

Synopsis
DOCK8 deficiency is an autosomal recessive combined immunodeficiency disease associated with elevated IgE, atopy, recurrent sinopulmonary and cutaneous viral infections, and malignancy. The DOCK8 protein is critical for cytoskeletal organization, and deficiency impairs dendritic cell transmigration, T cell survival, and NK cell cytotoxicity. Early hematopoietic stem cell transplant is gaining prominence as a definitive treatment given the potential for severe complications and mortality in this disease. Recently, DOCK2 deficiency has been identified in several patients with early-onset invasive bacterial and viral infections.
Keywords: Immunodeficiency, dedicator of cytokinesis 8, dedicator of cytokinesis 2, atopic dermatitis, cutaneous viral infection, malignancy
INTRODUCTION
Dedicator of cytokinesis 8 (DOCK8) deficiency is an autosomal recessive combined immunodeficiency syndrome characterized by recurrent sinopulmonary and cutaneous viral infections, as well as an increased IgE level and atopy. While patients with an autosomal recessive variant of hyper-IgE syndrome had been described as early as 2004, a genetic basis involving bi-allelic mutations often with large deletions was not established until 2009.1–3 In the intervening years, definitive treatment with early hematopoietic stem cell transplant (HSCT) has gained prominence, and advances have been made in understanding the functions of DOCK8 in dendritic cell and lymphocyte activity. Recently, another syndrome with differing phenotype but similar immunopathogenic basis, dedicator of cytokinesis 2 (DOCK2) deficiency, has been described.4
CLINICAL PRESENTATION OF DOCK8 DEFICIENCY
Atopy
Patients with DOCK8 deficiency demonstrate atopy early on. Nearly all patients exhibit atopic dermatitis (AD) which ranges from mild to very severe and difficult to treat (see Fig. 1a, 1b). Unlike patients with autosomal dominant hyper-IgE syndrome from STAT3 mutations, many have food allergies with anaphylaxis, as well as asthma. Eosinophilic esophagitis has also been seen with increased frequency.3,5–7
Fig. 1.
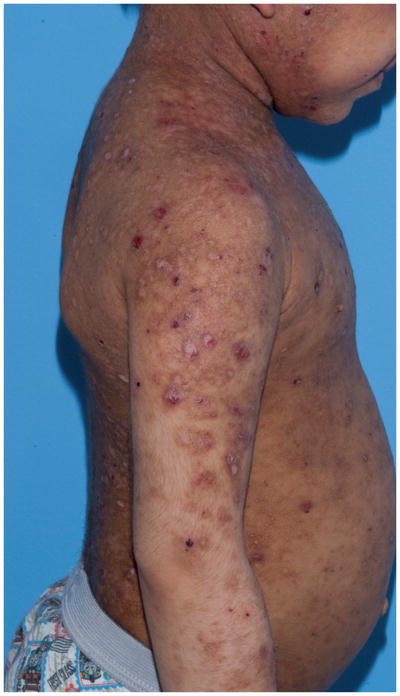

Fig. 1a, 1b: Chronic severe eczematous dermatitis in a 6 year old male with DOCK8 deficiency.
Infections
Cellulitis and skin abscesses are common, as is mucocutaneous candidiasis. There is a striking susceptibility to cutaneous infections by viruses, such as human papillomavirus (HPV) leading to widespread and recalcitrant warts, extensive and disfiguring molluscum contagiosum, herpes simplex virus (HSV) with recurrent or persistent lesions or herpes keratitis, and varicella zoster virus (VZV) with severe primary infection or recurrent zoster (see Fig. 2a, 2b, 2c). Chronic EBV viremia is frequent, and may be associated with transformation to malignancy. Interestingly, severe systemic viral infections are less common, although several patients have suffered from CMV disease, encephalitis and progressive multifocal leukoencephalopathy.3,5–7
Fig. 2.



Fig. 2a, 2b: Large, widespread warts as a manifestation of severe human papilloma virus infection in a 15 year old female with DOCK8 deficiency.
Fig. 2c: Extensive molluscum contagiosum in a patient with DOCK8 deficiency.
Most patients have a history of recurrent sinusitis and otitis media requiring tympanostomy tubes. Most also have had multiple pneumonias, with development of bronchiectasis in over a third but infrequent pneumatocele formation (see Fig. 3).3,5–7
Fig. 3.

Computed tomography of the chest of a 15 year old female with DOCK8 deficiency shows right middle lobe bronchiectasis with thickened bronchial walls.
Malignancy
Increased risk of neoplasms, especially hematological and epithelial, is an important feature of DOCK8 deficiency, and malignancy is often particularly aggressive and has early onset. In one large cohort of 136 patients, 17% of patients were diagnosed with malignancy at a median age of 12 years.6 Malignancy most frequently arises from poor control of viruses including squamous cell carcinomas from HPV infection and EBV related lymphomas. Microcystic adnexal carcinoma, aggressive cutaneous T-cell lymphoma, and diffuse large B cell lymphoma have been described.3,8 Of note, not all tumors noted have been associated with viral infection.
Other Clinical Manifestations
Vascular abnormalities have been recognized in over 10% of patients in two recently described large cohorts (see Fig. 4). Cerebral aneurysms and stenosis is seen, and has been associated with stroke. Vaccine strain varicella was identified as the etiologic agent in one case, but in others an infectious has not been identified. Aortic aneurysm and abdominal arterial vasculitis has also been described, without known etiology. Autoimmunity rarely has manifest in other forms such as hemolytic anemia.6,7 Liver disease, both associated with and without cryptosporidia has been described as well, and can be quite significant leading to liver transplant.9,10
Fig. 4.
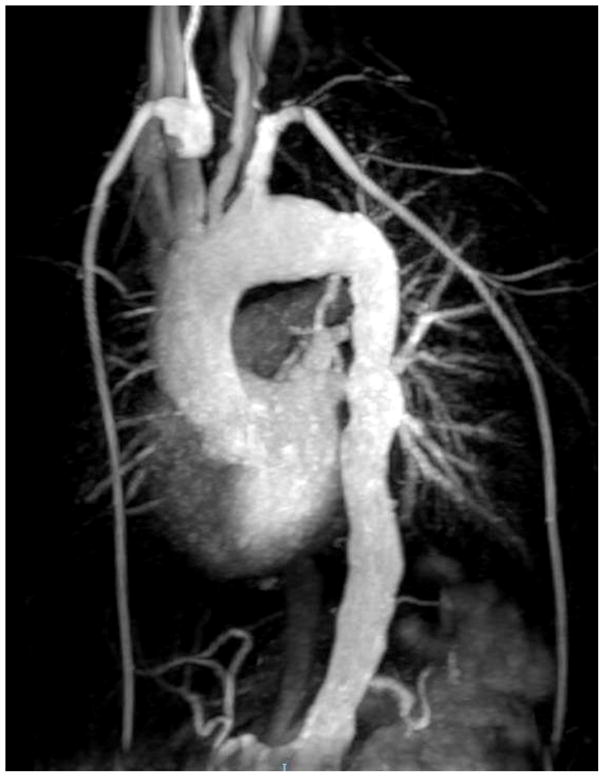
Magnetic resonance angiography demonstrates vasculopathy in a 19 year old patient with DOCK8 deficiency. Shown below are a dilated ascending and transverse thoracic aorta with diffuse irregularity and foci of narrowing in the descending aorta. Other findings (not shown) included narrowing at the bifurcation of the right brachial cephalic artery, dilatation of the right subclavian artery at its origin, narrowing of the left common carotid, narrowing of the common iliacs, and narrowing of the right external iliac artery.
Laboratory Features
DOCK8 deficiency is a combined T and B cell immunodeficiency. In the initial cohort of patients described by Zhang et al, all were noted to have normal neutrophils and monocytes. Ninety percent had low total T cells and CD8+ T cells, and all had low CD4+ T cells; 36% had low B and 60% low NK cells. In addition, there was poor CD8+ but not CD4+ T cell proliferation in response to stimulation.3 Engelhardt et al observed lower rates of T cell lymphopenia in their 2009 and 2015 cohorts (38% and 27% respectively), while Aydin et al observed low total lymphocyte counts in 20% of patients but low total T cells and CD4+ T cells in nearly half of patients.2,6,7
Elevated IgE and eosinophilia were nearly ubiquitous. Consistently, IgG levels were usually normal or elevated, IgA levels were variable, and IgM levels tended to be low and to decline with age.6 Vaccine responses to polysaccharide and protein antigens were variable, but the patients followed by Zhang et al showed protective titers to rubella and VZV.3 Half of patients had low or absent specific antibody responses to pneumococcus, diphtheria, tetanus, or Candida.7 Memory B cells in DOCK8-deficient patients were near absent, as were switched memory B cells.7,11 Memory T cell numbers were variable, but in one study most CD8+ cells had an exhausted CD45RA+/CCR7− phenotype.7,12 Caracciolo et al noted low numbers of naive and recent thymic emigrant T lymphocytes, along with Th2 skewing.11 This is consistent with low T cell receptor excision circles in 3 children with DOCK8 deficiency, a finding with ramifications for potential early detection of this disease.13
Making the Diagnosis
Given the potential for severe infection and malignancy, it is important to recognize DOCK8 deficiency before development of serious complications whenever possible. Diagnosis may be difficult, and, particularly in infants and young children, early presentation may significantly overlap with severe AD in both laboratory and clinical features. Genetic sequencing is key to making the diagnosis, but the expense makes this prohibitive for screening. Thus, several groups have sought to identify markers that can clue in the clinician to an underlying monogenetic disorder. Furthermore, distinguishing features of different monogenetic hyper-IgE syndromes on presentation is important for targeting subsequent evaluations.
When compared to severe AD patients, DOCK8-deficient patients were more likely to have low total T cells, low CD4+ T cells, and decreased naive CD8+ T cells in one small study. Total B lymphocyte numbers did not differ significantly between the two groups, but subsets revealed decreased memory and increased naive and transitional B cells in the DOCK8-deficient patients.14
When examining IgE-sensitization patterns in AD, STAT3 deficiency, and DOCK8 deficiency, Boos et al found that AD patients had the highest ratios of aeroallergen-specific IgE to total IgE, while patients with DOCK8 deficiency showed the highest serum specific IgE against food antigens, followed by AD patients.15
Using the NIH-developed Hyper-IgE Syndrome scoring system, Engelhardt et al compared clinical and laboratory scoring for DOCK8 and STAT3 patients and identified several objective features that were helpful in distinguishing the two syndromes: parenchymal lung abnormalities, retained primary teeth, and minor trauma fractures were deemed most consistent with STAT3 deficiency.7,16 Characteristic facies was also significantly associated with STAT3 deficiency but was considered a subjective assessment. By assigning negative points to the three features above and adding points for based on absolute eosinophil count and frequency of sinus and ear infections, the group developed a DOCK8 score that appears promising but has yet to be validated.7
Another study featured long-term follow-up of biomarker trends in individual DOCK8- and STAT3-deficient patients. DOCK8 patients demonstrated consistently lower total, CD4+, and CD8+ T cell numbers but normal Th17 cells as opposed to low Th17 cell but otherwise normal numbers of T cells in STAT3-deficient patients. In terms of clinical characteristics, the authors suggested that a history of recurrent viral infections, bronchial hyperreactivity, food allergies, and consanguinity should prompt greater concern for DOCK8 deficiency.17
In addition to severe atopic dermatitis and STAT3 deficiency, the differential diagnosis for a patient with atopic dermatitis, elevated IgE, and recurrent infections includes several other genetic disorders.
Wiskott-Aldrich syndrome (WAS) is characterized by T cell lymphopenia, poor lymphocyte proliferation, impaired NK cytotoxicity, autoimmunity, and malignancy. The WAS protein (WASp) coordinates cytoskeletal reorganization downstream from DOCK8, which accounts for some overlap in phenotype including recurrent bacterial and viral infections, eczema and vascular abnormalities. Distinguishing features of WAS include X-linked inheritance and microthrombocytopenia.18
Phosphoglucomutase 3 (PGM3) deficiency, a congenital disorder of glycosylation, has been recently identified in patients who, in addition to severe atopy and hypergammaglobulinemia, usually have lymphopenia and neutropenia. Developmental delay or neurologic impairment is common.19
Omenn syndrome is a form of severe combined immunodeficiency associated with several different genetic defects. Severe erythroderma and exfoliative dermatitis are evident in early infancy, along with elevated IgE, infections, lymphadenopathy and hepatosplenomegaly.20
STK4 or Macrophage Stimulating 1 (MST1) deficiency, discovered within the last few years, has a phenotype similar to DOCK8 deficiency, with cutaneous viral, bacterial, and fungal infections, recurrent respiratory infections, and CD4 lymphopenia. Atopic dermatitis seems to be milder, and IgG and IgA are elevated as well as IgE. Cardiac anomalies have been noted in multiple patients.21
Tyk2 deficiency was described in 2006 in a patient with elevated IgE, atopic dermatitis, recurrent skin staphylococcal abscesses, and mycobacterial infection. However, recently, seven new Tyk2-deficient patients were identified, all with normal IgE, calling into question the classification of Tyk2 deficiency as a hyper-IgE syndrome.22
FUNCTIONS OF THE DOCK8 PROTEIN
The DOCK8 protein is a member of the DOCK180-related family of atypical guanine nucleotide exchange factors that activate small Rho GTPases such as Rac and Cdc42.23,24 These GTPases interact with DOCK8 at actin projections called lamellipodia, which are found at the leading edge of motile cells such as endothelial cells, neurons, immune cells, and epithelial cells. Early studies suggested DOCK8 as an important player in dynamic actin reorganization because of its accumulation at lamellipodia and its ability to induce formation of vesicles containing filamentous actin.23 As DOCK8 is highly expressed only in the immune system (and at low levels in non-immune tissues such as the placenta, kidney, lung, and pancreas), more recent investigation has shed light on the protein’s specific role in the survival and function of dendritic cells (DCs) and lymphocytes.23,25
Dendritic Cells
DCs adapt their shape to facilitate amoeboid migration through the interstitium. In a DOCK8 knockout mouse model, DCs were unable to crawl through 3-dimensional (3D) fibrillar networks and transmigrate into the lymph node for T cell priming, due to impaired Cdc42 activation at the leading edge membrane.26 The peripheral blood of DOCK8-deficient patients shows severe deficiency of plasmacytoid dendritic cells and correspondingly low interferon alpha (IFN-α) levels.27,28
B Lymphocytes
The B cells of mice lacking DOCK8 cannot develop into marginal zone B cells, survive in germinal centers, and undergo affinity maturation, leading to normal initiation but poor persistence of antibody response post-immunization. DOCK8 appears to be required for organization of a B cell immunological synapse by recruiting the integrin ligand ICAM-1.29 In a study of DOCK8-deficient patients who showed either poor initial antibody responses to vaccination or poor persistence of protective titers, DOCK8 was shown to function as an adaptor linking TLR9 to MyD88 and downstream signaling pathways to effect B cell activation.30
T, NK, and NKT Lymphocytes
Recent studies of DOCK8-deficient mice show a lack of CD4+ T cell infiltration into HSV-infected skin, associated with poor control of primary cutaneous herpes simplex lesions and increased virus loads.31
T cell lymphopenia is a prominent feature in DOCK8-deficient mice, due to both decreased survival of CD4+ and CD8+ T cells and decreased egress of mature CD4+ T cells from the thymus.32 Primary T cell response to infection or immunization is near-normal, but there is poor secondary expansion given reduced survival of memory CD8+ T cells.12,32 Diminished recruitment of LFA-1 to the CD8+ T cell/DC synapse and delay in the first cell division likely result in impaired generation of long-lived CD8+ T cells.12
As mentioned previously, DOCK8 activates Cdc42, which via its effector WASp is necessary for reorganizing the F-actin cytoskeleton in NK and dendritic cells. WASp itself also interacts directly with DOCK8, and WASp function may thus also be reduced in DOCK8 patients.33,34 DOCK8-deficient NK cells show decreased natural and receptor-mediated cytotoxicity, with decreased polarization of LFA-1, F-actin, and cytolytic granules toward the cytotoxic synapse.33,35
Zhang et al examined lymphocyte migration through the dermis and found that DOCK8-deficient T and NK cells develop an abnormal shape when moving in confined spaces.36 In a 3D collagen gel matrix simulating the dermis, the cells were able to move, unlike the nonmotile dendritic cells described by Harada et al, and chemotaxis was not impaired.26 However, hours later, the cells underwent fragmentation (cytothripsis) and died, suggesting that DOCK8 is essential for coordinating lymphocyte cytoskeletal integrity in this milieu. The early cell death prevents the generation of long-lived skin-resident memory CD8+ T cells, which may explain the preponderance of severe cutaneous viral infections in these patients.
Low NKT cell numbers have been noted in DOCK8-deficient humans, while in mice ongoing NKT proliferative and cytokine responses are impaired.37
A recent analysis of the cytokine profile of DOCK8 deficiency shows that unstimulated DOCK8-deficient PBMCs secrete higher levels of inflammatory cytokines such as IFNgamma, IL1beta, IL4, and IL6 as compared to healthy control cells. Interestingly, stimulated PBMCs secreted less IFNgamma, suggesting impaired Th1 cell function. As Cdc42 is required for IFNgamma secretion at the immunological synapse, this finding is consistent with an intrinsic defect secondary to DOCK8 deficiency.17
DOCK8 Expression in Tumors
Many but not all of the malignancies described in DOCK8 deficiency are virus-associated, which has prompted the question of whether the DOCK8 protein may have some intrinsic tumor suppressive function. Loss of chromosome arm 9p, where the DOCK8 gene is also located, is common in lung cancer.38 Deletions and other chromosomal alterations encompassing the DOCK8 gene have been associated with lung, gastric, pancreatic, and head and neck squamous cell carcinomas, while decreased expression of DOCK8 has been noted in certain lung and liver tumors, as well as in high-grade gliomas.39–44 However, increased expression of DOCK8 has been noted in a radiosensitive esophageal cancer line and in hepatocellular carcinoma cells, so a consistent role for DOCK8 in tumorigenesis has not emerged.45,46
THERAPEUTIC APPROACHES TO DOCK8 DEFICIENCY
Nearly two thirds of patients receive immunoglobulin replacement therapy, as well as prophylactic antibiotics. Some patients also receive antiviral and antifungal prophylaxis.6 Even in patients who are otherwise well-controlled, HSV lesions, molluscum, and warts may be particularly recalcitrant, disfiguring, and problematic from a quality of life standpoint.
Systemic IFN-α 2b therapy, which may act by inhibiting viral replication and activating effector lymphocytes, has yielded dramatic improvement of viral infection in 3 published cases of DOCK8-deficient patients.27,28 However, significant side effects may be associated with IFN-α 2b therapy, and careful monitoring is essential.
Nevertheless, DOCK8 deficiency is associated with significant mortality, mainly due to infection or malignancy. In the cohort described by Engelhardt et al, mean age of death was 9 years and 3 months, while Aydin et al report probability of survival of 37% at age 30 years if not transplanted.6,7 Hematopoietic stem cell transplant (HSCT) has been repeatedly shown to be curative and more recently is being offered at an early stage. In their initial post-transplant course, patients may have a transient worsening of warts and chronic bacterial pretransplant infections. However, within several months, marked improvement or, more commonly, complete resolution of all skin manifestations has consistently been noted even in patients who previously experienced particularly severe or disfiguring skin disease. Complete immunological correction has generally been reported, even in several cases of mixed donor chimerism. Two deaths have been described in the literature, one considered transplant-related and one due to Klebsiella sepsis in the context of congenital asplenia, and unpublished experience has shown transplant-related mortality in the setting of pre-transplant significant end-organ disease.47–57
Somatic reversions were identified in 17 DOCK8 patients followed at the National Institutes of Health, and these patients demonstrated longer survival and a milder disease course; however, experience with these patients has shown that they may nevertheless have life-threatening complications and require HSCT.58
DOCK2 DEFICIENCY
DOCK2, like DOCK8, is a member of the DOCK180 superfamily of proteins. DOCK2-deficient mice were known to have immunological defects even before DOCK8 deficiency was described in humans. Notable features included T cell lymphopenia and decreased T cell proliferation, loss of marginal zone B cells, and decreased myeloid and lymphocyte migration, with the potential for developing hyper-IgE.25
Recently, biallelic DOCK 2 mutations were identified in 5 patients with invasive bacterial and viral infections, lymphopenia and impaired antibody responses.4 Three of the children were born to consanguineous parents. Infections included recurrent pneumonia, disseminated varicella, Mycobacterium avium, mumps meningoencephalitis, and Klebsiella pneumoniae sepsis. Unfortunately, only 3 of the patients survived to receive HSCT and attain clinical improvement. Further investigation of the patients’ T, B, and NK cells revealed defective chemotaxis, actin polymerization, and NK cell degranulation. Interestingly, viral replication and virus-induced cell death were increased in DOCK2-deficient fibroblasts, and inducing lentiviral-mediated DOCK2 expression in the presence of interferon alfa-2b protected the cells. Thus, DOCK2 may impair non-hematopoietic immunity as well. While some similarities exist between the DOCK2 and DOCK8 deficiency phenotypes, only one of the DOCK2 patients had elevated IgE, and severe eczema and allergies were not prominent features in these patients.
SUMMARY
In the years since DOCK8 deficiency was described, much progress has been made in delineating the phenotype, establishing the role of DOCK8 in leukocyte function, and defining HSCT as a necessary treatment. The phenotype of DOCK2 deficiency in humans remains to be further characterized beyond the index patients.
The growing experience with DOCK8-deficient patients highlights the concept that severe skin disease can be an indicator of underlying immunodeficiency, and thus dermatologists may be key to the early diagnosis of this disorder.
Key Points.
DOCK8 deficiency is an autosomal recessive hyper-IgE syndrome associated with atopy, recurrent sinopulmonary and cutaneous viral infections, and malignancies.
The DOCK8 protein plays an important role in cytoskeletal organization, impacting dendritic cell transmigration.
DOCK8 deficiency leads to persistence of B cells in germinal centers, early T cell death, and lowered NK cell cytotoxicity.
DOCK2 deficiency has been recently described in several patients with early-onset invasive bacterial and viral infections.
Abbreviations and Acronyms
- Cdc42
G protein activated by DOCK guanine exchange factors
- DOCK2
Dedicator of cytokinesis 2, one of a class of guanine nucleotide exchange factors whose role is to activate the G protein Rac.
- DOCK8
Dedicator of cytokinesis 8, one of a class of guanine nucleotide exchange factors whose role is to activate G proteins such as Rac and Cdc42. Deficiency leads to impaired cytoskeletal organization and a phenotype of combined immunodeficiency with eczema, elevated IgE, and malignancy.
- HSCT
Hematopoietic stem cell transplant, a treatment for primary immunodeficiencies that result from genetic defects in hematopoietic cells.
- ICAM-1
Intercellular adhesion molecule 1, a ligand for LFA-1 necessary for leukocyte endothelial transmigration.
- LFA-1
Lymphocyte function-associated antigen 1, binds to ICAM-1 and functions as an adhesion molecule.
- MST1
Macrophage stimulating 1 (also known as STK4). Deficiency results in a rare form of immunodeficiency wuth a phenotype similar to DOCK8 deficiency.
- MyD88
Myeloid differentiation primary response 88, a protein used by toll-like receptors to activate the transcription factor NF-κB.
- PGM3
Phosphoglucomutase 3, which mediates glycosylation. Deficiency results in a phenotype of severe atopy, hypergammaglobulinemia, leukopenia, and developmental delay.
- Rac
G protein activated by DOCK guanine exchange factors.
- Rho GTPase
G protein such as Cdc42 and Rac, activated by DOCK guanine exchange factors. These proteins regulate various aspects of cytoskeletal dynamics.
- STAT3
Signal transducer and activator of transcription 3, a transcription factor. Deficiency leads to autosomal dominant Hyper-IgE syndrome.
- TLR9
Toll-like receptor 9, important for activation of innate immunity via MyD88
- Tyk2
Tyrosine kinase 2, a protein involved in IL-10 and IFN-α signaling. Deficiency has been associated with a variable phenotype that includes susceptibility to mycobacterial infection.
- WAS
Wiskott-Aldrich syndrome is a rare X-linked recessive disease classically characterized by a triad of recurrent sinopulmonary infections, eczema, and thrombocytopenia with small platelets. Many patients do not exhibit the classic triad and may have autoimmune disease among other manifestations.
- WASp
Wiskott-Aldrich syndrome protein, a protein that coordinates cytoskeletal reorganization. Deficiency leads to WAS.
Footnotes
Neither author has any commercial or financial conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Renner ED, Puck JM, Holland SM, et al. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. The Journal of pediatrics. 2004;144:93–9. doi: 10.1016/S0022-3476(03)00449-9. [DOI] [PubMed] [Google Scholar]
- 2.Engelhardt KR, McGhee S, Winkler S, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2009;124:1289–302. e4. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Q, Davis JC, Lamborn IT, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046–55. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dobbs K, Dominguez Conde C, Zhang SY, et al. Inherited DOCK2 Deficiency in Patients with Early-Onset Invasive Infections. The New England journal of medicine. 2015;372:2409–22. doi: 10.1056/NEJMoa1413462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q, Davis JC, Dove CG, Su HC. Genetic, clinical, and laboratory markers for DOCK8 immunodeficiency syndrome. Disease markers. 2010;29:131–9. doi: 10.3233/DMA-2010-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aydin SE, Kilic SS, Aytekin C, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options - a review of 136 patients. Journal of clinical immunology. 2015;35:189–98. doi: 10.1007/s10875-014-0126-0. [DOI] [PubMed] [Google Scholar]
- 7.Engelhardt KR, Gertz ME, Keles S, et al. The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2015;136:402–12. doi: 10.1016/j.jaci.2014.12.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu EY, Freeman AF, Jing H, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch Dermatol. 2012;148:79–84. doi: 10.1001/archdermatol.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Herz W, Ragupathy R, Massaad MJ, et al. Clinical, immunologic and genetic profiles of DOCK8-deficient patients in Kuwait. Clinical immunology (Orlando, Fla) 2012;143:266–72. doi: 10.1016/j.clim.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah NN, Freeman AF, Parta M, et al. Haploidentical transplantation for DOCK8 deficiency. 57th Annual Meeting and Exposition of the American Society of Hematology; Orlando, FL. 2015. [Google Scholar]
- 11.Caracciolo S, Moratto D, Giacomelli M, et al. Expansion of CCR4+ activated T cells is associated with memory B cell reduction in DOCK8-deficient patients. Clin Immunol. 2014;152:164–70. doi: 10.1016/j.clim.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Randall KL, Chan SS, Ma CS, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. The Journal of experimental medicine. 2011;208:2305–20. doi: 10.1084/jem.20110345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dasouki M, Okonkwo KC, Ray A, et al. Deficient T Cell Receptor Excision Circles (TRECs) in autosomal recessive hyper IgE syndrome caused by DOCK8 mutation: implications for pathogenesis and potential detection by newborn screening. Clinical immunology (Orlando, Fla) 2011;141:128–32. doi: 10.1016/j.clim.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen E, Tsitsikov E, Al-Herz W, et al. Flow cytometry biomarkers distinguish DOCK8 deficiency from severe atopic dermatitis. Clin Immunol. 2014;150:220–4. doi: 10.1016/j.clim.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boos AC, Hagl B, Schlesinger A, et al. Atopic dermatitis, STAT3- and DOCK8- hyper-IgE syndromes differ in IgE-based sensitization pattern. Allergy. 2014;69:943–53. doi: 10.1111/all.12416. [DOI] [PubMed] [Google Scholar]
- 16.Grimbacher B, Schaffer AA, Holland SM, et al. Genetic linkage of hyper-IgE syndrome to chromosome 4. American journal of human genetics. 1999;65:735–44. doi: 10.1086/302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagl B, Heinz V, Schlesinger A, et al. Key findings to expedite the diagnosis of hyper-IgE syndromes in infants and young children. Pediatric allergy and immunology : official publication of the European Society of Pediatric Allergy and Immunology. 2015 doi: 10.1111/pai.12512. [DOI] [PubMed] [Google Scholar]
- 18.Moulding DA, Record J, Malinova D, Thrasher AJ. Actin cytoskeletal defects in immunodeficiency. Immunological reviews. 2013;256:282–99. doi: 10.1111/imr.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Yu X, Ichikawa M, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. The Journal of allergy and clinical immunology. 2014;133:1400–9. 9e1–5. doi: 10.1016/j.jaci.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chinn IK, Shearer WT. Severe Combined Immunodeficiency Disorders. Immunology and allergy clinics of North America. 2015;35:671–94. doi: 10.1016/j.iac.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Halacli SO, Ayvaz DC, Sun-Tan C, et al. STK4 (MST1) deficiency in two siblings with autoimmune cytopenias: A novel mutation. Clinical immunology (Orlando, Fla) 2015;161:316–23. doi: 10.1016/j.clim.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 22.Kreins AY, Ciancanelli MJ, Okada S, et al. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. The Journal of experimental medicine. 2015;212:1641–62. doi: 10.1084/jem.20140280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruusala A, Aspenstrom P. Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology. FEBS letters. 2004;572:159–66. doi: 10.1016/j.febslet.2004.06.095. [DOI] [PubMed] [Google Scholar]
- 24.Cote JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. Journal of cell science. 2002;115:4901–13. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 25.Su HC. Dedicator of cytokinesis 8 (DOCK8) deficiency. Current opinion in allergy and clinical immunology. 2010;10:515–20. doi: 10.1097/ACI.0b013e32833fd718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harada Y, Tanaka Y, Terasawa M, et al. DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood. 2012;119:4451–61. doi: 10.1182/blood-2012-01-407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Zahrani D, Raddadi A, Massaad M, et al. Successful interferon-alpha 2b therapy for unremitting warts in a patient with DOCK8 deficiency. Clin Immunol. 2014;153:104–8. doi: 10.1016/j.clim.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keles S, Jabara HH, Reisli I, et al. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: rescue of severe herpetic infections with IFN-alpha 2b therapy. J Allergy Clin Immunol. 2014;133:1753–5. e3. doi: 10.1016/j.jaci.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Randall KL, Lambe T, Johnson AL, et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat Immunol. 2009;10:1283–91. doi: 10.1038/ni.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jabara HH, McDonald DR, Janssen E, et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nature immunology. 2012;13:612–20. doi: 10.1038/ni.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flesch IE, Randall KL, Hollett NA, et al. Delayed control of herpes simplex virus infection and impaired CD4(+) T-cell migration to the skin in mouse models of DOCK8 deficiency. Immunol Cell Biol. 2015;93:517–21. doi: 10.1038/icb.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lambe T, Crawford G, Johnson AL, et al. DOCK8 is essential for T-cell survival and the maintenance of CD8+ T-cell memory. Eur J Immunol. 2011;41:3423–35. doi: 10.1002/eji.201141759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ham H, Guerrier S, Kim J, et al. Dedicator of cytokinesis 8 interacts with talin and Wiskott-Aldrich syndrome protein to regulate NK cell cytotoxicity. J Immunol. 2013;190:3661–9. doi: 10.4049/jimmunol.1202792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGhee SA, Chatila TA. DOCK8 immune deficiency as a model for primary cytoskeletal dysfunction. Disease markers. 2010;29:151–6. doi: 10.3233/DMA-2010-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizesko MC, Banerjee PP, Monaco-Shawver L, et al. Defective actin accumulation impairs human natural killer cell function in patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2013;131:840–8. doi: 10.1016/j.jaci.2012.12.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Q, Dove CG, Hor JL, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J Exp Med. 2014;211:2549–66. doi: 10.1084/jem.20141307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crawford G, Enders A, Gileadi U, et al. DOCK8 is critical for the survival and function of NKT cells. Blood. 2013;122:2052–61. doi: 10.1182/blood-2013-02-482331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato M, Takahashi K, Nagayama K, et al. Identification of chromosome arm 9p as the most frequent target of homozygous deletions in lung cancer. Genes, chromosomes & cancer. 2005;44:405–14. doi: 10.1002/gcc.20253. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi K, Kohno T, Ajima R, et al. Homozygous deletion and reduced expression of the DOCK8 gene in human lung cancer. International journal of oncology. 2006;28:321–8. [PubMed] [Google Scholar]
- 40.Takada H, Imoto I, Tsuda H, et al. Genomic loss and epigenetic silencing of very-low-density lipoprotein receptor involved in gastric carcinogenesis. Oncogene. 2006;25:6554–62. doi: 10.1038/sj.onc.1209657. [DOI] [PubMed] [Google Scholar]
- 41.Heidenblad M, Schoenmakers EF, Jonson T, et al. Genome-wide array-based comparative genomic hybridization reveals multiple amplification targets and novel homozygous deletions in pancreatic carcinoma cell lines. Cancer research. 2004;64:3052–9. doi: 10.1158/0008-5472.can-03-3159. [DOI] [PubMed] [Google Scholar]
- 42.Saelee P, Wongkham S, Puapairoj A, et al. Novel PNLIPRP3 and DOCK8 gene expression and prognostic implications of DNA loss on chromosome 10q25. 3 in hepatocellular carcinoma. Asian Pacific journal of cancer prevention : APJCP. 2009;10:501–6. [PubMed] [Google Scholar]
- 43.Idbaih A, Carvalho Silva R, Criniere E, et al. Genomic changes in progression of low-grade gliomas. Journal of neuro-oncology. 2008;90:133–40. doi: 10.1007/s11060-008-9644-z. [DOI] [PubMed] [Google Scholar]
- 44.Marescalco MS, Capizzi C, Condorelli DF, Barresi V. Genome-wide analysis of recurrent copy-number alterations and copy-neutral loss of heterozygosity in head and neck squamous cell carcinoma. Journal of oral pathology & medicine : official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology. 2014;43:20–7. doi: 10.1111/jop.12087. [DOI] [PubMed] [Google Scholar]
- 45.Ogawa R, Ishiguro H, Kuwabara Y, et al. Identification of candidate genes involved in the radiosensitivity of esophageal cancer cells by microarray analysis. Diseases of the esophagus : official journal of the International Society for Diseases of the Esophagus/ISDE. 2008;21:288–97. doi: 10.1111/j.1442-2050.2007.00759.x. [DOI] [PubMed] [Google Scholar]
- 46.Wang SJ, Cui HY, Liu YM, et al. CD147 promotes Src-dependent activation of Rac1 signaling through STAT3/DOCK8 during the motility of hepatocellular carcinoma cells. Oncotarget. 2015;6:243–57. doi: 10.18632/oncotarget.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDonald DR, Massaad MJ, Johnston A, et al. Successful engraftment of donor marrow after allogeneic hematopoietic cell transplantation in autosomal-recessive hyper-IgE syndrome caused by dedicator of cytokinesis 8 deficiency. The Journal of allergy and clinical immunology. 2010;126:1304–5. e3. doi: 10.1016/j.jaci.2010.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pai SY, de Boer H, Massaad MJ, et al. Flow cytometry diagnosis of dedicator of cytokinesis 8 (DOCK8) deficiency. The Journal of allergy and clinical immunology. 2014;134:221–3. doi: 10.1016/j.jaci.2014.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bittner TC, Pannicke U, Renner ED, et al. Successful long-term correction of autosomal recessive hyper-IgE syndrome due to DOCK8 deficiency by hematopoietic stem cell transplantation. Klin Padiatr. 2010;222:351–5. doi: 10.1055/s-0030-1265135. [DOI] [PubMed] [Google Scholar]
- 50.Gatz SA, Benninghoff U, Schutz C, et al. Curative treatment of autosomal-recessive hyper-IgE syndrome by hematopoietic cell transplantation. Bone Marrow Transplant. 2011;46:552–6. doi: 10.1038/bmt.2010.169. [DOI] [PubMed] [Google Scholar]
- 51.Metin A, Tavil B, Azik F, et al. Successful bone marrow transplantation for DOCK8 deficient hyper IgE syndrome. Pediatr Transplant. 2012;16:398–9. doi: 10.1111/j.1399-3046.2011.01641.x. [DOI] [PubMed] [Google Scholar]
- 52.Boztug H, Karitnig-Weiss C, Ausserer B, et al. Clinical and immunological correction of DOCK8 deficiency by allogeneic hematopoietic stem cell transplantation following a reduced toxicity conditioning regimen. Pediatr Hematol Oncol. 2012;29:585–94. doi: 10.3109/08880018.2012.714844. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh S, Schuster FR, Fuchs I, Laws HJ, Borkhardt A, Meisel R. Treosulfan-based conditioning in DOCK8 deficiency: complete lympho-hematopoietic reconstitution with minimal toxicity. Clinical immunology (Orlando, Fla) 2012;145:259–61. doi: 10.1016/j.clim.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 54.Cuellar-Rodriguez J, Freeman AF, Grossman J, et al. Matched related and unrelated donor hematopoietic stem cell transplantation for DOCK8 deficiency. Biol Blood Marrow Transplant. 2015;21:1037–45. doi: 10.1016/j.bbmt.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Purcell C, Cant A, Irvine AD. DOCK8 primary immunodeficiency syndrome. Lancet. 2015;386:982. doi: 10.1016/S0140-6736(14)62039-0. [DOI] [PubMed] [Google Scholar]
- 56.Ghosh S, Schuster FR, Adams O, et al. Haploidentical stem cell transplantation in DOCK8 deficiency - Successful control of pre-existing severe viremia with a TCRass/CD19-depleted graft and antiviral treatment. Clinical immunology (Orlando, Fla) 2014;152:111–4. doi: 10.1016/j.clim.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 57.Barlogis V, Galambrun C, Chambost H, et al. Successful allogeneic hematopoietic stem cell transplantation for DOCK8 deficiency. The Journal of allergy and clinical immunology. 2011;128:420–22. e2. doi: 10.1016/j.jaci.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 58.Jing H, Zhang Q, Zhang Y, et al. Somatic reversion in dedicator of cytokinesis 8 immunodeficiency modulates disease phenotype. The Journal of allergy and clinical immunology. 2014;133:1667–75. doi: 10.1016/j.jaci.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]