

Abstract
BACKGROUND
Multiple sclerosis is a chronic inflammatory demyelinating disease of the central nervous system. Many findings suggest that the disease has an autoimmune pathogenesis; the target of the immune response is not yet known.
METHODS
We screened serum IgG from persons with multiple sclerosis to identify antibodies that are capable of binding to brain tissue and observed specific binding of IgG to glial cells in a subgroup of patients. Using a proteomic approach focusing on membrane proteins, we identified the ATP-sensitive inward rectifying potassium channel KIR4.1 as the target of the IgG antibodies. We used a multifaceted validation strategy to confirm KIR4.1 as a target of the autoantibody response in multiple sclerosis and to show its potential pathogenicity in vivo.
RESULTS
Serum levels of antibodies to KIR4.1 were higher in persons with multiple sclerosis than in persons with other neurologic diseases and healthy donors (P<0.001 for both comparisons). We replicated this finding in two independent groups of persons with multiple sclerosis or other neurologic diseases (P<0.001 for both comparisons). Analysis of the combined data sets indicated the presence of serum antibodies to KIR4.1 in 186 of 397 persons with multiple sclerosis (46.9%), in 3 of 329 persons with other neurologic diseases (0.9%), and in none of the 59 healthy donors. These antibodies bound to the first extracellular loop of KIR4.1. Injection of KIR4.1 serum IgG into the cisternae magnae of mice led to a profound loss of KIR4.1 expression, altered expression of glial fibrillary acidic protein in astrocytes, and activation of the complement cascade at sites of KIR4.1 expression in the cerebellum.
CONCLUSIONS
KIR4.1 is a target of the autoantibody response in a subgroup of persons with multiple sclerosis. (Funded by the German Ministry for Education and Research and Deutsche Forschungsgemeinschaft.)
Multiple Sclerosis, the most common chronic inflammatory disease of the central nervous system (CNS), causes disability in the majority of affected patients.1,2 The cause of this disease is unknown, but epidemiologic evidence suggests that there is a complex interplay between genetic and environmental factors.3,4 An uncertain pathogenic mechanism, clinical heterogeneity, and unpredictable therapeutic response add to the complexity of the disease.5
One hypothesis that has been suggested is that autoreactive T cells are key to the pathogenesis of multiple sclerosis.5 However, histopathological studies have revealed prominent deposition of immunoglobulins and complement activation in acute demyelinating lesions.6–8 Some patients with multiple sclerosis who have these lesions have a response to therapeutic plasma exchange.9 Moreover, depletion of B cells by therapeutic monoclonal antibodies has an effect on inflammatory activity in patients with multiple sclerosis.10 It would therefore seem that, at least in a subgroup of patients with multiple sclerosis, B cells and antibodies contribute substantially to the disease.11,12 However, direct proof of clinically relevant antibodies in multiple sclerosis has not been established, and the molecular targets for humoral responses in the disease are not known.
A specific serum autoantibody against the water channel aquaporin-4 (AQP4), which is expressed on astrocytes, has been described previously in persons with neuromyelitis optica.13,14 The antibody seems to exert pathogenic effects in vivo and in vitro.15–17 Historically, neuromyelitis optica was considered to be a variant of multiple sclerosis. However, the identification of the AQP4 autoantibody provides evidence that neuromyelitis optica is a distinct disease entity18 and has reinvigorated the search for specific autoantibody responses in multiple sclerosis. We undertook this study to identify the target of the autoantibody response in multiple sclerosis.
METHODS
PATIENTS
The multiple sclerosis cohort comprised persons with multiple sclerosis or with a clinically isolated syndrome. In all persons with multiple sclerosis, the disease was diagnosed according to the 2005 McDonald criteria. Persons with a clinically isolated syndrome had at least one episode compatible with a relapse of multiple sclerosis and two or more lesions on magnetic resonance imaging (MRI), oligoclonal bands in the cerebrospinal fluid, or both. There were two control groups: one consisted of age-matched healthy donors, and the second comprised persons with other neurologic diseases. Analyses were performed on a discovery series of patients with multiple sclerosis or a clinically isolated syndrome and were confirmed in two validation series. Details of the multiple sclerosis and control groups, and the extent to which patients and controls from each group were included in the various analyses, are provided in Figure S1 and Tables S1 and S2 in the Supplementary Appendix, available with the full text of this article at NEJM.org).
IMMUNOPRECIPITATION, ELECTROPHORESIS, AND WESTERN BLOTTING
Details of the specific antibodies and peptides that were used in the study are provided in the Supplementary Appendix. We derived IgG antibodies specific to membrane-expressed proteins in the CNS from the pooled serum specimens from 12 persons with multiple sclerosis and purified them using a protein A/G bead-based approach (GE Healthcare Life Sciences): we used gentle antigen–antibody binding and elution buffers (Pierce) to enable the purification of IgG under nondenaturating conditions (high salt concentration and nearly neutral pH). We enriched IgG with reactivity against CNS membrane antigen using a cyanogen bromide–activated Sepharose CNS membrane-protein enrichment column (see the Methods section in the Supplementary Appendix). Magnetic protein G beads (Invitrogen) were used according to the manufacturer’s protocol to immunoprecipitate antigen–antibody complexes from the eluate of the CNS membrane-protein enrichment column. The eluted molecules were precipitated with chloroform–methanol and then further solubilized with a two-dimensional protein solubilizer (Invitrogen). The solubilized fractions were run on pH 3–10 isoelectric focusing strips (Invitrogen). Two-dimensional gel electrophoresis was performed with two-dimensional Benchtop technology (Invitrogen). Spots were picked and subjected to matrix-assisted laser desorption–tandem mass spectrometry (Alphalyse) for identification. As a control, we purified parallel samples of pooled serum IgG antibodies from 12 persons with other neurologic diseases. To determine whether the purified antibodies bind KIR4.1, we used purified IgG antibodies to immunoprecipitate rat kidney lysate,19 human brain lysate, and a sample of in vitro–translated KIR4.1 protein. We eluted the protein and then subjected it to Western blotting using rabbit antihuman KIR4.1 antibody (see the Supplementary Appendix).
ENZYME-LINKED IMMUNOSORBENT ASSAYS
We used plate-bound purified recombinant KIR4.1 (see the Supplementary Appendix) to screen for anti-KIR4.1 reactivity in serum samples. We used two negative controls: plates coated with effluent (“flow-through”) obtained after KIR4.1 purification with the use of HisPur cobalt agarose beads (Thermo Scientific) and plates coated with bovine serum albumin. Purified protein was diluted in phosphate-buffered saline (PBS) to a final concentration of 6 μg per milliliter, and 100 μl was added to each well of Nunc Immobilizer amino plates (Thermo Scientific). Plates were left overnight at 4°C on a rotary shaker. Coated plates were washed twice with PBS with Tween 20 detergent and blocked for 1 hour with 10 mM ethanolamine in 100 mM sodium bicarbonate, pH 9.6. All assays were performed on blinded samples, were run in duplicate, and were controlled by standard serum samples at two dilutions. Anti-KIR4.1 activity in the serum of patients and controls from the discovery series and the two validation series was determined with the use of this assay.
INTRATHECAL INJECTION OF IgG IN MICE
To show the pathogenicity of serum IgG KIR4.1-specific antibody in vivo, serum IgG from persons with multiple sclerosis who had high antibody titers against KIR4.1 was purified. We split the purified IgG into two fractions and depleted KIR4.1-specific antibodies from one of these fractions using a preabsorption column containing bead-bound recombinant KIR4.1 protein. IgG fractions were injected into mice intracisternally, as described previously.20 Further details and validation of prepared IgG fractions are provided in the Supplementary Appendix.
STATISTICAL ANALYSIS
We considered serum specimens to be antibody-positive when the optical density exceeded the cutoff value, which was set at 5 SD above the mean optical density in serum specimens from healthy donors. In the screening experiments, anti-KIR4.1 serum reactivity was compared with the use of the Kruskal–Wallis test for one-way analysis of variance, followed by Dunn’s multiple-comparison test or the Mann–Whitney t-test, for which P values are shown. P values of less than 0.05 were considered to indicate statistical significance.
RESULTS
BINDING OF CNS MEMBRANE ANTIGENS BY IgG FROM PERSONS WITH MULTIPLE SCLEROSIS
For the immunohistochemical analyses, we purified IgG antibodies from serum samples of 19 persons with multiple sclerosis and 24 with other neurologic diseases and tested for reactivity with brain-tissue sections. We observed that 11 of 19 samples (58%) from persons with multiple sclerosis showed glia-specific immunoreactivity when they were tested on human-brain sections, and 7 of those samples (37% of the 19 samples) showed immunoreactivity when they were tested on rat cerebellar sections (representative micrograph shown in Fig. S2A in the Supplementary Appendix). We did not observe glia-specific immunoreactivity when we used serum IgG from persons with other neurologic diseases. Next, we designed an enzyme-linked immunosorbent assay (ELISA) using plate-bound human cerebellar protein fractions enriched for either membrane or cytoplasmic antigens. We tested serum specimens from 56 persons with multiple sclerosis and from 29 with other neurologic diseases (including the serum specimens used in the immunohistochemical analyses described above). The serum from patients with multiple sclerosis bound plates coated with membrane proteins — but not plates coated with cytoplasmic proteins — more avidly than did the serum from persons with other neurologic diseases (P = 0.006 for the comparison with respect to membrane proteins and P = 0.42 for the comparison with respect to cytoplasmic proteins) (Fig. S2B in the Supplementary Appendix and data not shown). Therefore, in subsequent immunoprecipitation studies, we used CNS tissue enriched for membrane proteins.
IDENTIFICATION OF KIR4.1 AS A TARGET IN MULTIPLE SCLEROSIS
We purified and enriched IgG reactive against CNS membrane proteins from the pooled serum specimens of persons with multiple sclerosis using a column consisting of the membrane protein fraction from human brain. The enriched IgG fraction was used for subsequent antigen immunoprecipitation assays from human brain-tissue lysate. Immunoprecipitated CNS antigens were then analyzed by means of sodium dodecyl sulfate–polyacrylamide-gel electrophoresis (SDS-PAGE) and separated by means of two-dimensional gel electrophoresis (Fig. 1A). Seven protein spots were excised and analyzed with the use of matrix-assisted laser desorption–tandem mass spectrometry. One of the spots contained KIR4.1. We confirmed the identity of KIR4.1 as the target of serum IgG from persons with multiple sclerosis by means of immunoprecipitation and Western blotting, using extracts from rat kidney lysate, human brain lysate (Fig. 1B), and in vitro–translated KIR4.1 protein (Fig. 1C). The proteins recovered from the other spots did not specifically bind IgG from persons with multiple sclerosis.
Figure 1. Identification of KIR4.1 as a Target of Serum IgG in Multiple Sclerosis (MS).
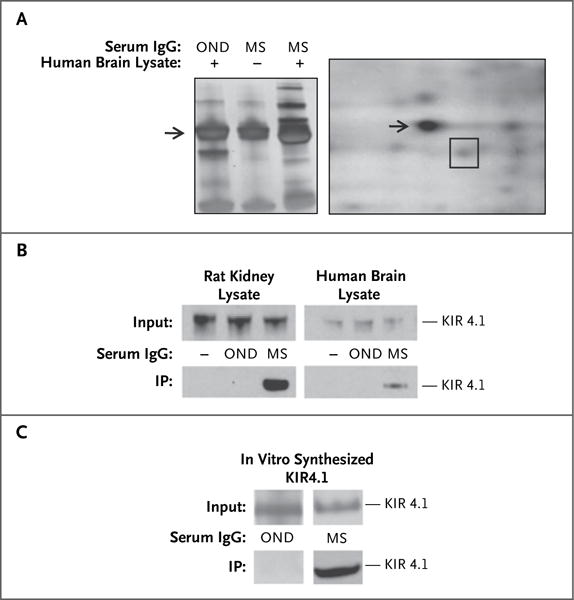
Panel A, left, shows a one-dimensional SDS-PAGE of human brain lysate precipitated with IgG from the pooled serum of 12 patients with MS and 12 with other neurologic diseases (OND). (For the middle lane [marked –], no brain lysate was used in the precipitation.) Unique bands (third lane) above and below the IgG heavy chain band (arrow) are observed after immunoprecipitation with purified IgG from the pooled serum of patients with MS. Panel A, right, shows a two-dimensional electrophoresis of brain antigens obtained after immunoprecipitation with serum IgG from patients with MS. The spot containing the KIR4.1 protein, identified by means of matrix-assisted laser desorption–tandem mass spectrometry, is outlined with a square. The arrow shows the IgG heavy chain spot. Panel B shows KIR4.1 detection by Western blot analysis in two different immunoprecipitation (IP) assays, as indicated. IP assays were performed with serum IgG from patients with OND and from patients with MS on membrane-protein– enriched fractions of rat kidney and human brain tissue lysates. The lanes on the left (marked –) of the blots are negative controls (no IgG used for IP). Panel C shows a Western blot analysis of KIR4.1 immunoprecipitation from in vitro–translated human KIR4.1 protein with serum IgG from patients with OND and from patients with MS.
BINDING OF KIR4.1 ON GLIAL CELLS BY SERUM IgG FROM PERSONS WITH MULTIPLE SCLEROSIS
Using rat-brain sections, we performed double immunofluorescence labeling with an anti-KIR4.1 monoclonal antibody and purified IgG antibodies from the serum specimens of persons with multiple sclerosis, selectively enriched for CNS membrane reactivity. As a control, we used purified IgG from the serum specimens of persons with other neurologic diseases. We observed a colocalization of the immunofluorescence signal when we used the monoclonal anti-KIR4.1 antibody and purified IgG antibodies from the serum specimens of persons with multiple sclerosis on rat cerebellar sections. We did not observe colocalization of the signal when we used purified IgG from the serum specimens of persons with other neurologic diseases (Fig. 2A). Next, we performed immunolabeling of cerebellar sections from 10-day-old wild-type mice and Kir4.1 knockout mice with IgG from the serum specimens of persons with multiple sclerosis. Kir4.1 is expressed in high amounts in the mouse brain 10 days after birth.21 KIR4.1 antibody-positive serum specimens from persons with multiple sclerosis stained glial cells in cerebellar sections from wild-type mice but not in sections from the knockout mice (Fig. 2B). We then prepared mixed glial primary cultures from mice. Purified IgG antibodies from the serum specimens of persons with multiple sclerosis stained the membranes of glial cells as detected by immunofluorescence and flow cytometry (Fig. S3 in the Supplementary Appendix), and anti-KIR4.1 antibodies isolated from purified IgG antibodies from the serum specimens of persons with multiple sclerosis bound to KIR4.1-transfected, but not mock-transfected, cells (Fig. S4 in the Supplementary Appendix). We concluded that anti-KIR4.1 antibodies exist in some persons with multiple sclerosis and that these antibodies react with KIR4.1 protein expressed on glial cells.
Figure 2. Validation of KIR4.1 as the Target of the Serum IgG Reactivity in Patients with MS.
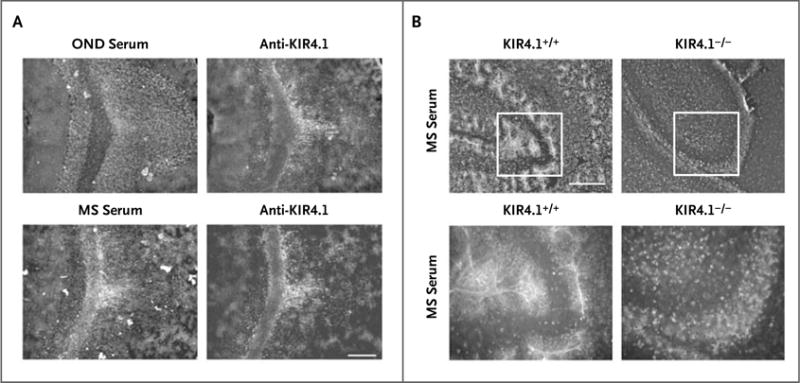
Panel A shows double immunofluorescence labeling revealing colocalization of monoclonal anti-KIR4.1 with serum IgG from a patient with MS in cerebellar sections of rat brain. Staining with serum of a patient with other neurologic diseases (OND) is shown as a control (scale bar, 200 μm for all parts of Panel A). Panel B shows immunofluorescence labeling of cerebellar sections of wild-type (left) and Kir4.1−/− (right) mice with purified serum IgG from a patient with MS (scale bar, 100 μm for the upper panels and 50 μm for the lower panels). KIR4.1-antibody–negative serum specimens did not stain central nervous system (CNS) tissue from wild-type mice or Kir4.1−/− mice (data not shown).
SPECIFICITY OF ANTI-KIR4.1 ANTIBODIES FOR MULTIPLE SCLEROSIS
To quantify anti-KIR4.1 reactivity, we used an ELISA with solid-phase recombinant KIR4.1 protein (Fig. S5 in the Supplementary Appendix). We detected no antibodies to KIR4.1 in the serum specimens from 37 healthy donors (dilution 1:100, data not shown). Next, we analyzed the serum specimens from healthy donors and from persons with multiple sclerosis or other neurologic diseases from the discovery series. Antibody titers were significantly higher in the serum specimens from persons with multiple sclerosis than in those from healthy donors and those from persons with other neurologic diseases (P<0.001 for both comparisons) (Fig. 3A). We obtained similar results in two validation series (Fig. 3A). Combining the results, we observed antibodies to KIR4.1 in 186 of 397 persons with multiple sclerosis (46.9%), in 3 of 329 persons with other neurologic diseases (0.9%), and in none of the 59 healthy donors. We found no significant differences in the prevalences or titers of serum anti-KIR4.1 antibodies among persons with a clinically isolated syndrome, those with relapsing–remitting multiple sclerosis, and those with progressive multiple sclerosis (Fig. S6A in the Supplementary Appendix) and observed no correlation between KIR4.1 antibody positivity and age, clinical characteristics, or characteristics of the cerebrospinal fluid (Table S3 in the Supplementary Appendix), although the study was neither designed nor powered to provide a test of correlation. We also detected anti-KIR4.1 antibodies in the cerebrospinal fluid of 19 of 30 patients with multiple sclerosis whose cerebrospinal fluid was tested for anti-KIR4.1 reactivity. We observed evidence for intrathecal antibody synthesis in 2 of these 19 patients (Fig. S6B in the Supplementary Appendix).
Figure 3. High-Titer Serum Reactivity to the KIR4.1 Protein in a Subgroup of Patients with MS.
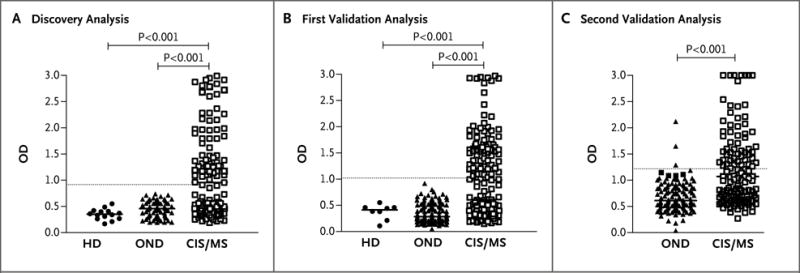
A protein-based enzyme-linked immunosorbent assay (ELISA) was used to detect anti-KIR4.1 serum autoantibodies. Purified recombinant KIR4.1 from HEK293 cells was covalently coupled to ELISA plates. Serum antibody binding to KIR4.1 was determined in healthy donors (HD), in patients with other neurologic diseases (OND), and in patients with MS or a clinically isolated syndrome (CIS) for the discovery series (Panel A), the first validation series (Panel B), and the second validation series (Panel C). For the first two series, KIR4.1 antibody titers of healthy persons, of patients with OND, and of patients with MS or a CIS were compared with the use of the Kruskal–Wallis test of one-way analysis of variance followed by Dunn’s multiple comparison test, for which P values are shown. In the third series, KIR4.1 antibody titers in patients with OND and those with MS or a CIS were compared with the use of the Mann–Whitney t-test. The threshold for anti-KIR4.1 antibody positivity (5 SD above the mean optical density [OD] for healthy persons) is indicated by a dashed horizontal line. For further details, see the Supplementary Appendix.
MAPPING THE KIR4.1 EPITOPE
Analysis of the membrane topology of the KIR4.1 protein has predicted two extracellular loops (KIR4.190–114 and KIR4.1134–142) (Fig. S7A in the Supplementary Appendix). We synthesized peptides representing the extracellular loops of KIR4.1 and the adjacent intramembrane regions (KIR4.183–120 and KIR4.1128–148, respectively), tagged them with biotin, and layered them onto streptavidin-coated ELISA plates. We observed serum reactivity to KIR4.1128–148 in 4% of the persons with multiple sclerosis but in none of the healthy donors or persons with other neurologic diseases (P = 0.08) (data not shown). In contrast, antibody titers to KIR4.183–120 were significantly higher in serum specimens from persons with multiple sclerosis than in those from healthy donors (P<0.001) or those from persons with other neurologic diseases (P<0.001) (Fig. S7B in the Supplementary Appendix).
We observed a strong correlation between the serum antibody reactivity measured by means of ELISA with the use of recombinant KIR4.1 and the antibody reactivity measured by ELISA with the use of the peptide fragment KIR4.183–120 (two-tailed Pearson’s correlation coefficient, 0.93; P<0.001) (Fig. S7C in the Supplementary Appendix). We therefore performed an ELISA-based competition assay using the KIR4.183–120 peptide and the recombinant KIR4.1 protein. Binding of serum anti-KIR4.1 antibodies to the KIR4.1 protein was outcompeted by the KIR4.183–120 peptide but not by a control peptide derived from the C-terminal sequence of KIR4.1 (KIR4.1356–375) (Fig. S7D in the Supplementary Appendix). Similarly, binding of a monoclonal antibody to the C-terminal part of the protein was outcompeted by the C-terminal peptide but not by the KIR4.183–120 peptide (data not shown). We made similar observations using a cell-based competitive binding assay. Antibodies against the KIR4.1 protein (obtained from persons with multiple sclerosis) were outcompeted by KIR4.183–120 but not by the control peptide (Fig. S8 in the Supplementary Appendix). Thus, serum reactivity to KIR4.1 is present in a significant percentage of patients with multiple sclerosis and is directed against determinants of the first extracellular loop of the protein.
EFFECT OF ANTI-KIR4.1 IgG IN VIVO
The IgG isotype is critical to complement activation: IgG1 and IgG3, but not IgG2 and IgG4, can activate the complement cascade. We determined IgG isotypes in the serum of 20 persons with multiple sclerosis who were positive for anti-KIR4.1 antibodies and observed that all 20 had IgG1 antibodies, IgG3 antibodies, or both (Fig. S9 in the Supplementary Appendix).
To determine the pathogenic potency of KIR4.1-specific serum antibodies, we prepared KIR4.1 containing IgG from persons with multiple sclerosis. As a control, we used KIR4.1-depleted IgG, in which KIR4.1 reactive antibodies had been preabsorbed (Fig. S10 in the Supplementary Appendix). We supplemented aliquots of these preparations with human complement and injected them into the cisternae magnae of wild-type mice. We used PBS supplemented with complement as a further control. The mice were killed after 24 hours, and we assayed expression of glial fibrillary acidic protein (Gfap), Kir4.1, and C9neo (which is a marker of complement activation) in sections of the cerebellum. PBS-injected mice and mice injected with serum IgG depleted of KIR4.1 antibodies lacked C9neo reactivity and showed a normal pattern of Gfap and Kir4.1 expression; Kir4.1 was expressed in astrocytes and oligodendrocytes (Fig. 4). In contrast, the mice injected with KIR4.1-reactive serum IgG showed alteration of Gfap expression, loss of Kir4.1 expression, and C9neo deposits. The loss of parenchymal Kir4.1 was most pronounced in the vicinity of the subarachnoid space and less pronounced with increasing distance from the sub-arachnoid space. C9neo deposits were locally restricted to regions of Kir4.1 depletion. These results suggest that multiple sclerosis–specific anti-KIR4.1 serum IgG can recognize its target antigen in the CNS and induce structural damage to glial cells.
Figure 4. KIR4.1-Specific MS Serum IgG Antibodies and Loss of Kir4.1 Staining, Disruption of Gfap Architecture, and Activation of Complement in Vivo in Mice.
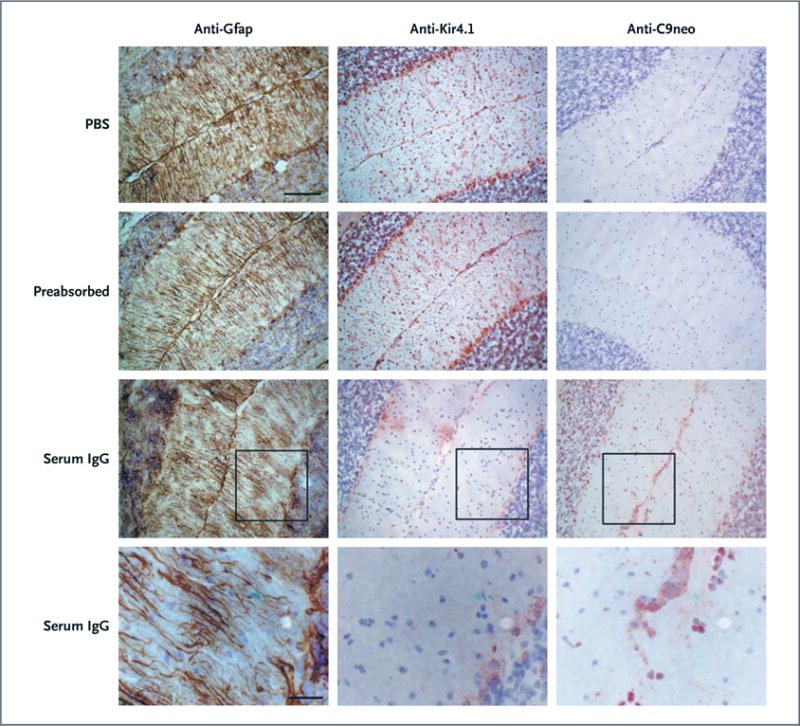
Phosphate-buffered saline (PBS; first row), serum IgG from a patient with MS depleted of KIR4.1-specific antibodies (preabsorbed, second row), or serum IgG with preserved anti-KIR4.1 reactivity (third and fourth rows) was injected into the cisternae magnae of C57BL/6 mice together with human complement. Twenty-four hours after injection, the mice were killed and brain sections were assessed for glial fibrillary acidic protein (Gfap) expression (left column), Kir4.1 expression (middle column), and C9neo reactivity (right column) by means of immunohistochemical analysis. The monoclonal anti-Kir4.1 antibody, which was used to visualize Kir4.1 expression, binds the intracellular domain of the protein and does not compete with the KIR4.1 reactive serum IgG for the same epitope of the KIR4.1 protein (Fig. S7 in the Supplementary Appendix and data not shown). Moreover, the lack of Kir4.1 immunoreactivity in mice that were injected with serum IgG with preserved anti-KIR4.1 specific antibodies is not caused by masking of the Kir4.1 antigen by nonspecific serum IgG antibodies, because the monoclonal antibody used to detect glial Kir4.1 expression was not blocked when it was applied together with serum IgG that had been preabsorbed with KIR4.1 (see second row) (scale bars, 50 μm in the upper three rows and 20 μm in the bottom row).
DISCUSSION
Conventional strategies to uncover autoantibodies in patients with multiple sclerosis have focused largely on serologic screening for immunoglobulins to candidate target molecules preselected on the basis of their relevance to myelin biology and to autoimmune encephalitis in animal models.22 These screenings have been performed with the use of proteins expressed by Escherichia coli, through phage display, and through the use of peptide libraries,23–26 but none have yielded potential multiple sclerosis–specific targets of the humoral immune response.27,28 Ideally, screens should embrace the entire antigenic repertoire of the CNS and allow for the recognition of conformationally dependent determinants.
It was with these considerations in mind that we designed our study and were able to identify and characterize a specific serum IgG directed against KIR4.1. The antibody is present in a subgroup of persons with multiple sclerosis — 47% of the group that we analyzed — and it has biologic effects in vivo.
In the rat brain, Kir4.1 expression is restricted to glial cells, specifically to oligodendrocyte cell bodies and to astrocyte processes surrounding synapses and blood vessels.29–31 We detected a similar astrocytic localization of Kir4.1 in mouse-brain sections. In human brain, we observed a perivascular localization of KIR4.1 (similar to Kir4.1 expression in rat brain) when we used either monoclonal anti-KIR4.1 antibody or IgG antibodies purified from the serum specimens of persons with multiple sclerosis. Various functions have been attributed to astroglial expression of KIR4.1, including maintenance of the electrochemical gradient across the cell membrane of perisynaptic astrocytes, which is critical to the efficient potassium buffering and uptake of glutamate.30,32 It has been proposed that there is a functional interaction between KIR4.1 and AQP4 at perivascular astrocyte processes and that this interaction regulates water homeostasis.33,34 In addition, studies of Kir4.1 knockout mice have shown that development of oligodendrocytes and myelination depend on Kir4.1 expression.35 Mutations in KCNJ10 (the gene encoding KIR4.1) in humans are associated with the EAST or SeSAME syndrome, characterized by epilepsy, ataxia, tubulopathy, and sensorineural deafness.36–39 These observations, combined with the findings we describe here, suggest that KIR4.1 is a candidate autoantigen in multiple sclerosis.
How could antibodies against KIR4.1 contribute to the pathogenesis of multiple sclerosis? As we have shown, serum anti-KIR4.1 antibodies from persons with multiple sclerosis can deplete KIR4.1 on glial cells and alter expression of GFAP in astrocytes, possibly through activation of complement at sites of KIR4.1 expression. KIR4.1-specific antibodies may also induce antibody-dependent cell-mediated cytotoxicity. C9neo deposits (indicating complement activation) have been observed in acute demyelinating multiple sclerosis lesions.6 In addition, astroglial damage seems to occur in a subset of active multiple sclerosis lesions.40 It is also possible that anti-KIR4.1 antibodies may interfere with the channel function of KIR4.1, resulting in a disruption of potassium buffering and neurotransmitter homeostasis,32–34 and thus tissue injury or impaired remyelination.
Supplementary Material
Acknowledgments
Supported by grants from the German Competence Network Multiple Sclerosis through the German Ministry for Education and Research (Control-MS, 01GI0917) and from the Deutsche Forschungsgemeinschaft (DFG) (He2386/7-1). Dr. Korn is supported by grants from the DFG and the Gemeinnützige Hertie-Stiftung; Dr. Aslam, by grants from the German Academic Exchange Service; and Dr. Bennett, by grants from the National Multiple Sclerosis Society (RG4320) and the Guthy-Jackson Charitable Foundation.
We thank Jürgen Schlegel, Technische Universität Munich, for providing the human brain tissues; Gerald Seifert, University of Bonn, for providing the Kir4.1−/− mice; Christine Stadelmann, University of Göttingen, for providing the C9neo antibody; and Verena Grummel and Stephanie Wirth for technical support.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–52. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 2.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 3.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol. 2007;61:288–99. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- 4.The International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genome-wide study. N Engl J Med. 2007;357:851–62. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 5.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–9. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 6.Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol. 1998;43:465–71. doi: 10.1002/ana.410430409. [DOI] [PubMed] [Google Scholar]
- 7.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 8.Breij EC, Brink BP, Veerhuis R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. 2008;63:16–25. doi: 10.1002/ana.21311. [DOI] [PubMed] [Google Scholar]
- 9.Keegan M, König F, McClelland R, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet. 2005;366:579–82. doi: 10.1016/S0140-6736(05)67102-4. [DOI] [PubMed] [Google Scholar]
- 10.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. 2008;358:676–88. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 11.Uccelli A, Aloisi F, Pistoia V. Unveiling the enigma of the CNS as a B-cell fostering environment. Trends Immunol. 2005;26:254–9. doi: 10.1016/j.it.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Meinl E, Krumbholz M, Hohlfeld R. B lineage cells in the inflammatory central nervous system environment: migration, maintenance, local antibody production, and therapeutic modulation. Ann Neurol. 2006;59:880–92. doi: 10.1002/ana.20890. [DOI] [PubMed] [Google Scholar]
- 13.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–12. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 15.Bennett JL, Lam C, Kalluri SR, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66:617–29. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–43. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- 17.Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intracerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–61. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roemer SF, Parisi JE, Lennon VA, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130:1194–205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- 19.Berglund L, Björling E, Oksvold P, et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019–27. doi: 10.1074/mcp.R800013-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Klein M, Koedel U, Pfister HW, Kastenbauer S. Meningitis-associated hearing loss: protection by adjunctive antioxidant therapy. Ann Neurol. 2003;54:451–8. doi: 10.1002/ana.10684. [DOI] [PubMed] [Google Scholar]
- 21.Seifert G, Hüttmann K, Binder DK, et al. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci. 2009;29:7474–88. doi: 10.1523/JNEUROSCI.3790-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quintana FJ, Farez MF, Viglietta V, et al. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci U S A. 2008;105:18889–94. doi: 10.1073/pnas.0806310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cortese I, Tafi R, Grimaldi LM, Martino G, Nicosia A, Cortese R. Identification of peptides specific for cerebrospinal fluid antibodies in multiple sclerosis by using phage libraries. Proc Natl Acad Sci U S A. 1996;93:11063–7. doi: 10.1073/pnas.93.20.11063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Archelos JJ, Trotter J, Previtali S, Weissbrich B, Toyka KV, Hartung HP. Isolation and characterization of an oligodendrocyte precursor-derived B-cell epitope in multiple sclerosis. Ann Neurol. 1998;43:15–24. doi: 10.1002/ana.410430107. [DOI] [PubMed] [Google Scholar]
- 25.Cepok S, Zhou D, Srivastava R, et al. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest. 2005;115:1352–60. doi: 10.1172/JCI23661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Somers V, Govarts C, Somers K, Hupperts R, Medaer R, Stinissen P. Autoantibody profiling in multiple sclerosis reveals novel antigenic candidates. J Immunol. 2008;180:3957–63. doi: 10.4049/jimmunol.180.6.3957. [DOI] [PubMed] [Google Scholar]
- 27.Berger T, Reindl M. Multiple sclerosis: disease biomarkers as indicated by pathophysiology. J Neurol Sci. 2007;259:21–6. doi: 10.1016/j.jns.2006.05.070. [DOI] [PubMed] [Google Scholar]
- 28.Derfuss T, Linington C, Hohlfeld R, Meinl E. Axo-glial antigens as targets in multiple sclerosis: implications for axonal and grey matter injury. J Mol Med. 2010;88:753–61. doi: 10.1007/s00109-010-0632-3. [DOI] [PubMed] [Google Scholar]
- 29.Poopalasundaram S, Knott C, Shamotienko OG, et al. Glial heterogeneity in expression of the inwardly rectifying K(+) channel, Kir4.1, in adult rat CNS. Glia. 2000;30:362–72. doi: 10.1002/(sici)1098-1136(200006)30:4<362::aid-glia50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 30.Tang X, Taniguchi K, Kofuji P. Heterogeneity of Kir4.1 channel expression in glia revealed by mouse transgenesis. Glia. 2009;57:1706–15. doi: 10.1002/glia.20882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higashi K, Fujita A, Inanobe A, et al. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–C931. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- 32.Kucheryavykh YV, Kucheryavykh LY, Nichols CG, et al. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. 2007;55:274–81. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- 33.Nagelhus EA, Horio Y, Inanobe A, et al. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26:47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 34.Amiry-Moghaddam M, Williamson A, Palomba M, et al. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003;100:13615–20. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21:5429–38. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960–70. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scholl UI, Choi M, Liu T, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106:5842–7. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang X, Hang D, Sand A, Kofuji P. Variable loss of Kir4.1 channel function in SeSAME syndrome mutations. Biochem Biophys Res Commun. 2010;399:537–41. doi: 10.1016/j.bbrc.2010.07.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sala-Rabanal M, Kucheryavykh LY, Skatchkov SN, Eaton MJ, Nichols CG. Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1 (KCNJ10) J Biol Chem. 2010;285:36040–8. doi: 10.1074/jbc.M110.163170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharma R, Fischer MT, Bauer J, et al. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol. 2010;120:223–36. doi: 10.1007/s00401-010-0704-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.