

Abstract
One of the most remarkable manifestations of plant evolution is the diversity for floral branching systems. These “inflorescences” arise from stem cell populations in shoot meristems that mature gradually to reproductive states in response to environmental and endogenous signals. The morphology of the shoot meristem maturation process is conserved across distantly related plants, raising the question of how diverse inflorescence architectures arise from seemingly common maturation programs. In tomato and related nightshades (Solanaceae), inflorescences range from solitary flowers to highly branched structures bearing hundreds of flowers. Since reproductive barriers between even closely related Solanaceae have precluded a genetic dissection, we captured and compared meristem maturation transcriptomes from five domesticated and wild species reflecting the evolutionary continuum of inflorescence complexity. We find these divergent species share hundreds of dynamically expressed genes, enriched for transcription factors. Meristem stages are defined by distinct molecular states and point to modified maturation schedules underlying architectural variation. These modified schedules are marked by a peak of transcriptome expression divergence during the reproductive transition, driven by heterochronic shifts of dynamic genes, including transcriptional regulators with known roles in flowering. Thus, evolutionary diversity in Solanaceae inflorescence complexity is determined by subtle modifications of transcriptional programs during a critical transitional window of meristem maturation, which we propose underlies similar cases of plant architectural variation. More broadly, our findings parallel the recently described transcriptome “inverse hourglass” model for animal embryogenesis, suggesting both plant and animal morphological variation is guided by a mid-development period of transcriptome divergence.
The foundation for the extensive shoot architectural diversity found in flowering plants lies in the developmental programs controlling the maturation of apical meristems, populations of stem cells located at the tips of shoots (Barton 2010). Decades of research in model and crop species have revealed genetic pathways that dictate the reproductive transition to flowering and the development of diverse inflorescence forms (Pautler et al. 2013; Tanaka et al. 2013; Kyozuka et al. 2014; Benlloch et al. 2015). Upon perception and integration of environmental and endogenous cues, such as day length and the flowering hormone florigen (Benlloch et al. 2007; Shalit et al. 2009), meristems gradually mature from a small flat structure into a large domed reproductive inflorescence meristem (IM) (Kwiatkowska 2008). There are striking similarities in meristem ontogeny during the reproductive transitions of distantly related plants, suggesting that the process of meristem maturation is a prerequisite for inflorescence development and flower production shared by all flowering plants. Yet, this generic ontogeny belies the remarkable diversity between and within species for shoot architecture, most evident in inflorescences. For example, not all doming meristems become flowers, and those that achieve floral fate often do so at different rates. Furthermore, there can be extensive variation for the number, position, and timing at which additional IMs form as the inflorescence develops, all of which contribute to architectural diversity.
Mathematical modeling supports a theory of inflorescence diversity united under a common principle of meristem maturation, in which meristems repeatedly undergo a reduction of a vegetative promoting program and an increase of a floral promoting program (Frijters 1978; Prusinkiewicz et al. 2007). Whereas the florigen flowering pathway is universally responsible for initiating the maturation process (Corbesier et al. 2007; Turck et al. 2008), other regulators of flowering and inflorescence development represent a strikingly large and variable collection of genes. For example, in maize, the zinc finger transcription factor gene INDETERMINATE1 (ID1) promotes flowering, and the RAMOSA (RA) genes are major regulators of ear and tassel inflorescence development (Colasanti et al. 1998; Gallavotti et al. 2010). In contrast, critical players in tomato are the unrelated transcriptional regulators TERMINATING FLOWER (TMF), COMPOUND INFLORESCENCE (S), and the floral identity gene ANANTHA (AN) (Lippman et al. 2008; MacAlister et al. 2012). In rice, the functions of ID1 and TMF are conserved (Park et al. 2008; Yoshida et al. 2013), but the RAs have no known role in panicle development (Vollbrecht et al. 2005), and the homolog of S primarily controls tillering (Wang et al. 2014). Nevertheless, all these genes share a common function of modulating the process of meristem maturation.
Our previous genetic and genomic analyses on tomato mutants and a wild relative with variable inflorescence architectures have suggested that the rate of meristem maturation drives inflorescence complexity and flower production. Specifically, a delay in meristem maturation can prolong the transitional state of inflorescence meristems to increase complexity (Park et al. 2012, 2014). However, the extent to which divergence in the heterochronic regulation of meristem maturation, including both the identity and timing of expression of key factors, underlies inflorescence diversity in a broader evolutionary context remains unclear and has been hindered by genetic incompatibilities between species. Here, using a systems genetics approach, we have taken advantage of the continuum of inflorescence architectures among members of the Solanaceae family, along with their uniquely large and accessible meristems, as a model to dissect the relationship between meristem maturation and evolutionary variation in inflorescence complexity and flower production.
Results
A continuum of inflorescence complexity in the Solanaceae family
We surveyed inflorescence architecture in a collection of 96 diverse Solanaceae species (Supplemental Table S1) representing the breadth of the Solanaceae phylogeny. To choose species amenable for transcriptome profiling across meristem maturation, we evaluated several developmental factors including plastochron index and time to flowering. We also prioritized species with readily accessible meristems and relatively early and narrow ranges of flowering times. Most importantly, we sought to maximize the range of inflorescence architecture diversity. The selected species represented important model and nonmodel systems and consisted of two species with single-flowered inflorescences (Capsicum annuum: cultivated pepper; Nicotiana benthamiana: a model tobacco species), two species with linear, multiflowered inflorescences (Solanum lycopersicum: cultivated tomato; S. prinophyllum: forest nightshade), and a previously profiled wild tomato species (S. peruvianum) with branched inflorescences (Fig. 1).
Figure 1.

Phylogeny and meristem ontogeny from five Solanaceae species representing a quantitative range of inflorescence complexity. (A) Phylogeny of S. lycopersicum (Sl, domesticated tomato), S. peruvianum (Spe, wild tomato species), S. prinophyllum (Spr, forest nightshade), N. benthamiana (Nb, tobacco), and C. annuum (Ca, pepper) estimated by the Orthologous MAtrix (OMA) pipeline, with distances in PAM units (Dessimoz et al. 2005; Roth et al. 2008). (B) Stereoscope images of the Middle Vegetative Meristem (MVM), Transition Meristem (TM), Floral Meristem (FM), and inflorescence from the five species arranged from most complex (top) to least complex (bottom) inflorescence architecture. Days after germination (DAG) and leaf number (L) at point of harvest are indicated. Dashed lines mark incisions for microdissection of meristems for RNA-seq transcriptome analysis. Average number of flowers produced (±SD) by the mature inflorescence is shown in the inflorescence frame. White scale bars represent 100 µm. Staging for meristem collection was based on the plastochron index and a time scale based on DAG to flowering (Supplemental Figs. S1, S2; Supplemental Table S2). Reference images of meristem stages for all species except S. prinohyllum and C. annuum were adapted from MacAlister et al. (2012) and Park et al. (2012).
To ensure consistent collection and analysis across meristem maturation, we characterized flowering time in each species, defined as the number of leaves initiated before the first inflorescence, which ranged from six to 13 leaves (Supplemental Fig. S1; Supplemental Table S2). Following our previous analysis of S. lycopersicum and S. peruvianum (Park et al. 2012), we completed a detailed study of meristem ontogeny and plastochron index to enable accurate collection of maturation stages for the remaining three species. Notably, meristem ontogeny was nearly indistinguishable between species, with vegetative stages appearing as small flat structures that gradually increased in size to large tall domes at the reproductive transition stage, subtended by the final leaf before becoming a floral meristem (Fig. 1; Supplemental Fig. S2A). Three developmentally distinct meristem stages were harvested in all species: Middle Vegetative Meristem (MVM, a vegetative stage prior to floral transition), Transition Meristem (TM, tall dome subtended by final leaf), and Floral Meristem (FM, initiation of the first flower). An additional Late Vegetative Meristem (LVM) stage was collected in N. benthamiana to complement preexisting S. lycopersicum and S. peruvianum data (Supplemental Table S3). Meristem staging was validated by semiquantitative RT-PCR using known meristem stage specific marker genes (Supplemental Fig. S2B).
Transcriptome profiling reveals dynamic gene expression across meristem maturation stages
Following our optimized protocol previously developed to generate meristem transcriptomes for S. lycopersicum and S. peruvianum (Park et al. 2012), we extracted mRNA from pooled meristems of the same stage for the other three species and performed Illumina sequencing. In total, we analyzed 1.38 billion paired-end reads across the five species, averaging 31.3 million reads per biological replicate. Read quality was high, with an average of ∼92.9% of reads passing quality-based read trimming filters and used in subsequent alignment and assembly analyses (Supplemental Table S3).
Reference genome or transcriptome sequences were available for all species except for the nonmodel plant, S. prinophyllum. To obtain a transcriptome for comparative analyses, we performed a de novo assembly for S. prinophyllum using Trinity (Grabherr et al. 2011). We refined the initial set of 189,729 assembled contigs into 103,896 assembled transcripts with an N50 size of 2.1 kb. To assess the depth and quality of our de novo assembly, we used BLAST to compare the S. prinophyllum transcriptome with annotations of the closely related tomato, potato, and pepper genomes (Potato Genome Sequencing Consortium et al. 2011; Tomato Genome Consortium 2012; Kim et al. 2014). Notably, 12,609 tomato, 12,590 potato, and 11,340 pepper genes were at least 90% reconstructed by raw de novo contigs, representing 36.3%, 36.0%, and 32.5% of total annotated genes from tomato, potato, and pepper, respectively (Supplemental Table S4). When considering the 17,492 genes expressed with at least one count per million (CPM) in the tomato meristem, the S. prinophyllum assembly included 72% of the meristem expressed tomato transcriptome. The assembled contigs were next scanned for open reading frames (ORFs), and 47,900 ORFs with high quality matches to tomato proteins were quantified.
To identify differentially expressed genes, expression levels were quantified as raw counts and CPM (Supplemental Tables S5, S6). Since our main interest was relative changes in expression dynamics between species rather than absolute expression, we followed the method used by Law et al. (2014) and used CPM as a simple expression metric. This quantification showed that biological replicates were highly consistent (Supplemental Fig. S3). Differential expression between stages within each species (dynamic expression) was determined using edgeR and identified 2015 to 7198 genes depending on FDR, CPM, and fold-change cutoffs. The number of dynamic genes was virtually unaffected using 0.10 or no FDR, while removing an average one CPM threshold or reducing the fold-change cutoff from 2 to 1.5 approximately doubled the number of dynamic genes. Thus, to exclude lowly expressed and low fold-change genes, which are more likely to be false positives and introduce noise, we focused analyses on one CPM, twofold change, and 0.10 FDR cutoffs (Supplemental Fig. S4; Supplemental Table S7). These cutoffs identified dynamic expression for 2015–3272 genes, or 4%–7% of the transcriptome, indicating substantial transcriptional changes during meristem maturation.
Dynamic orthologous gene groups are highly enriched for transcription factors and define meristem maturation states
Dynamically expressed genes are expected to include both core components of the transcriptional networks directing the overall process of meristem maturation, as well as species-specific dynamics that may not be conserved across the Solanaceae. To compare expression profiles between species, we developed a set of orthologous gene groups (orthogroups) using the Orthologous MAtrix (OMA) pipeline (Dessimoz et al. 2005; Roth et al. 2008). Orthologous genes were found for 20,038, 18,363, and 20,559 of the 34,725 reference tomato genes in S. prinophyllum, C. annuum, and N. benthamiana, respectively. Given the limited divergence between S. peruvianum and S. lycopersicum, we used the reference S. lycopersicum genome and transcriptome for both species (Park et al. 2012). We found 13,252 complete orthogroups (members from all four transcriptomes) accounting for 38.2% of the tomato transcriptome. Incomplete orthogroups were much less common, with an average of 2437 three-species and 1533 two-species orthogroups. No orthologous genes were identified for 9561 of the reference tomato gene set (Supplemental Fig. S5; Supplemental Table S8).
Dynamic orthogroups, in which orthologs were dynamically expressed in one or more of the species (3646 orthogroups total) were highly variable, with the majority of orthogroups (60.7%) having dynamic expression in only a single species (Supplemental Fig. S6). Since orthogroups with only a single dynamic member likely represent species-specific divergence or errors in orthogroup assignment, we focused comparative meristem maturation analyses on 282 orthogroups dynamically expressed in at least four species, providing a conservative set of markers to compare maturation programs. Hierarchical clustering of within species Z-score normalized expression profiles showed matched stages across species grouped together, indicating this set of dynamic orthogroups is sufficient to describe distinct molecular profiles for vegetative, transition, and floral stages (Fig. 2A). Notably, the expression profile of the S. peruvianum TM closely resembled that of the S. lycopersicum LVM, supporting our previous findings that meristem maturation in S. peruvianum is severely delayed (Park et al. 2012). This delay is also evident in other stages, particularly the S. peruvianum FM, which displays a mixed transition and floral state.
Figure 2.

Clustering and principal component analysis (PCA) of dynamically expressed (DE) genes during meristem maturation. RNA-seq was used to quantify gene expression in the EVM, MVM, LVM, TM, and FM stages. (A) EdgeR was used to identify dynamically expressed genes between meristem stages within each species (Methods). Expression was Z-score normalized within species, and hierarchical clustering was visualized by heatmap, with hot colors (yellow) indicating higher expression and cold colors (blue) lower expression. Clustering generally grouped vegetative, transition, and floral meristem stages together, with the exception of the Spe TM and FM, which associated with vegetative and transition clusters, respectively, reflecting the severe delay in meristem maturation compared to Sl (Park et al. 2012). (B) PCA of normalized expression from DE genes. The first two PCs account for ∼52% of the overall variation, with PC1 primarily accounting for differences between vegetative and floral meristem stages. PC2 primarily separates the transition stage from vegetative and floral. Regions where the majority of vegetative, transition, and floral samples cluster together are indicated by green, blue, and red dashed ovals, respectively. Three notable outliers from the main vegetative, transition, and floral groups include the Spe TM and FM, and the Nb LVM, reflecting heterochronic shifts in expression that imply a delay (Spe) and acceleration (Nb) in meristem maturation. Heatmaps (C) and PCA (D) based on 63 DE transcription factors also reflect heterochronic shifts in expression and maturation schedules.
Given the clear delay of S. peruvianum expression profiles, we hypothesized species with simplified inflorescence complexity relative to S. lycopersicum would show strong evidence of more rapid maturation. However, expression profiles hinted at only subtle acceleration of maturation schedules in single-flowered species. For example, the tobacco LVM showed high expression of some orthogroups associated with the TM stage, but retained a mostly vegetative profile and clustered with other vegetative meristems. To summarize the comparison of maturation stages between species, we performed principal component analysis (PCA) of dynamic orthogroups. As with the clustering and heatmap analysis, meristems grouped together by developmental stage with the first two PCs explaining ∼52% of the variation, further corroborating the comparable molecular states of vegetative, transition, and floral meristems from our five species (Fig. 2B). There were several notable outliers in the PCA, most prominently the S. peruvianum TM and FM, which associated strongly with vegetative and mixed transition/floral expression profiles, respectively. Significantly, we found the N. benthamiana LVM was positioned between the vegetative and transition clusters, indicating a mixed molecular state and accelerated maturation schedule. However, evidence of accelerated maturation in C. annuum and S. prinophyllum was less apparent, perhaps because LVM profiles were not collected. Importantly, clustering (Supplemental Fig. S7) and PCA (Supplemental Fig. S8) using more permissive orthogroup gene sets with dynamic expression in one species (3646 genes; 12.4% transcription factors), two species (1728 genes; 15.3% transcription factors), and three species (782 genes; 17.9% transcription factors) were similar to the four species analysis.
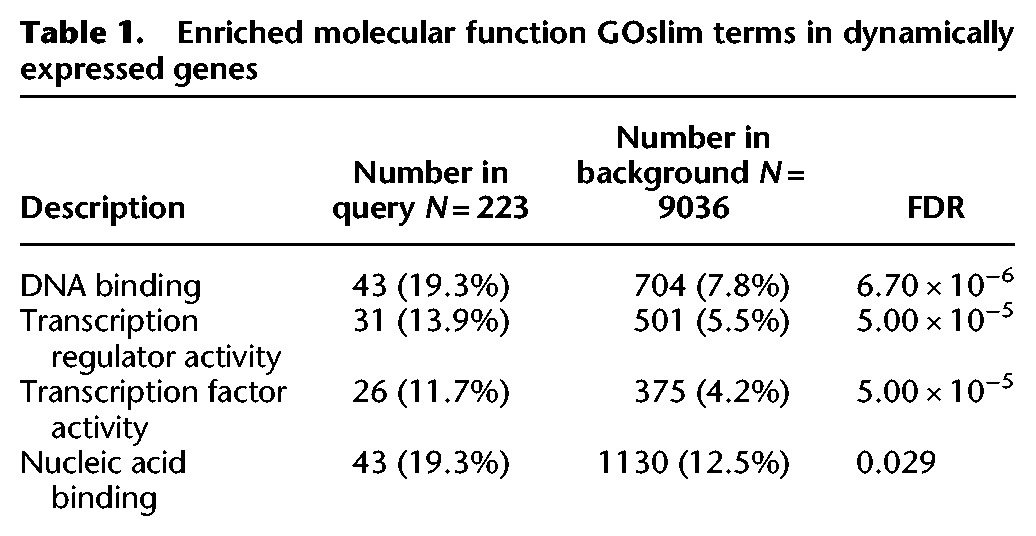
We explored the functional composition of the dynamic orthogroups by performing gene ontology (GO) analysis and found significant enrichment for multiple transcription factor related terms (Table 1). As with the full set of 282 dynamic orthogroups, a heatmap of the 63 annotated transcription factors clustered by developmental stage, suggesting transcription factors are major drivers of the vegetative, transition, and floral states (Fig. 2C). In support of this, PCA of transcription factors captured a greater proportion of variation (∼70%) in the first two principal components, and grouping of maturation stages remained virtually unchanged (Fig. 2D).
Table 1.
Enriched molecular function GOslim terms in dynamically expressed genes
Heterochronic shifts in expression during the reproductive transition in single-flowered species reveals modified maturation schedules
Clustering of matched vegetative, transition, and floral stages suggested similar expression dynamics characterize these stages among species. The most notable exception to this pattern was S. peruvianum, which showed prominent delays in all three maturation stages, most evident in the TM profile (Fig. 2; Park et al. 2012). Furthermore, PCA showed only modest acceleration of maturation in the single-flowered N. benthamiana, marked by transition stage identity in the tobacco LVM, although there were few indications of accelerated maturation in C. annuum and S. prinophyllum. One possibility is that the molecular hallmarks of accelerated meristem maturation in species with simpler inflorescences are reflected in a smaller set of genes compared to the widespread delays in S. peruvianum. These data suggested the LVM and TM stages—the transitional stages of maturation—define when maturation schedules shift to determine final inflorescence architecture. Indeed, upon examining the extent of variation in the dynamic meristem transcriptome between matched stages, we found transcriptome expression divergence was at the threshold of significance. These significance levels improved when the combined transitional LVM and TM stages were compared to the MVM and FM stages (Kolmogorov-Smirnov test) (Fig. 3A). Specifically, we found N. benthamiana had higher divergence in the LVM, whereas S. peruvianum was more divergent in the TM (Supplemental Fig. S9G). Thus, a peak of expression divergence between species with different inflorescence complexities occurred just prior to flower initiation. Notably, elevation of transcriptome expression divergence in transitional stages was more pronounced when considering dynamic transcription factors (Fig. 3B) and consistent across sets of more liberal dynamic orthogroups (Supplemental Fig. S9).
Figure 3.

Transcriptome expression divergence during meristem maturation and precocious gene expression in single-flowered species. Pairwise transcriptome distance measured by Euclidean distance between species and within stage showed elevated transcriptome expression divergence in the combined transitional stages (LVM and TM) for all 282 dynamic orthogroups (LVM/TM vs. MVM P < 0.026; LVM/TM vs. FM P < 0.012; Kolmogorov-Smirnov test) (A) and 63 dynamic transcription factor orthogroups (LVM/TM vs. MVM P < 0.038; LVM/TM vs. FM P < 0.065; Kolmogorov-Smirnov test) (B). K-means clustering of Z-score normalized tomato (Sl) dynamic genes (12 clusters) was performed. Orthologs from the single-flowered tobacco (Nb) and pepper (Ca) were then subclustered (10 subclusters) for each of the 12 tomato clusters. (C,D) Representative clusters and subclusters showing tomato FM-peaking (C) and TM-peaking (D) genes in tomato (red lines) compared to orthologs from single-flowered species (blue lines). Shown are orthogroups with conserved (Cons) expression (top), precocious expression peaking one stage early (middle), and precocious expression peaking in the same meristem stage as tomato (bottom). The number of genes (N) in each specific Cluster:Subcluster (C:S) is indicated above. Gene expression was compared on a gene-by-gene basis to more precisely identify precocious and conserved genes (Methods). (E,F) Venn diagrams showing overlap between tobacco and pepper precocious orthogroups from the floral (E) and transition (F) stages identified in the gene-by-gene analysis. A substantial proportion of orthogroups expressed precociously in both tobacco and pepper are transcription factors (50% and 23%, respectively). (G,H) Representative precocious floral (G) and transitional (H) orthogroups, scaled from 0 to 1, in tomato, pepper, tobacco, and S. peruvianum. Notably, precocious orthogroups include the known key floral identity F-box gene, ANANTHA, and the transition homeobox gene, COMPOUND INFLORESCENCE (Lippman et al. 2008).
The peak of transcriptome expression divergence during the reproductive transition suggested a critical window for maturation schedule modification, likely driven by a subset of dynamic orthogroups. Since we previously showed (Park et al. 2012), and validated here (Fig. 2), that S. peruvianum displays a dramatically delayed maturation schedule involving hundreds of dynamic genes, we used k-means clustering to identify genes underlying accelerated maturation during transitional stages of pepper and tobacco. Beginning with 12 reference clusters of tomato dynamic genes, orthogroup expression from the single-flowered species in each original tomato cluster was divided into 10 subclusters to identify genes showing precocious expression. Although many clusters had conserved expression dynamics between species (Supplemental Fig. S10), a number of orthogroups displayed precocious expression (Fig. 3C,D). Based on the number of genes in each k-means cluster and subcluster, precociously expressed genes comprised ∼15% and 60% of tomato TM enriched genes in pepper and tobacco, respectively, with the lower percentage in pepper likely reflecting absence of the LVM stage. Similarly, 45% of tomato FM enriched genes were precociously expressed in tobacco and pepper. This precocious expression of TM and FM identity in single-flowered species suggests an accelerated maturation schedule, marked by heterochronic shifts in expression of a smaller subset of dynamic genes compared to S. peruvianum.
The stochastic nature of k-means clustering can cause grouping of conserved and subtly precocious genes. We therefore performed a gene-by-gene search for precocious expression, defined by slightly elevated (1.5-fold) expression in the previous maturation stage. Of 56 transition and 86 floral tomato marker genes, 60% of transition and 75% of floral marker genes were precociously expressed in at least one of the single-flowered species. We further found 26 floral and eight tomato transition genes were precociously expressed in both tobacco and pepper (Fig. 3E,F). Among these (Fig. 3G,H) is the tomato FM marker gene AN (Lippman et al. 2008; Souer et al. 2008), whose orthologs begin climbing in expression at the TM stage of single-flowered species (Fig. 3G, bottom). This high proportion of precocious expression is maintained across several expression level cutoffs (Supplemental Table S9), and these precocious genes are again enriched for transcription factor-related GO terms (Supplemental Tables S10, S11). Furthermore, 50% of FM and 90% of TM marker genes precociously expressed in pepper and tobacco are delayed in S. peruvianum, indicating many of the same factors define delayed and accelerated maturation schedules (Supplemental Table S10). Together, these analyses indicate heterochronic shifts in a handful of key regulators during a critical transitional window of meristem maturation dictate inflorescence complexity.
Modified maturation schedules underlie quantitative variation in flower production
In tomato, additional flowers arise from specialized reproductive axillary meristems known as sympodial inflorescence meristems (SIMs). SIMs form iteratively after termination of the previous SIM, and each SIM is born in an advanced stage of mixed TM and FM identity reflected in marker genes for both stages (Park et al. 2012). The tomato SIM maturation schedule allows only a single SIM to form during each cycle, and SIM cycling eventually ends, resulting in an inflorescence with 5–15 flowers depending on genotype (Park et al. 2012). In other species, differences in SIM cycling result in as few as two flowers on each inflorescence (Supplemental Table S1). One explanation for this quantitative variation in flower production is species-specific variation in SIM maturation schedules. To test this idea, we took advantage of SIM transcriptome data from the three multiflowered species representing a continuum of flower production. These species allowed comparison of SIM maturation schedules from S. lycopersicum with species that develop less (S. prinophyllum) and more (S. peruvianum) complex inflorescences.
We asked if the three SIM transcriptomes reflected quantitative shifts in the proportion of TM to FM meristem identity using PCA of 172 TM and FM tomato marker genes. Using tomato TM and FM stages as reference points, the first PC accounted for 55% of the variation, with the tomato TM and FM having the lowest and highest PC1 values, respectively (Fig. 4). Notably, SIMs from the three multiflowered species were arrayed between the tomato TM and FM and were ordered from most to least complex inflorescence. The correlation between maturation state and inflorescence complexity strongly suggested quantitative variation in SIM maturation rate is responsible for producing the broad range of Solanaceae inflorescences. This result was consistent across more liberal sets of TM and FM marker genes (Supplemental Fig. S11). Thus, the mixed molecular state of the SIM is predictive of the quantitative level of flower production and inflorescence complexity in multiflowered species (Fig. 4).
Figure 4.
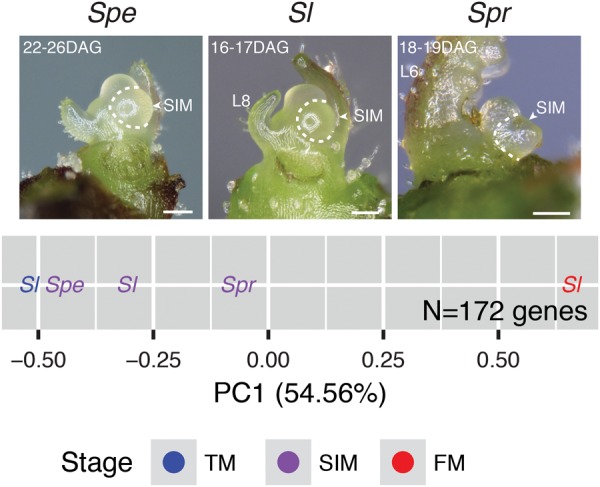
PCA of sympodial inflorescence meristem (SIM) transcriptomes from multiflowered species. The maturation state of each SIM was assessed by PCA of normalized gene expression. The tomato TM and FM were included as reference stages. The analysis was restricted to TM and FM marker genes (twofold higher expression in the TM or FM than any other stage) (Methods). PC1 accounted for 55% of the variation, with the tomato TM and FM representing the minimum and maximum values, respectively. As expected, all SIMs were intermediate between the tomato TM and FM. Notably, PC1 was positively correlated with inflorescence complexity with the Spe SIM closest to the tomato TM, Sl SIM intermediate, and Spr SIM closest to the tomato FM, indicating PC1 predicts SIM maturation state and flower production potential in multiflowered species. White scale bars represent 100 µm. Reference images of SIM for S. lycopersicum and S. peruvianum adapted from Park et al. (2012).
Heterochronic expression of two key meristem maturation regulators define inflorescence complexity
Our previous work showed tomato inflorescence development depends on precise timing of the homeobox transcription factor gene S, followed by the F-box floral specification gene AN. Expression of S promotes floral termination, beginning in the LVM and peaking at the TM, just before activation of AN in the FM. When mutated, floral identity is delayed (s mutant) or never achieved (an mutant), causing SIM overproliferation and highly branched inflorescences (Lippman et al. 2008; Park et al. 2012). Parallel functions for S and AN in pepper and petunia (Lippman et al. 2008; Rebocho et al. 2008; Souer et al. 2008; Cohen et al. 2014) suggest they can serve as specific markers of meristem maturation schedules. To obtain a more precise assessment of when and how maturation schedules vary, we performed in situ hybridization of S and AN in TM through FM stages in S. lycopersicum, S. peruvianum, S. prinophyllum, and N. benthamiana (Fig. 5; Supplemental Figs. S12–S15).
Figure 5.

Four species comparative mRNA in situ hybridization of the inflorescence architecture genes COMPOUND INFLORESCENCE (S) and ANANTHA (AN). Scaled expression values for S and AN are shown in Figure 3, G and H. (A) S is expressed in the tomato TM, late TM (LTM), and SIM, and shows delayed expression in the Spe LTM. Conversely, Spr and Nb have strong expression of S in the TM, with Spr also expressing S in the sympodial shoot meristem (SYM). Earliest expression of S is indicated by a red arrowhead. (B) AN is expressed weakly in the tomato TM and increases in the LTM, reaching peak expression in the FM. Expression in Spe only begins in the LTM and FM/SIM. In contrast, AN is expressed strongly in the TM stages of Spr and Nb. The earliest detected expression is indicated by a red arrowhead. (C) Cartoons of meristem maturation stages depicting expression dynamics and patterns of S and AN across the four species. Black scale bars represent 100 µm.
In S. lycopersicum, S expression in the TM stage persisted strongly through a late transition meristem (LTM) stage, but was absent by the FM (Fig. 5A). Consistent with previous findings (Park et al. 2012), the S. peruvianum homolog of S was delayed until the LTM and maintained in the FM, supporting a strong delay in maturation. Expression in tomato contrasted with simple inflorescence species, in which S expressed strongly in the TM and weakly in the LTM. Likewise, S was strongly expressed in the SIM of S. lycopersicum and S. peruvianum, but was less expressed in the S. prinophyllum SIM (Fig. 5A). Thus, activation of S homologs occurred earlier in meristem maturation for species with simple inflorescences.
Similar to S, expression of AN also reflected modified maturation schedules associated with inflorescence complexity. In all four species, AN was initially lowly or not expressed during transitional stages before peaking in the FM (Fig. 5B). AN was minimally expressed in the TM of S. peruvianum and S. lycopersicum. In contrast, the AN homologs of N. benthamiana and S. prinophyllum showed stronger expression in the TM, indicating precocious activation. The subtle shifts in S and AN expression were especially clear in serial sections from multiple in situ experiments (Supplemental Figs. S12–S15). These results provide two complementary examples of heterochronic shifts in gene expression, strongly supporting our transcriptome analysis.
Discussion
Inflorescence complexity in various plant systems is controlled by diverse factors that regulate meristem maturation, emphasizing a common theme in the evolution of floral branching systems (Benlloch et al. 2007; Kyozuka et al. 2014; Park et al. 2014). Our study established a strong association between modified meristem maturation schedules and quantitative variation in inflorescence complexity and further revealed a defining period of expression divergence during transitional maturation stages in the Solanaceae (Fig. 6). This divergence is reflected by heterochronic shifts of a relatively small number of dynamically expressed transcription factors and other genes. Some of these factors are likely drivers of modified meristem maturation schedules, explaining both qualitative and quantitative differences in inflorescence complexity.
Figure 6.

A meristem maturation model for inflorescence diversity in the Solanaceae. (A) Schematics depicting the relationship between variable rates of meristem maturation and inflorescence complexity. Bars and color gradients reflect maturation of the various meristems, with red dots marking a terminal floral meristem. Variation in maturation rate, reflected in shifting color gradients, determines the number of axillary meristems—thus, branches/flowers—that can form before termination. Depending on initial maturation state and rate of maturation, an axillary inflorescence meristem (SIM) can give rise to a single flower, a linear arrangement of a few to many flowers, or a few to many branches that will give rise to dozens or hundreds of flowers. The result is a quantitative range of inflorescence complexity and flower production. High transcriptome expression divergence during transitional stages, variable maturation rate, and evolutionary divergence in inflorescence complexity is ultimately driven by heterochronic shifts in the expression of key regulators that occur primarily in the late vegetative and transition meristem stages. (B) Selected species representing the continuum of Solanaceae inflorescence complexity and flower production that can be explained by the model.
The small number of genes that capture variation in primary and axillary inflorescence meristem maturation implies that the genetic basis for modified maturation schedules is relatively simple. In support of this, we previously showed that precociously activating AN in the tomato TM using S promoter sequences was sufficient to produce single and few-flowered inflorescences in tomato, mimicking N. benthamiana and S. prinophyllum (MacAlister et al. 2012). This led us to propose heterochronic differences in AN expression due to variation in cis-regulatory elements might explain simple inflorescences (MacAlister et al. 2012; Park et al. 2014). However, we found expressing either the tobacco or tomato AN CDS under tobacco AN regulatory sequences in an mutants was insufficient to produce simplified inflorescences (Supplemental Fig. S16), suggesting a complex interaction between cis- and trans-regulatory factors are required to explain the evolutionary shift from single-flowered tobacco to multiflowered tomato. This is perhaps not surprising given the amount of heterochronic changes in expression observed in our analyses, as well as numerous other documented variation in gene regulatory sequences that affect shoot architecture diversity across flowering plants (Studer et al. 2011; Meyer and Purugganan 2013; Kusters et al. 2015; Xu et al. 2015).
In the case of the Solanaceae, genes with known functions in inflorescence development, such as S and AN, could be partly responsible for altered maturation schedules. However, control of this critical maturation process clearly requires additional factors, some of which were likely identified in this study. Thus, a genomic approach benefits our understanding of shoot architecture diversity by elucidating the broader impact of subtle changes in gene regulatory networks. This is especially relevant to other plant systems such as grasses, in which mutant studies have revealed many shoot architecture genes. Notably, transcriptome profiling of maize ra mutants, which mimic the complex sorghum inflorescence, revealed changes in meristem identity and determinacy (Eveland et al. 2014), parallel to the delays in tomato meristem maturation caused by s and an (MacAlister et al. 2012; Park et al. 2012). The two systems share striking similarities, despite spanning the monocot–eudicot divide and having adopted different terminologies, suggesting our observation of modified maturation schedules is widely applicable. The principle of modified meristem maturation schedules and inflorescence complexity in the grasses is evident in many recent studies, including defects in the florigen pathway in wheat (Boden et al. 2015) and maize (Danilevskaya et al. 2010; Meng et al. 2011), delays in ra1 ortholog expression in sorghum (Vollbrecht et al. 2005), and delays in rice inflorescence meristem specification (Kyozuka et al. 2014). Thus, genomic studies of meristem maturation in diverse grasses could establish the role of modified meristem identity and determinacy (i.e., maturation) in the evolution of grass inflorescence diversity.
Our findings indicate that regardless of the specific genes, pathways, and networks controlling inflorescence architecture, achieving morphological diversity by fine-tuning meristem maturation during a critical transitional window is likely conserved in flowering plants. This provides a useful framework for a unifying model of meristem maturation and inflorescence complexity, consistent with mathematical modeling of inflorescence diversity rooted in a hypothetical “vegetativeness” variable (Frijters 1978; Prusinkiewicz et al. 2007), in which coevolution of cis- and trans-regulatory modules explain subtle heterochronic shifts in expression of key regulators. These shifts in expression alter the acquisition of floral fate, modifying the window of meristem renewal and differentiation to ultimately drive the extensive diversity for shoot branching systems. Furthermore, this unifying principle provides a foundation for understanding the origin of inflorescence diversity, by discarding boundaries imposed by botanical terminology and highlighting the quantitative nature of meristem maturation.
The concept that morphological diversity originates from heterochrony in development is not new, neither is the notion that progression of maturation and developmental morphology can be dissociated (Gould 1977). These ideas have been refreshed by recent transcriptome studies of the hourglass model of embryogenesis in animals (Richardson 1999; Domazet-Lošo and Tautz 2010; Kalinka et al. 2010) and plants (Quint et al. 2012), in which common morphologies during embryogenesis associate with evolutionarily ancient genes with conserved expression. In particular, our work strongly parallels the recently described transcriptome “inverse hourglass” of animal embryogenesis (Levin et al. 2016), in which cross-phyla transcriptome expression divergence peaks during the “mid-developmental transition” stage. This similarity suggests mid-development transcriptional networks are particularly flexible, perhaps providing a simple means to fine-tune developmental progression that may have been repeatedly favored by evolution to produce morphological diversity with minimal selective constraints. Notably, our model has implications for the existence of inverse hourglasses in additional development contexts, such as leaves (Freeling 1992; Ori et al. 2007; Ichihashi et al. 2014; Alvarez et al. 2016). This further raises the exciting possibility that transcriptome inverse hourglasses exist in other developmental programs beyond plants, such as in animal lung and limb development, in which tissue ontogeny is associated with organ complexity (Metzger et al. 2008; Zuniga 2015).
Methods
Plant materials and growth conditions
Ninety-six Solanaceae were phenotyped in Rehovot, Israel, and Cold Spring Harbor, New York. The species used for transcriptome profiling included: (1) Solanum peruvianum (LA0103), a wild tomato species with a single sympodial shoot and branched, multiflowered inflorescence; (2) S. lycopersicum cv. M82 (LA3475), a domesticated tomato with a single sympodial shoot and unbranched inflorescence; (3) S. prinophyllum, a species with short multiflowered inflorescences; (4) Capsicum annuum cv. MAOR (pepper), a single-flowered species with two sympodial shoots; and (5) Nicotiana benthamiana accession Nb-1 (tobacco), a single-flowered single sympodial shoot species. Seeds were pregerminated, sown in flats, and grown in a Cold Spring Harbor Laboratory greenhouse. Flowering time, flowers per inflorescence, and sympodial index were characterized for all species (Supplemental Fig. S1) as previously described (Park et al. 2012). Additional details are available in the Supplemental Methods.
Tissue collection and RNA sequencing
Plastochron index, morphological markers, meristem size and position relative to the last leaf formed, and a time scale based on days after germination (DAG) were used to define meristem stages (Fig. 1; Supplemental Fig. S2). Stages used for mRNA sequencing included Early, Middle, and Late Vegetative Meristem (EVM, MVM, and LVM) as well as Transition Meristem (TM), Floral Meristem (FM), and Sympodial Inflorescence Meristem (SIM). Notably, only the MVM, TM, FM, and SIM from multiflowered species were collected for all species, with additional stages profiled as project resources allowed. Generally, vegetative stages present as small, flat domes and the TM stages as broad, tall, and more swollen meristem domes. In multiflowered species, the FM and SIM were harvested after the last leaf formed as apical and lateral domes, respectively. Single-flowered species do not produce a SIM, and only FM samples were collected.
Meristems were collected and RNA extracted using previously published protocols (Park et al. 2012). In brief, seedlings of ∼3 cm long were fixed and meristems were collected by microdissection. Each biological replicate consisted of 50 or more pooled meristems from individual plants. Total RNA was extracted and checked for quality by gel electrophoresis or Bioanalyzer 2100 (Agilent). Stage and meristem specificity were validated with semiquantitative RT-PCR using stage specific marker genes (Park et al. 2012). Primer sequences are available in Supplemental Table S12.
Two biological replicates per stage and species were profiled. Libraries were produced using Epicentre or New England Biolabs library preparation kits (Supplemental Table S3), quality checked by Bioanalyzer 2100 (Agilent), and quantified with the KAPA Library Quantification Kit (Kapa Biosystems). Sequencing for N. benthamiana, S. prinophyllum, and C. annuum was performed on the HiSeq 2000 or GAIIx Illumina platforms (Supplemental Table S3). Reads for S. peruvianum and S. lycopersicum were obtained from a previous study that sequenced one sample per lane of Illumina GAIIx (Park et al. 2012). Raw sequencing reads used in this study are available from the NCBI Sequence Read Archive (Leinonen et al. 2011) under accession number SRP090200. Additional details on meristem staging and RNA sequencing are available in the Supplemental Methods.
Transcriptome assembly and quantification
Reads were assessed for quality, trimmed, and aligned to reference or de novo sequences for quantification. Paired reads for wild tomato, domesticated tomato, and tobacco were aligned to their respective reference genome sequences (Bombarely et al. 2012; Tomato Genome Consortium 2012) using TopHat2 (Kim et al. 2013). S. peruvianum was aligned to the tomato genome (SL2.50) with four mismatches allowed to accommodate the cross-species alignment. Unique fragments aligned to gene features were counted with HTSeq-count (Anders et al. 2015).
Species lacking complete genome sequences were quantified by aligning to transcriptome sequences. A transcriptome de novo assembly was obtained for S. prinophyllum using Trinity (Grabherr et al. 2011). Open reading frames were extracted (Trinity TransDecoder), and CDS sequences detected by the orthology pipeline were quantified. Quantification of C. annuum used the reference transcriptome v1.55 (Kim et al. 2014). Reads were aligned to transcriptomes using Bowtie 2 (Langmead et al. 2009; Langmead and Salzberg 2012), and concordant read pair alignments counted with a bash script.
Orthologous gene groups (orthogroups) were defined using the Orthologous MAtrix (OMA) pipeline (Dessimoz et al. 2005; Roth et al. 2008) with reference tomato (ITAG2.4) proteins (Tomato Genome Consortium 2012), pepper (v1.55) proteins (Kim et al. 2014), tobacco (v0.4.4) proteins (Bombarely et al. 2012), and S. prinophyllum protein sequences translated from the de novo assembly. S. lycopersicum genes were used as a foundation for orthogroups by considering all pairwise hits with other species. Cumulative orthogroup expression in CPM was calculated by averaging transcripts with at least 10% of the expression of the maximally expressed transcript. Specific orthogroup content can be found in Supplemental Table S8. Additional details on read processing and expression quantification are available in the Supplemental Methods.
Statistical analyses for heterochrony and modified maturation schedules
All statistical analyses were conducted in R (R Core Team 2015) except for GO term enrichment analyses, which used the agriGO toolkit (Du et al. 2010). For cross-species comparisons, orthogroup expression was examined after Z-score normalization (normalized expression) or scaling from 0 to 1 (scaled expression) within species. Hierarchical clustering, heatmaps, PCA, transcriptome expression divergence, and k-means clustering used normalized expression values. Gene-by-gene comparisons of precocious expression used scaled values.
Significant differential expression between meristem stages within species was determined with edgeR (Robinson et al. 2010) using twofold change, average 1 CPM, and 0.10 FDR cutoffs. Consistent dynamic orthogroup expression was defined as differential expression in four of five species. For orthogroups with multiple genes in a given species, differential expression was only required for one geneid. Heatmaps, PCA, and pairwise transcriptome distance (Euclidean distance) were done using normalized expression. A one-sided Kolmogorov-Smirnov test was used to compare the combined transitional stages (LVM and TM) transcriptome distances versus MVM and FM. Heatmaps, PCA, and transcriptome distance for more liberal and conservative definitions of dynamic expression were consistent with the main text (Supplemental Figs. S7–S9).
Analyses of heterochronic expression used k-means clustering of normalized orthogroup expression in a two-step process. Twelve clusters were defined for tomato expression, and then 10 subclusters for non-tomato orthogroup expression were calculated within each of the 12 original tomato clusters for a total of 120 cluster:subcluster groups for each species (Supplemental Fig. S10). Gene-by-gene analyses in tobacco and pepper used scaled expression. Tomato marker orthogroups were chosen where expression was twofold higher than any other developmental stage to focus the analysis on stage specific genes. Precocious expression of marker orthogroups in non-tomato species was defined as higher expression in the preceding developmental stage by several fold change and average CPM cutoffs. Additional details on statistical analyses are available in the Supplemental Methods.
In situ hybridization
RNA in situ hybridization was performed according to standard protocols (Jackson 1992). According to stage definitions described above, shoot apices were dissected and fixed. Late transition meristems (LTM), intermediate to TM and FM stages, were collected for a more granular dissection of S and AN expression patterns. In vitro transcribed RNA probes for AN orthologs were generated from full-length cDNA, whereas probes for S orthologs were synthesized from 5′ CDS fragments (∼600 bp length). Transcripts were detected using the DIG in situ hybridization system (Roche). Primer sequences are provided in Supplemental Table S12. Additional information on in situ hybridization is available in the Supplemental Methods.
AN cross-species transgenic complementation
Two transgenic constructs for expression of tomato and tobacco AN CDS under tobacco regulatory sequences were constructed. Tobacco regulatory sequences (3475 bp upstream and 1303 bp downstream) were fused with the tobacco and tomato AN CDS and transformed into a segregating tomato an mutant (an-e1546) background (Supplemental Fig. S16; Lippman et al. 2008). Sixteen first-generation transgenic (T0) plants were obtained and genotyped for the presence of the transgene and the an-e1546 allele, then phenotyped for inflorescence rescue. Progeny of an-e1546 heterozygotes were also genotyped and screened, and a similar range of partial rescue was observed. Transformation was performed according to Brooks et al. (2014).
Data access
Raw data from this study have been submitted to the Sol Genomics Network ftp site (ftp://ftp.solgenomics.net) and the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP090200. The S. prinophyllum Trinity de novo assembly is available at the NCBI Transcriptome Shotgun Assembly (TSA; https://www.ncbi.nlm.nih.gov/genbank/tsa/) database under accession number GEZT00000000. The version described in this paper is the first version, GEZT01000000.
Supplementary Material
Acknowledgments
We thank Sandra Knapp and the Botanical Gardens at Nijmegen (the Netherlands) for helpful discussion and providing seed for many of the species used in the initial screen and John (Stan) Alvarez for discussion and assistance in phenotyping the 96 species. We also thank Tim Mulligan and Sarah Vermylen from the CSHL Uplands Farm for assistance with plant care and Andrew Krainer and Justin Dalrymple for technical assistance. This material is based upon work supported by the National Science Foundation Postdoctoral Research Fellowship in Biology Grant No. 1523423 to Z.H.L., the Next-Generation BioGreen 21 Program (SSAC, PJ0118832016) to S.J.P., and National Science Foundation Plant Genome Research Project Grant No. 1237880 to M.C.S., J.V.E., and Z.B.L.
Author contributions: Z.B.L. conceived the project; S.J.P. collected plant material and performed Illumina sequencing; Z.H.L. and K.J. were responsible for transcriptome assembly and quantification; Z.H.L. was responsible for maturation and heterochrony analysis; S.J.P. performed in situ hybridization; Z.H.L., S.J.P., and J.V.E. performed the cross-species transformation experiment; and Z.H.L., S.J.P., K.J., M.C.S., and Z.B.L. wrote and prepared the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.207837.116.
References
- Alvarez JP, Furumizu C, Efroni I, Eshed Y, Bowman JL. 2016. Active suppression of a leaf meristem orchestrates determinate leaf growth. eLife 5: e15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton MK. 2010. Twenty years on: the inner workings of the shoot apical meristem, a developmental dynamo. Dev Biol 341: 95–113. [DOI] [PubMed] [Google Scholar]
- Benlloch R, Berbel A, Serrano-Mislata A, Madueño F. 2007. Floral initiation and inflorescence architecture: a comparative view. Ann Bot 100: 659–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benlloch R, Berbel A, Ali L, Gohari G, Millán T, Madueño F. 2015. Genetic control of inflorescence architecture in legumes. Front Plant Sci 6: 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden SA, Cavanagh C, Cullis BR, Ramm K, Greenwood J, Jean Finnegan E, Trevaskis B, Swain SM. 2015. Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat Plants 1: 14016. [DOI] [PubMed] [Google Scholar]
- Bombarely A, Rosli HG, Vrebalov J, Moffett P, Mueller LA, Martin GB. 2012. A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol Plant Microbe Interact 25: 1523–1530. [DOI] [PubMed] [Google Scholar]
- Brooks C, Nekrasov V, Lippman ZB, Van Eck J. 2014. Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system. Plant Physiol 166: 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen O, Borovsky Y, David-Schwartz R, Paran I. 2014. Capsicum annuum S (CaS) promotes reproductive transition and is required for flower formation in pepper (Capsicum annuum). New Phytol 202: 1014–1023. [DOI] [PubMed] [Google Scholar]
- Colasanti J, Yuan Z, Sundaresan V. 1998. The indeterminate gene encodes a zinc finger protein and regulates a leaf-generated signal required for the transition to flowering in maize. Cell 93: 593–603. [DOI] [PubMed] [Google Scholar]
- Corbesier L, Vincent C, Jang S, Fornara F, Fan Q, Searle I, Giakountis A, Farrona S, Gissot L, Turnbull C, et al. 2007. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316: 1030–1033. [DOI] [PubMed] [Google Scholar]
- Danilevskaya ON, Meng X, Ananiev EV. 2010. Concerted modification of flowering time and inflorescence architecture by ectopic expression of TFL1-like genes in maize. Plant Physiol 153: 238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessimoz C, Cannarozzi G, Gil M, Margadant D, Roth A, Schneider A, Gonnet GH. 2005. OMA, a comprehensive, automated project for the identification of orthologs from complete genome data: introduction and first achievements. In Comparative genomics (ed. McLysaght A, Huson DH), Vol. 3678, pp. 61–72. Springer, Berlin. [Google Scholar]
- Domazet-Lošo T, Tautz D. 2010. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468: 815–818. [DOI] [PubMed] [Google Scholar]
- Du Z, Zhou X, Ling Y, Zhang Z, Su Z. 2010. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38: W64–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eveland AL, Goldshmidt A, Pautler M, Morohashi K, Liseron-Monfils C, Lewis MW, Kumari S, Hiraga S, Yang F, Unger-Wallace E, et al. 2014. Regulatory modules controlling maize inflorescence architecture. Genome Res 24: 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M. 1992. A conceptual framework for maize leaf development. Dev Biol 153: 44–58. [DOI] [PubMed] [Google Scholar]
- Frijters D. 1978. Principles of simulation of inflorescence development. Ann Bot 42: 549–560. [Google Scholar]
- Gallavotti A, Long JA, Stanfield S, Yang X, Jackson D, Vollbrecht E, Schmidt RJ. 2010. The control of axillary meristem fate in the maize ramosa pathway. Development 137: 2849–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould SJ. 1977. Ontogeny and phylogeny, 1st ed Belknap Press of Harvard University Press, Cambridge, MA. [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihashi Y, Aguilar-Martínez JA, Farhi M, Chitwood DH, Kumar R, Millon LV, Peng J, Maloof JN, Sinha NR. 2014. Evolutionary developmental transcriptomics reveals a gene network module regulating interspecific diversity in plant leaf shape. Proc Natl Acad Sci 111: E2616–E2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DP. 1992. In-situ hybridisation in plants. In Molecular plant pathology: a practical approach (ed. Gurr SJ, et al. ), pp. 163–174. Oxford University Press, Oxford, UK. [Google Scholar]
- Kalinka AT, Varga KM, Gerrard DT, Preibisch S, Corcoran DL, Jarrells J, Ohler U, Bergman CM, Tomancak P. 2010. Gene expression divergence recapitulates the developmental hourglass model. Nature 468: 811–814. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Park M, Yeom SI, Kim YM, Lee JM, Lee HA, Seo E, Choi J, Cheong K, Kim KT, et al. 2014. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet 46: 270–278. [DOI] [PubMed] [Google Scholar]
- Kusters E, Della Pina S, Castel R, Souer E, Koes R. 2015. Changes in cis-regulatory elements of a key floral regulator are associated with divergence of inflorescence architectures. Development 142: 2822–2831. [DOI] [PubMed] [Google Scholar]
- Kwiatkowska D. 2008. Flowering and apical meristem growth dynamics. J Exp Bot 59: 187–201. [DOI] [PubMed] [Google Scholar]
- Kyozuka J, Tokunaga H, Yoshida A. 2014. Control of grass inflorescence form by the fine-tuning of meristem phase change. Curr Opin Plant Biol 17: 110–115. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. 2014. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H, Shumway M. 2011. The Sequence Read Archive. Nucleic Acids Res 39: D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M, Anavy L, Cole AG, Winter E, Mostov N, Khair S, Senderovich N, Kovalev E, Silver DH, Feder M, et al. 2016. The mid-developmental transition and the evolution of animal body plans. Nature 531: 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman ZB, Cohen O, Alvarez JP, Abu-Abied M, Pekker I, Paran I, Eshed Y, Zamir D. 2008. The making of a compound inflorescence in tomato and related nightshades. PLoS Biol 6: e288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlister CA, Park SJ, Jiang K, Marcel F, Bendahmane A, Izkovich Y, Eshed Y, Lippman ZB. 2012. Synchronization of the flowering transition by the tomato TERMINATING FLOWER gene. Nat Genet 44: 1393–1398. [DOI] [PubMed] [Google Scholar]
- Meng X, Muszynski MG, Danilevskaya ON. 2011. The FT-like ZCN8 gene functions as a floral activator and is involved in photoperiod sensitivity in maize. Plant Cell 23: 942–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger RJ, Klein OD, Martin GR, Krasnow MA. 2008. The branching programme of mouse lung development. Nature 453: 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RS, Purugganan MD. 2013. Evolution of crop species: genetics of domestication and diversification. Nat Rev Genet 14: 840–852. [DOI] [PubMed] [Google Scholar]
- Ori N, Cohen AR, Etzioni A, Brand A, Yanai O, Shleizer S, Menda N, Amsellem Z, Efroni I, Pekker I, et al. 2007. Regulation of LANCEOLATE by miR319 is required for compound-leaf development in tomato. Nat Genet 39: 787–791. [DOI] [PubMed] [Google Scholar]
- Park SJ, Kim SL, Lee S, Je BI, Piao HL, Park SH, Kim CM, Ryu CH, Park SH, Xuan Y, et al. 2008. Rice Indeterminate 1 (OsId1) is necessary for the expression of Ehd1 (Early heading date 1) regardless of photoperiod. Plant J 56: 1018–1029. [DOI] [PubMed] [Google Scholar]
- Park SJ, Jiang K, Schatz MC, Lippman ZB. 2012. Rate of meristem maturation determines inflorescence architecture in tomato. Proc Natl Acad Sci 109: 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Eshed Y, Lippman ZB. 2014. Meristem maturation and inflorescence architecture—lessons from the Solanaceae. Curr Opin Plant Biol 17: 70–77. [DOI] [PubMed] [Google Scholar]
- Pautler M, Tanaka W, Hirano HY, Jackson D. 2013. Grass meristems I: shoot apical meristem maintenance, axillary meristem determinacy and the floral transition. Plant Cell Physiol 54: 302–312. [DOI] [PubMed] [Google Scholar]
- Potato Genome Sequencing Consortium, Xu X, Pan S, Cheng S, Zhang B, Mu D, Ni P, Zhang G, Yang S, Li R, et al. 2011. Genome sequence and analysis of the tuber crop potato. Nature 475: 189–195. [DOI] [PubMed] [Google Scholar]
- Prusinkiewicz P, Erasmus Y, Lane B, Harder LD, Coen E. 2007. Evolution and development of inflorescence architectures. Science 316: 1452–1456. [DOI] [PubMed] [Google Scholar]
- Quint M, Drost HG, Gabel A, Ullrich KK, Bönn M, Grosse I. 2012. A transcriptomic hourglass in plant embryogenesis. Nature 490: 98–101. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Rebocho AB, Bliek M, Kusters E, Castel R, Procissi A, Roobeek I, Souer E, Koes R. 2008. Role of EVERGREEN in the development of the cymose petunia inflorescence. Dev Cell 15: 437–447. [DOI] [PubMed] [Google Scholar]
- Richardson MK. 1999. Vertebrate evolution: the developmental origins of adult variation. Bioessays 21: 604–613. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth ACJ, Gonnet GH, Dessimoz C. 2008. Algorithm of OMA for large-scale orthology inference. BMC Bioinformatics 9: 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalit A, Rozman A, Goldshmidt A, Alvarez JP, Bowman JL, Eshed Y, Lifschitz E. 2009. The flowering hormone florigen functions as a general systemic regulator of growth and termination. Proc Natl Acad Sci 106: 8392–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souer E, Rebocho AB, Bliek M, Kusters E, de Bruin RAM, Koes R. 2008. Patterning of inflorescences and flowers by the F-Box protein DOUBLE TOP and the LEAFY homolog ABERRANT LEAF AND FLOWER of petunia. Plant Cell 20: 2033–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer A, Zhao Q, Ross-Ibarra J, Doebley J. 2011. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat Genet 43: 1160–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka W, Pautler M, Jackson D, Hirano HY. 2013. Grass meristems II: inflorescence architecture, flower development and meristem fate. Plant Cell Physiol 54: 313–324. [DOI] [PubMed] [Google Scholar]
- Tomato Genome Consortium. 2012. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485: 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turck F, Fornara F, Coupland G. 2008. Regulation and identity of florigen: FLOWERING LOCUS T moves center stage. Annu Rev Plant Biol 59: 573–594. [DOI] [PubMed] [Google Scholar]
- Vollbrecht E, Springer PS, Goh L, Buckler ES, Martienssen R. 2005. Architecture of floral branch systems in maize and related grasses. Nature 436: 1119–1126. [DOI] [PubMed] [Google Scholar]
- Wang W, Li G, Zhao J, Chu H, Lin W, Zhang D, Wang Z, Liang W. 2014. DWARF TILLER1, a WUSCHEL-related homeobox transcription factor, is required for tiller growth in rice. PLoS Genet 10: e1004154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Liberatore KL, MacAlister CA, Huang Z, Chu YH, Jiang K, Brooks C, Ogawa-Ohnishi M, Xiong G, Pauly M, et al. 2015. A cascade of arabinosyltransferases controls shoot meristem size in tomato. Nat Genet 47: 784–792. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Sasao M, Yasuno N, Takagi K, Daimon Y, Chen R, Yamazaki R, Tokunaga H, Kitaguchi Y, Sato Y, et al. 2013. TAWAWA1, a regulator of rice inflorescence architecture, functions through the suppression of meristem phase transition. Proc Natl Acad Sci 110: 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga A. 2015. Next generation limb development and evolution: old questions, new perspectives. Development 142: 3810–3820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.