriatal BDNF expression. J Neurochem. 2009;109:1459–1468. doi: 10.1111/j.1471-4159.2009.06073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ahmadiantehrani S, Barak S, Ron D. GDNF is a novel ethanol-responsive gene in the VTA: implications for the development and persistence of excessive drinking. Addict Biol. 2014;19:623–633. doi: 10.1111/adb.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hensler JG, Ladenheim EE, Lyons WE. Ethanol consumption and serotonin-1A (5-HT1A) receptor function in heterozygous BDNF (+/−) mice. J Neurochem. 2003;85:1139–1147. doi: 10.1046/j.1471-4159.2003.01748.x. [DOI] [PubMed] [Google Scholar]
- 78.Logrip ML, Barak S, Warnault V, Ron D. Corticostriatal BDNF and alcohol addiction. Brain Res. 2015;1628:60–67. doi: 10.1016/j.brainres.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carnicella S, Ahmadiantehrani S, Janak PH, Ron D. GDNF is an endogenous negative regulator of ethanol-mediated reward and of ethanol consumption after a period of abstinence. Alcohol Clin Exp Res. 2009;33:1012–1024. doi: 10.1111/j.1530-0277.2009.00922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jeanblanc J, et al. Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. J Neurosci. 2009;29:13494–13502. doi: 10.1523/JNEUROSCI.2243-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jeanblanc J, Logrip ML, Janak PH, Ron D. BDNF-mediated regulation of ethanol consumption requires the activation of the MAP kinase pathway and protein synthesis. Eur J Neurosci. 2013;37:607–612. doi: 10.1111/ejn.12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darcq E, et al. MicroRNA-30a-5p in the prefrontal cortex controls the transition from moderate to excessive alcohol consumption. Mol Psychiatry. 2014;20:1219–1231. doi: 10.1038/mp.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Warnault V, et al. The BDNF valine 68 to methionine polymorphism increases compulsive alcohol drinking in mice that is reversed by tropomyosin receptor kinase B activation. Biol Psychiatry. 2015;79:463–473. doi: 10.1016/j.biopsych.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pandey SC, Roy A, Zhang H, Xu T. Partial deletion of the cAMP response element-binding protein gene promotes alcohol-drinking behaviors. J Neurosci. 2004;24:5022–5030. doi: 10.1523/JNEUROSCI.5557-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pandey SC, Zhang H, Roy A, Misra K. Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J Neurosci. 2006;26:8320–8331. doi: 10.1523/JNEUROSCI.4988-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barak S, et al. Glial cell line-derived neurotrophic factor (GDNF) is an endogenous protector in the mesolimbic system against excessive alcohol consumption and relapse. Addict Biol. 2015;20:629–642. doi: 10.1111/adb.12152. [DOI] [PubMed] [Google Scholar]
- 87.Carnicella S, Kharazia V, Jeanblanc J, Janak PH, Ron D. GDNF is a fast-acting potent inhibitor of alcohol consumption and relapse. Proc Natl Acad Sci USA. 2008;105:8114–8119. doi: 10.1073/pnas.0711755105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Logrip ML, Janak PH, Ron D. Dynorphin is a downstream effector of striatal BDNF regulation of ethanol intake. FASEB J. 2008;22:2393–2404. doi: 10.1096/fj.07-099135. [DOI] [PubMed] [Google Scholar]
- 89.Jeanblanc J, et al. The dopamine D3 receptor is part of a homeostatic pathway regulating ethanol consumption. J Neurosci. 2006;26:1457–1464. doi: 10.1523/JNEUROSCI.3786-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moonat S, Sakharkar AJ, Zhang H, Pandey SC. The role of amygdaloid brain-derived neurotrophic factor, activity-regulated cytoskeleton-associated protein and dendritic spines in anxiety and alcoholism. Addict Biol. 2011;16:238–250. doi: 10.1111/j.1369-1600.2010.00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.You C, Zhang H, Sakharkar AJ, Teppen T, Pandey SC. Reversal of deficits in dendritic spines, BDNF and Arc expression in the amygdala during alcohol dependence by HDAC inhibitor treatment. Int J Neuropsychopharmacol. 2014;17:313–322. doi: 10.1017/S1461145713001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pandey SC, et al. Effector immediate-early gene Arc in the amygdala plays a critical role in alcoholism. J Neurosci. 2008;28:2589–2600. doi: 10.1523/JNEUROSCI.4752-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stragier E, et al. Ethanol-induced epigenetic regulations at the Bdnf gene in C57BL/6J mice. Mol Psychiatry. 2014;20:405–412. doi: 10.1038/mp.2014.38. [DOI] [PubMed] [Google Scholar]
- 94.Barak S, Carnicella S, Yowell QV, Ron D. Glial cell line-derived neurotrophic factor reverses alcohol-induced allostasis of the mesolimbic dopaminergic system: implications for alcohol reward and seeking. J Neurosci. 2011;31:9885–9894. doi: 10.1523/JNEUROSCI.1750-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Barak S, Ahmadiantehrani S, Kharazia V, Ron D. Positive autoregulation of GDNF levels in the ventral tegmental area mediates long-lasting inhibition of excessive alcohol consumption. Transl Psychiatry. 2011;1:e60. doi: 10.1038/tp.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tapocik JD, et al. microRNA-206 in rat medial prefrontal cortex regulates BDNF expression and alcohol drinking. J Neurosci. 2014;34:4581–4588. doi: 10.1523/JNEUROSCI.0445-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Joe KH, et al. Decreased plasma brain-derived neurotrophic factor levels in patients with alcohol dependence. Alcohol Clin Exp Res. 2007;31:1833–1838. doi: 10.1111/j.1530-0277.2007.00507.x. [DOI] [PubMed] [Google Scholar]
- 98.Heberlein A, et al. BDNF and GDNF serum levels in alcohol-dependent patients during withdrawal. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:1060–1064. doi: 10.1016/j.pnpbp.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 99.Prakash A, Zhang H, Pandey SC. Innate differences in the expression of brain-derived neurotrophic factor in the regions within the extended amygdala between alcohol preferring and nonpreferring rats. Alcohol Clin Exp Res. 2008;32:909–920. doi: 10.1111/j.1530-0277.2008.00650.x. [DOI] [PubMed] [Google Scholar]
- 100.Egan MF, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 101.Chen ZY, et al. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci. 2004;24:4401–4411. doi: 10.1523/JNEUROSCI.0348-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matsushita S, et al. Association study of brain-derived neurotrophic factor gene polymorphism and alcoholism. Alcohol Clin Exp Res. 2004;28:1609–1612. doi: 10.1097/01.alc.0000145697.81741.d2. [DOI] [PubMed] [Google Scholar]
- 103.Wojnar M, et al. Association between Val66Met brain-derived neurotrophic factor (BDNF) gene polymorphism and post-treatment relapse in alcohol dependence. Alcohol Clin Exp Res. 2009;33:693–702. doi: 10.1111/j.1530-0277.2008.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mon A, et al. Brain-derived neurotrophic factor genotype is associated with brain gray and white matter tissue volumes recovery in abstinent alcohol-dependent individuals. Genes Brain Behav. 2013;12:98–107. doi: 10.1111/j.1601-183X.2012.00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Agoglia AE, et al. Alcohol alters the activation of ERK1/2, a functional regulator of binge alcohol drinking in adult C57BL/6J mice. Alcohol Clin Exp Res. 2015;39:463–475. doi: 10.1111/acer.12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Faccidomo S, Salling MC, Galunas C, Hodge CW. Operant ethanol self-administration increases extracellular-signal regulated protein kinase (ERK) phosphorylation in reward-related brain regions: selective regulation of positive reinforcement in the prefrontal cortex of C57BL/6J mice. Psychopharmacology. 2015;232:3417–3430. doi: 10.1007/s00213-015-3993-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Faccidomo S, Besheer J, Stanford PC, Hodge CW. Increased operant responding for ethanol in male C57BL/6J mice: specific regulation by the ERK1/2, but not JNK, MAP kinase pathway. Psychopharmacology. 2009;204:135–147. doi: 10.1007/s00213-008-1444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Karatsoreos IN. Links between circadian rhythms and psychiatric disease. Front Behav Neurosci. 2014;8:162. doi: 10.3389/fnbeh.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spanagel R, Rosenwasser AM, Schumann G, Sarkar DK. Alcohol consumption and the body’s biological clock. Alcohol Clin Exp Res. 2005;29:1550–1557. doi: 10.1097/01.alc.0000175074.70807.fd. [DOI] [PubMed] [Google Scholar]
- 110.Dong L, et al. Effects of the circadian rhythm gene period 1 (Per1) on psychosocial stress-induced alcohol drinking. Am J Psychiatry. 2011;168:1090–1098. doi: 10.1176/appi.ajp.2011.10111579. [DOI] [PubMed] [Google Scholar]
- 111.Spanagel R, et al. The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nat Med. 2005;11:35–42. doi: 10.1038/nm1163. [DOI] [PubMed] [Google Scholar]
- 112.Blomeyer D, et al. Association of PER2 genotype and stressful life events with alcohol drinking in young adults. PLoS ONE. 2013;8:e59136. doi: 10.1371/journal.pone.0059136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ozburn AR, et al. The role of clock in ethanol-related behaviors. Neuropsychopharmacology. 2013;38:2393–2400. doi: 10.1038/npp.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Eide EJ, et al. Control of mammalian circadian rhythm by CKIε-regulated proteasome-mediated PER2 degradation. Mol Cell Biol. 2005;25:2795–2807. doi: 10.1128/MCB.25.7.2795-2807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Perreau-Lenz S, et al. Inhibition of the casein-kinase-1-epsilon/delta/prevents relapse-like alcohol drinking. Neuropsychopharmacology. 2012;37:2121–2131. doi: 10.1038/npp.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim KS, Kim H, Baek IS, Lee KW, Han PL. Mice lacking adenylyl cyclase type 5 (AC5) show increased ethanol consumption and reduced ethanol sensitivity. Psychopharmacology. 2011;215:391–398. doi: 10.1007/s00213-010-2143-x. [DOI] [PubMed] [Google Scholar]
- 117.Thiele TE, et al. High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci. 2000;20:RC75. doi: 10.1523/JNEUROSCI.20-10-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pandey SC. The gene transcription factor cyclic AMP-responsive element binding protein: role in positive and negative affective states of alcohol addiction. Pharmacol Ther. 2004;104:47–58. doi: 10.1016/j.pharmthera.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 119.Pandey SC, Roy A, Zhang H. The decreased phosphorylation of cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein in the central amygdala acts as a molecular substrate for anxiety related to ethanol withdrawal in rats. Alcohol Clin Exp Res. 2003;27:396–409. doi: 10.1097/01.ALC.0000056616.81971.49. [DOI] [PubMed] [Google Scholar]
- 120.Pandey SC, Zhang H, Roy A, Xu T. Deficits in amygdaloid cAMP-responsive element-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J Clin Invest. 2005;115:2762–2773. doi: 10.1172/JCI24381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Moonat S, Starkman BG, Sakharkar A, Pandey SC. Neuroscience of alcoholism: molecular and cellular mechanisms. Cell Mol Life Sci. 2010;67:73–88. doi: 10.1007/s00018-009-0135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Heilig M. The NPY system in stress, anxiety and depression. Neuropeptides. 2004;38:213–224. doi: 10.1016/j.npep.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 123.Thiele TE, Marsh DJ, Ste Marie L, Bernstein IL, Palmiter RD. Ethanol consumption and resistance are inversely related to neuropeptide Y levels. Nature. 1998;396:366–369. doi: 10.1038/24614. [DOI] [PubMed] [Google Scholar]
- 124.Bell RL, et al. Ibudilast reduces alcohol drinking in multiple animal models of alcohol dependence. Addict Biol. 2015;20:38–42. doi: 10.1111/adb.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wen RT, et al. The phosphodiesterase-4 (PDE4) inhibitor rolipram decreases ethanol seeking and consumption in alcohol-preferring Fawn-Hooded rats. Alcohol Clin Exp Res. 2012;36:2157–2167. doi: 10.1111/j.1530-0277.2012.01845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Franklin KM, et al. Reduction of alcohol drinking of alcohol-preferring (P) and high-alcohol drinking (HAD1) rats by targeting phosphodiesterase-4 (PDE4) Psychopharmacology. 2015;232:2251–2262. doi: 10.1007/s00213-014-3852-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Logrip ML, Vendruscolo LF, Schlosburg JE, Koob GF, Zorrilla EP. Phosphodiesterase 10A regulates alcohol and saccharin self-administration in rats. Neuropsychopharmacology. 2014;39:1722–1731. doi: 10.1038/npp.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Logrip ML, Zorrilla EP. Stress history increases alcohol intake in relapse: relation to phosphodiesterase 10A. Addict Biol. 2012;17:920–933. doi: 10.1111/j.1369-1600.2012.00460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Logrip ML, Zorrilla EP. Differential changes in amygdala and frontal cortex Pde10a expression during acute and protracted withdrawal. Front Integr Neurosci. 2014;8:30. doi: 10.3389/fnint.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee AM, et al. Deletion of Prkcz increases intermittent ethanol consumption in mice. Alcohol Clin Exp Res. 2014;38:170–178. doi: 10.1111/acer.12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee AM, et al. Prkcz null mice show normal learning and memory. Nature. 2013;493:416–419. doi: 10.1038/nature11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–423. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Savarese A, Zou ME, Kharazia V, Maiya R, Lasek AW. Increased behavioral responses to ethanol in Lmo3 knockout mice. Genes Brain Behav. 2014;13:777–783. doi: 10.1111/gbb.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lasek AW, et al. An evolutionary conserved role for anaplastic lymphoma kinase in behavioral responses to ethanol. PLoS ONE. 2011;6:e22636. doi: 10.1371/journal.pone.0022636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lasek AW, Giorgetti F, Berger KH, Tayor S, Heberlein U. Lmo genes regulate behavioral responses to ethanol in Drosophila melanogaster and the mouse. Alcohol Clin Exp Res. 2011;35:1600–1606. doi: 10.1111/j.1530-0277.2011.01506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 137.Repunte-Canonigo V, et al. Nf1 regulates alcohol dependence-associated excessive drinking and gamma-aminobutyric acid release in the central amygdala in mice and is associated with alcohol dependence in humans. Biol Psychiatry. 2015;77:870–879. doi: 10.1016/j.biopsych.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nestler EJ. Epigenetic mechanisms of drug addiction. Neuropharmacology. 2014;76:259–268. doi: 10.1016/j.neuropharm.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bali P, Kenny PJ. MicroRNAs and drug addiction. Front Genet. 2013;4:43. doi: 10.3389/fgene.2013.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Krishnan HR, Sakharkar AJ, Teppen TL, Berkel TD, Pandey SC. The epigenetic landscape of alcoholism. Int Rev Neurobiol. 2014;115:75–116. doi: 10.1016/B978-0-12-801311-3.00003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Warnault V, Darcq E, Levine A, Barak S, Ron D. Chromatin remodeling — a novel strategy to control excessive alcohol drinking. Transl Psychiatry. 2013;3:e231. doi: 10.1038/tp.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci. 2008;28:3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC. Aberrant histone deacetylase2-mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism. Biol Psychiatry. 2013;73:763–773. doi: 10.1016/j.biopsych.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Barbier E, et al. DNA methylation in the medial prefrontal cortex regulates alcohol-induced behavior and plasticity. J Neurosci. 2015;35:6153–6164. doi: 10.1523/JNEUROSCI.4571-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang R, et al. Genome-wide DNA methylation analysis in alcohol dependence. Addict Biol. 2013;18:392–403. doi: 10.1111/adb.12037. [DOI] [PubMed] [Google Scholar]
- 146.Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J Neurosci. 2012;32:1884–1897. doi: 10.1523/JNEUROSCI.3136-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ruggeri B, et al. Association of protein phosphatase PPM1G with alcohol use disorder and brain activity during behavioral control in a genome-wide methylation analysis. Am J Psychiatry. 2015;172:543–552. doi: 10.1176/appi.ajp.2014.14030382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Heberlein A, et al. Epigenetic down regulation of nerve growth factor during alcohol withdrawal. Addict Biol. 2013;18:508–510. doi: 10.1111/j.1369-1600.2010.00307.x. [DOI] [PubMed] [Google Scholar]
- 149.Heberlein A, et al. Do changes in the BDNF promoter methylation indicate the risk of alcohol relapse? Eur Neuropsychopharmacol. 2015;25:1892–1897. doi: 10.1016/j.euroneuro.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 150.Bothwell M. In: Neurotrophic Factors. Lewin GR, Carter BD, editors. Springer; 2014. pp. 3–15. [Google Scholar]
- 151.Miranda RC, et al. MicroRNAs: master regulators of ethanol abuse and toxicity? Alcohol Clin Exp Res. 2010;34:575–587. doi: 10.1111/j.1530-0277.2009.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Most D, Workman E, Harris RA. Synaptic adaptations by alcohol and drugs of abuse: changes in microRNA expression and mRNA regulation. Front Mol Neurosci. 2014;7:85. doi: 10.3389/fnmol.2014.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nunez YO, Mayfield RD. Understanding alcoholism through microRNA signatures in brains of human alcoholics. Front Genet. 2012;3:43. doi: 10.3389/fgene.2012.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pietrzykowski AZ, et al. Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron. 2008;59:274–287. doi: 10.1016/j.neuron.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li J, et al. MicroRNA expression profile and functional analysis reveal that miR-382 is a critical novel gene of alcohol addiction. EMBO Mol Med. 2013;5:1402–1414. doi: 10.1002/emmm.201201900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Beech RD, et al. Stress-related alcohol consumption in heavy drinkers correlates with expression of miR-10a, miR-21, and components of the TAR-RNA-binding protein-associated complex. Alcohol Clin Exp Res. 2014;38:2743–2753. doi: 10.1111/acer.12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tapocik JD, et al. Coordinated dysregulation of mRNAs and microRNAs in the rat medial prefrontal cortex following a history of alcohol dependence. Pharmacogenomics J. 2013;13:286–296. doi: 10.1038/tpj.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lewohl JM, et al. Up-regulation of microRNAs in brain of human alcoholics. Alcohol Clin Exp Res. 2011;35:1928–1937. doi: 10.1111/j.1530-0277.2011.01544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Most D, Leiter C, Blednov YA, Harris RA, Mayfield RD. Synaptic microRNAs coordinately regulate synaptic mRNAs: perturbation by chronic alcohol consumption. Neuropsychopharmacology. 2016;41:538–548. doi: 10.1038/npp.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pickens CL, et al. Neurobiology of the incubation of drug craving. Trends Neurosci. 2011;34:411–420. doi: 10.1016/j.tins.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Berridge KC, Kringelbach ML. Pleasure systems in the brain. Neuron. 2015;86:646–664. doi: 10.1016/j.neuron.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Volkow ND, Morales M. The brain on drugs: from reward to addiction. Cell. 2015;162:712–725. doi: 10.1016/j.cell.2015.07.046. [DOI] [PubMed] [Google Scholar]
- 163.Gonzales RA, Job MO, Doyon WM. The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacol Ther. 2004;103:121–146. doi: 10.1016/j.pharmthera.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 164.Pierce RC, Kumaresan V. The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev. 2006;30:215–238. doi: 10.1016/j.neubiorev.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 165.Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 166.Wise RA. Roles for nigrostriatal—not just mesocorticolimbic—dopamine in reward and addiction. Trends Neurosci. 2009;32:517–524. doi: 10.1016/j.tins.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Everitt BJ, Robbins TW. From the ventral to the dorsal striatum: devolving views of their roles in drug addiction. Neurosci Biobehav Rev. 2013;37:1946–1954. doi: 10.1016/j.neubiorev.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 168.Corbit LH, Nie H, Janak PH. Habitual alcohol seeking: time course and the contribution of subregions of the dorsal striatum. Biol Psychiatry. 2012;72:389–395. doi: 10.1016/j.biopsych.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Heimer L, Alheid GF. In: The Basal Forebrain. Napier TC, Kalivas PW, Hanin I, editors. Springer; 1991. pp. 1–42. [Google Scholar]
- 170.Koob GF. Brain stress systems in the amygdala and addiction. Brain Res. 2009;1293:61–75. doi: 10.1016/j.brainres.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang H, Pandey SC. Effects of PKA modulation on the expression of neuropeptide Y in rat amygdaloid structures during ethanol withdrawal. Peptides. 2003;24:1397–1402. doi: 10.1016/j.peptides.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 172.Do-Monte FH, Quinones-Laracuente K, Quirk GJ. A temporal shift in the circuits mediating retrieval of fear memory. Nature. 2015;519:460–463. doi: 10.1038/nature14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kaplan GB, Heinrichs SC, Carey RJ. Treatment of addiction and anxiety using extinction approaches: neural mechanisms and their treatment implications. Pharmacol Biochem Behav. 2011;97:619–625. doi: 10.1016/j.pbb.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 174.Toettcher JE, Weiner OD, Lim WA. Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell. 2013;155:1422–1434. doi: 10.1016/j.cell.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Doupe DP, Perrimon N. Visualizing and manipulating temporal signaling dynamics with fluorescence-based tools. Sci Signal. 2014;7:re1. doi: 10.1126/scisignal.2005077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wend S, et al. Optogenetic control of protein kinase activity in mammalian cells. ACS Synth Biol. 2014;3:280–285. doi: 10.1021/sb400090s. [DOI] [PubMed] [Google Scholar]
- 177.Lobo MK, et al. ΔFosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. J Neurosci. 2013;33:18381–18395. doi: 10.1523/JNEUROSCI.1875-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Trudell JR, Messing RO, Mayfield J, Harris RA. Alcohol dependence: molecular and behavioral evidence. Trends Pharmacol Sci. 2014;35:317–323. doi: 10.1016/j.tips.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Holmes A, Spanagel R, Krystal JH. Glutamatergic targets for new alcohol medications. Psychopharmacology. 2013;229:539–554. doi: 10.1007/s00213-013-3226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- 181.Eisenberg S, Shvartsman DE, Ehrlich M, Henis YI. Clustering of raft-associated proteins in the external membrane leaflet modulates internal leaflet H-Ras diffusion and signaling. Mol Cell Biol. 2006;26:7190–7200. doi: 10.1128/MCB.01059-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chin JH, Goldstein DB. Electron paramagnetic resonance studies of ethanol on membrane fluidity. Adv Exp Med Biol. 1977;85A:111–122. doi: 10.1007/978-1-4899-5181-6_8. [DOI] [PubMed] [Google Scholar]
- 183.Chin JH, Parsons LM, Goldstein DB. Increased cholesterol content of erythrocyte and brain membranes in ethanol-tolerant mice. Biochim Biophys Acta. 1978;513:358–363. doi: 10.1016/0005-2736(78)90204-3. [DOI] [PubMed] [Google Scholar]
- 184.Pascual-Lucas M, Fernandez-Lizarbe S, Montesinos J, Guerri C. LPS or ethanol triggers clathrin- and rafts/caveolae-dependent endocytosis of TLR4 in cortical astrocytes. J Neurochem. 2014;129:448–462. doi: 10.1111/jnc.12639. [DOI] [PubMed] [Google Scholar]
- 185.Tobin SJ, et al. Nanoscale effects of ethanol and naltrexone on protein organization in the plasma membrane studied by photoactivated localization microscopy (PALM) PLoS ONE. 2014;9:e87225. doi: 10.1371/journal.pone.0087225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Petit-Paitel A, et al. Prion protein is a key determinant of alcohol sensitivity through the modulation of N-methyl-d-aspartate receptor (NMDAR) activity. PLoS ONE. 2012;7:e34691. doi: 10.1371/journal.pone.0034691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bettinger JC, Leung K, Bolling MH, Goldsmith AD, Davies AG. Lipid environment modulates the development of acute tolerance to ethanol in Caenorhabditis elegans. PLoS ONE. 2012;7:e35192. doi: 10.1371/journal.pone.0035192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Vilpoux C, Warnault V, Pierrefiche O, Daoust M, Naassila M. Ethanol-sensitive brain regions in rat and mouse: a cartographic review, using immediate early gene expression. Alcohol Clin Exp Res. 2009;33:945–969. doi: 10.1111/j.1530-0277.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- 189.Barson JR, Ho HT, Leibowitz SF. Anterior thalamic paraventricular nucleus is involved in intermittent access ethanol drinking: role of orexin receptor 2. Addict Biol. 2015;20:469–481. doi: 10.1111/adb.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Dayas CV, Liu X, Simms JA, Weiss F. Distinct patterns of neural activation associated with ethanol seeking: effects of naltrexone. Biol Psychiatry. 2007;61:979–989. doi: 10.1016/j.biopsych.2006.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Millan EZ, Furlong TM, McNally GP. Accumbens shell–hypothalamus interactions mediate extinction of alcohol seeking. J Neurosci. 2010;30:4626–4635. doi: 10.1523/JNEUROSCI.4933-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cruz FC, et al. New technologies for examining the role of neuronal ensembles in drug addiction and fear. Nat Rev Neurosci. 2013;14:743–754. doi: 10.1038/nrn3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Finegersh A, Homanics GE. Paternal alcohol exposure reduces alcohol drinking and increases behavioral sensitivity to alcohol selectively in male offspring. PLoS ONE. 2014;9:e99078. doi: 10.1371/journal.pone.0099078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Carnicella S, et al. Cabergoline decreases alcohol drinking and seeking behaviors via glial cell line-derived neurotrophic factor. Biol Psychiatry. 2009;66:146–153. doi: 10.1016/j.biopsych.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7:209–219. doi: 10.1038/nrclinonc.2010.21. [DOI] [PubMed] [Google Scholar]
- 196.Sanchis-Segura C, Spanagel R. Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol. 2006;11:2–38. doi: 10.1111/j.1369-1600.2006.00012.x. [DOI] [PubMed] [Google Scholar]
- 197.Carnicella S, Ron D, Barak S. Intermittent ethanol access schedule in rats as a preclinical model of alcohol abuse. Alcohol. 2014;48:243–252. doi: 10.1016/j.alcohol.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hwa LS, et al. Persistent escalation of alcohol drinking in C57BL/6J mice with intermittent access to 20% ethanol. Alcohol Clin Exp Res. 2011;35:1938–1947. doi: 10.1111/j.1530-0277.2011.01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Griffin WC., III Alcohol dependence and free-choice drinking in mice. Alcohol. 2014;48:287–293. doi: 10.1016/j.alcohol.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Thiele TE, Navarro M. “Drinking in the dark” (DID) procedures: a model of binge-like ethanol drinking in non-dependent mice. Alcohol. 2014;48:235–241. doi: 10.1016/j.alcohol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF. Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology. 2000;22:581–594. doi: 10.1016/S0893-133X(99)00167-0. [DOI] [PubMed] [Google Scholar]
- 202.Vendruscolo LF, Roberts AJ. Operant alcohol self-administration in dependent rats: focus on the vapor model. Alcohol. 2014;48:277–286. doi: 10.1016/j.alcohol.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Griffin WC, III, Lopez MF, Becker HC. Intensity and duration of chronic ethanol exposure is critical for subsequent escalation of voluntary ethanol drinking in mice. Alcohol Clin Exp Res. 2009;33:1893–1900. doi: 10.1111/j.1530-0277.2009.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vengeliene V, Bilbao A, Spanagel R. The alcohol deprivation effect model for studying relapse behavior: a comparison between rats and mice. Alcohol. 2014;48:313–320. doi: 10.1016/j.alcohol.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 205.Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology. 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- 206.Costin BN, Dever SM, Miles MF. Ethanol regulation of serum glucocorticoid kinase 1 expression in DBA2/J mouse prefrontal cortex. PLoS ONE. 2013;8:e72979. doi: 10.1371/journal.pone.0072979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wolen AR, et al. Genetic dissection of acute ethanol responsive gene networks in prefrontal cortex: functional and mechanistic implications. PLoS ONE. 2012;7:e33575. doi: 10.1371/journal.pone.0033575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sommer W, et al. Differential expression of diacylglycerol kinase iota and L18A mRNAs in the brains of alcohol-preferring AA and alcohol-avoiding ANA rats. Mol Psychiatry. 2001;6:103–108. doi: 10.1038/sj.mp.4000823. [DOI] [PubMed] [Google Scholar]
- 209.Ojelade SA, et al. Rsu1 regulates ethanol consumption in Drosophila and humans. Proc Natl Acad Sci USA. 2015;112:E4085–E4093. doi: 10.1073/pnas.1417222112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gelernter J, et al. Genome-wide association study of alcohol dependence: significant findings in African- and European-Americans including novel risk loci. Mol Psychiatry. 2014;19:41–49. doi: 10.1038/mp.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Le Roy F, Silhol M, Salehzada T, Bisbal C. Regulation of mitochondrial mRNA stability by RNase L is translation-dependent and controls IFNα-induced apoptosis. Cell Death Differ. 2007;14:1406–1413. doi: 10.1038/sj.cdd.4402130. [DOI] [PubMed] [Google Scholar]
- 212.von der Goltz C, et al. Cue-induced alcohol-seeking behaviour is reduced by disrupting the reconsolidation of alcohol-related memories. Psychopharmacology. 2009;205:389–397. doi: 10.1007/s00213-009-1544-1. [DOI] [PubMed] [Google Scholar]
- 213.Kitamura T, et al. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008;10:329–337. doi: 10.1038/ncb1695. [DOI] [PubMed] [Google Scholar]
- 214.Mathies LD, et al. SWI/SNF chromatin remodeling regulates alcohol response behaviors in Caenorhabditis elegans and is associated with alcohol dependence in humans. Proc Natl Acad Sci USA. 2015;112:3032–3037. doi: 10.1073/pnas.1413451112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kadrmas JL, Beckerle MC. The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol. 2004;5:920–931. doi: 10.1038/nrm1499. [DOI] [PubMed] [Google Scholar]
- 216.Kapoor M, et al. A meta-analysis of two genome-wide association studies to identify novel loci for maximum number of alcoholic drinks. Hum Genet. 2013;132:1141–1151. doi: 10.1007/s00439-013-1318-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Guevara-Aguirre J, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3:70ra13. doi: 10.1126/scitranslmed.3001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.