

Abstract
Objective
Sjögren's syndrome (SS) is a complex multisystem autoimmune disease that results in progressive destruction of the exocrine glands. The purpose of this study was to characterize epigenetic changes in affected gland tissue and describe the relationship of these changes to known inflammatory processes.
Methods
A genome‐wide DNA methylation study was performed on human labial salivary gland (LSG) biopsy samples obtained from 28 female members of the Sjögren's International Collaborative Clinical Alliance (SICCA) Registry. Gland tissue was methylotyped using the Illumina HumanMethylation450 BeadChip platform, followed by rigorous probe‐filtering and data‐normalization procedures.
Results
A genome‐wide case–control study of 26 of the 28 subjects revealed 7,820 differentially methylated positions (DMPs) associated with disease status, including 5,699 hypomethylated and 2,121 hypermethylated DMPs. Further analysis identified 57 genes that were enriched for DMPs in their respective promoters; many are involved in immune response, including 2 previously established SS genetic risk loci. Bioinformatics analysis highlighted an extended region of hypomethylation surrounding PSMB8 and TAP1, consistent with an increased frequency of antigen‐presenting cells in LSG tissue from the SS cases. Transcription factor motif enrichment analysis revealed the specific nature of the genome‐wide methylation differences, demonstrating colocalization of SS‐associated DMPs with stress‐ and immune response–related motifs.
Conclusion
Our findings underscore the utility of CpG methylotyping as an independent probe of active disease processes in SS, offering unique insights into the composition of disease‐relevant tissue. Methylation profiling implicated several genes and pathways previously thought to be involved in disease‐related processes, as well as a number of new candidates.
Sjögren's syndrome (SS) is a chronic multisystem autoimmune disease with potential to cause substantial morbidity 1, 2. Primary SS is characterized by progressive destruction of the exocrine glands, with subsequent mucosal and conjunctival dryness 1. Although the precise cause of SS remains unknown, it is understood to be a complex and heterogeneous disease, with important contributions from both genetic and environmental factors 3, 4. It is likely that widespread clinical heterogeneity in SS reflects differences in underlying disease mechanisms, and current approaches to research and clinical care in SS are compromised by such phenotypic heterogeneity. Elucidation of how genetic and nongenetic factors contribute to disease heterogeneity should significantly affect how we diagnose, manage, and treat this complex disorder.
A growing body of evidence has implicated epigenetic factors, in particular, altered patterns of DNA methylation, in models of autoimmune disease 5, 6. Furthermore, recent studies characterizing the DNA methylation profiles of naive CD4+ T cells, B cells, and salivary gland epithelial cells provide evidence of aberrant DNA methylation profiles in SS patients 7, 8, 9, 10. While it is unknown which differences, if any, reflect causal determinants of risk, it is likely that many of these patterns reflect subtle differences in subpopulation composition 11 downstream of true causal risk factors or disease processes. One of the most important compartments for analyzing immunoregulatory heterogeneity in SS is whole labial salivary glands (LSGs), a prominent target of the disease‐specific processes.
We report our findings of a genome‐wide study of DNA methylation in LSG tissue biopsied from 13 SS cases, 13 controls, and 2 subjects with intermediate phenotypes; all are women of genetically confirmed European descent who are participants in the Sjögren's International Collaborative Clinical Alliance (SICCA) Registry. We identified thousands of DNA methylation differences across the genome associated with case status, implicating immune‐related and cell lineage–specific pathways in disease pathogenesis. In addition to highlighting a large number of genes involved in general immune system processes (including known genetic risk loci associated with SS), we also observed enrichment for DNA methylation differences around specific transcription factor motifs. In total, our results demonstrate both widespread and targeted DNA methylation differences marking LSG‐specific immune processes in SS.
SUBJECTS AND METHODS
Study subjects and clinical evaluation
Our study used samples of LSG tissue biopsied from 28 female subjects of European descent who were participants in the SICCA Registry (Table 1). As part of the enrollment into the SICCA Registry, subjects were evaluated for clinical criteria of SS at 1 or 2 time points; LSG tissue was biopsied during at least 1 of these visits, frozen, and stored using standard procedures.
Table 1.
Phenotype and covariates across study groupsa
| Cases (n = 13) | Controls (n = 13) | Noncases (n = 15) | P (cases vs. controls) | |
|---|---|---|---|---|
| Focus score | 3.4 ± 2 | 0.07 ± 0.13 | 0.09 ± 0.17 | 9.1 × 10−6 |
| Ocular staining score | 6.1 ± 2.8 | 1.2 ± 0.7 | 1.7 ± 1.9 | 1.5 × 10−5 |
| SSA seropositive (indicator) | 0.92 | 0 | 0 | – |
| SSB seropositive (indicator) | 0.54 | 0 | 0 | – |
| Age | 55 ± 13 | 53 ± 7.9 | 56 ± 10 | 0.84 |
| Ancestry PC1 | 0.005 ± 0.003 | −0.014 ± 0.026 | −0.012 ± 0.024 | 0.035 |
Noncases are a combined set of subjects with intermediate phenotype (n = 2) and controls (n = 13), who were included to emphasize the contrast in phenotype between the cases and the other subjects. Values are the mean ± SD of covariates, except for SSA/SSB seropositivity data, which are shown as indicator variables (1 = positive), and only the mean values (proportions) are reported for these phenotypes. P values were determined by Wilcoxon's rank sum test. PC1 = first principal component.
Case–control status was evaluated according to the American College of Rheumatology (ACR) criteria for SS 12. Our study targeted cases with severe SS, requiring that cases meet all 3 of the following criteria: seropositivity (SSA and/or SSB autoantibodies), an ocular staining score (OSS) of ≥3 in at least 1 eye, and a focus score ≥1 (no subjects had a focus score of 1). Controls did not meet any of these criteria. Samples were designated as case or control based on clinical evaluation at the time of biopsy. Two of the study subjects met only the high OSS criterion at time of sample collection (Table 1) and are referred to herein as “intermediate phenotype” subjects. Neither cases nor controls were disqualified based on an additional systemic autoimmune disease diagnose (e.g., rheumatoid arthritis, Hashimoto's disease). Self‐reported medication data for the study participants are shown in Supplementary Table 1 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39792/abstract). Univariate testing to compare variable distributions in cases and controls was conducted using Fisher's exact test implemented in R. The Institutional Review Boards at the University of California, San Francisco and the University of California, Berkeley approved our study protocol.
Genotyping and principal components (PCs) analysis
Prior to this study, the 28 SICCA subjects were genotyped using the HumanOmni2.5‐Quad BeadChip array (Illumina), as part of a genome‐wide association study (GWAS) 13. In addition to sample verification and other quality control assessments, these data were used to evaluate the genetic ancestry of the study subjects. EigenStrat analysis 14 was applied to genotypes from the full GWAS dataset in order to derive PCs reflecting global genetic variation. The 28 study subjects fell within 2 SD of the mean of the first 2 PCs in self‐identified Europeans; GWAS subjects within this range were deemed “European candidates.”
In order to examine the effects of intra‐European ancestry on LSG DNA methylation, we applied EigenStrat analysis to genotypes from all European candidates. The first 4 PCs were retained for downstream analysis. We saw no significant evidence of association between case status and age in our study population, and only weak association with the first ancestry PC (Table 1). Given the small study size, we chose not to adjust for these factors when comparing DNA methylation patterns between cases and controls, but rather to screen disease‐associated DNA methylation differences for any effects of ancestry PC1 and age.
DNA methylotyping
DNA methylation data were obtained for each sample using the Illumina 450K Infinium Methylation BeadChip (450K chip) platform. The 450K chip allows for high‐throughput interrogation of more than 450,000 highly informative CpG sites spanning ∼22,000 genes across the genome. The primary measure of DNA methylation at each CpG site is β, which is the ratio of the intensities of fluorescent signals from methylated and unmethylated alleles. Sample identity was verified by comparing genome‐wide genotypes to the genotypes derived from 35 single nucleotide polymorphism (SNP) probes on the 450K chip. Three of the DNA samples were subdivided into 2 intrabatch technical replicates, contributing to a total of 31 samples for subsequent DNA methylation analysis.
Data normalization and filtering
Our data preprocessing pipeline was implemented entirely in R 15 and used the methylumi data representation in Bioconductor 16, 17. We applied the normal‐exponential convolution method on out‐of‐band probe intensities (“noob”) to correct each sample for technical variation in background fluorescence 18. The 2 color channels on the 450K chip were normalized using the all‐sample mean normalization method, which is a natural extension of the Illumina GenomeStudio protocol 19.
The 450K chip includes 3,091 CpH (non‐CpG) probes and 65 SNP probes, all of which were removed prior to analysis. We also removed 16,177 cross‐reactive CpG probes 20. In order to avoid direct effects of genotype variation, we removed 1,213 CpG probes targeting variable SNPs genotyped in our study sample. We also considered the set of SNPs from the 1000 Genomes project that lie within the probe‐hybridizing sequence as tabulated by Chen et al 20. Using the University of California, Santa Cruz (UCSC) Genome Browser SNP138 track 21, we identified and removed 62,220 CpG probes neighboring SNPs. An additional 3,392 CpG probes were removed from the analysis due to high‐detection P values (P > 0.05) in 1 or more samples, as computed by Illumina's GenomeStudio software. A total of 404,353 CpG probes were therefore used for the primary analysis. After probe filtering, we corrected each sample for type I/II probe design bias using the beta‐mixture quantile normalization method 22.
Principal components analysis of DNA methylation data
We computed PCs of the normalized β value matrix, centering and scaling per CpG. After averaging the PC values of replicates, the top 5 PCs were tested for association with several continuous covariates (focus score, mean OSS, age, and genetic ancestry PCs) and categorical covariates (SSA/SSB seropositivity and assay plate), applying Z tests (to Fisher‐transformed Spearman's rho) and Kruskal Wallis tests, respectively.
Nonlinear adjustment for technical variation
Despite our efforts to normalize the data using standard methods, the first PC of the β‐matrix clearly separated samples according to the assay plate (Supplementary Figures 1A and B, available at http://onlinelibrary.wiley.com/doi/10.1002/art.39792/abstract). We adjusted the data against proxies of known batch effects to remove technical bias (see Supplementary materials, available at http://onlinelibrary.wiley.com/doi/10.1002/art.39792/abstract).
Single CpG site tests for differentially methylated positions and global DNA methylation analysis
Wilcoxon's rank sum test was used to test each CpG β value for association with case–control status, followed by the Benjamini‐Yekutieli adjustment for multiple comparisons. The Benjamini‐Yekutieli adjustment is a more conservative version of the Benjamini‐Hochberg false discovery rate (FDR) procedure, which may be preferable when test statistics are correlated 23. Given the complex and often strong correlations between CpG methylation levels, we chose to use this more conservative FDR procedure. No thresholds were placed on mean or median β‐differences between cases and controls—specifically, we set no constraints on the magnitude of significant differences in methylation. We refer to disease‐associated CpGs (q < 0.01) as differentially methylated positions (DMPs). The β values for replicate samples were averaged prior to single CpG–site association tests.
Global DNA methylation was evaluated using 2 methods. First, Wilcoxon's rank sum tests were applied to evaluate differences in mean genome DNA methylation status between cases and controls; for each subject, mean genome DNA methylation is defined as the mean β value across all probes passing our stringent quality filtering. Second, we applied Fisher's exact test to determine whether the fraction of hypermethylated CpGs (as determined by the sign of the mean difference between cases and controls) varied significantly between DMPs and non‐DMPs.
Identification of differentially methylated promoters
CpGs were mapped to promoters (or, more generally, upstream regulatory regions) using the BEDTools suite 24. For each RefSeq entry in the UCSC RefGene track 21, we defined a promoter region as the genomic interval spanning 2,500 bp upstream and 500 bp downstream of the annotated transcription start site, similar to the definition described by Whitaker et al 25. RefSeq identifiers were mapped to gene symbols using the org.Hs.eg.db package in Bioconductor 26; all unmapped RefSeq entries were excluded from the analysis.
We tested for differentially methylated promoters using hypergeometric tests for DMP enrichment, as described by Nakano et al 27. Enrichment P values were adjusted for multiple testing using the Benjamini‐Hochberg correction, with a q value threshold of 0.05. To avoid promoter‐specific bias, we excluded all CpGs that did not fall within promoters; enrichment tests were performed solely on promoter CpGs. Furthermore, to protect against biases associated with double‐counting CpGs sitting in the intersection of multiple loci, we excluded any CpGs mapping to 2 or more promoters.
Gene set enrichment analysis
After identifying the set of genes with significantly differentially methylated promoters, we considered whether this gene set is enriched for categories of biologic function or genomic position. Hypergeometric gene set enrichment analysis was used to test 2,666 gene sets from the Molecular Signature Database 28 for enrichment of differentially methylated promoters, including “hallmark” gene sets, positional gene sets, motif gene sets, and gene ontology gene sets, with a Benjamini‐Hochberg q value cutoff of 0.05.
We further tested 2 candidate gene sets for enrichment of genes possessing differentially methylated promoters: 1) genes encoding the 50 transcripts showing the greatest fold‐change in LSG expression between SS cases and controls in the microarray study by Hjelmervik et al 29, and 2) genes highlighted in recent SS GWAS: GTF2I, TNFAIP3, IRF5, STAT4, IL12A, BLK, CXCR5, TNIP1, HLA–DRA, HLA–DQB1, HLA–DRB1, HLA–DPB1, and COL11A2 30, 31.
CpG set enrichment analysis
Although gene set enrichment analysis is a valuable tool for understanding the distribution of differentially methylated promoters, the DMPs on which this analysis is based are called at single‐basepair resolution; therefore, some information is lost when the analysis is applied to broad genomic regions such as promoters. This discrepancy can even lead to bias due to the variation in promoter coverage across the 450K chip platform; some promoters contain far more probed CpGs than others, giving us greater power to resolve extended differences in those regions. Some of this bias of differential power can be avoided by considering CpG sets rather than gene sets. For each of the differentially methylated gene sets identified in the gene set enrichment analysis, as well as the 2 candidate gene sets, a CpG set was also defined, containing all of the CpGs mapping to promoters of the corresponding gene set. DMP enrichment was performed using hypergeometric tests, as before, although CpGs mapping to multiple sets were included in this analysis. The CpG set enrichment analysis was adjusted for multiple testing, accounting for the 2,668 gene set enrichment tests used to select CpG sets. CpG sets with a Bonferroni‐adjusted P value less than 0.01 were considered enriched for DMPs.
Transcription factor motif enrichment analysis
Given the intimate relationship between transcription‐factor binding and chromatin state, we considered whether disease‐associated DNA methylation changes colocalize with specific transcription factor binding motifs (TFBMs), using the Analysis of Motif Enrichment (AME) tool 32 to identify enriched TFBMs in the sequence surrounding disease‐associated DMPs. For each DMP, we extracted a window of the UCSC hg19 reference genome within 150 bp of the annotated CpG position. Overlapping intervals were merged, producing a set of DMP‐associated sequences. A “control” set of CpG‐neighboring sequences was generated using the same procedure applied to all non‐DMPs passing our quality filter.
Using the AME, we tested DMP‐associated sequences for enrichment of 205 TFBMs from the JASPAR CORE 2014 vertebrates set 33, adjusting for sequence length and using the “control” set as a sequence control. AME was performed using 3 motif affinity options that use different scoring methods to evaluate motif matches: total number of matches above a threshold (“totalhits”), sum of motif scores (“sum”), and average motif score (“avg”). Default thresholds were used for all choices of motif affinity function, and observed enrichment was evaluated for statistical significance using Fisher's exact tests. Motifs were considered enriched if the corresponding Bonferroni‐adjusted P value fell below 0.01 (correcting for 615 tests) for any of the 3 affinity functions.
RESULTS
Different global methylation patterns in LSGs from SS cases and controls
After adjusting for technical effects, none of the top 5 DNA methylation PCs (60% of variance) showed significant association with the sample batch (Supplementary Figures 1C and D). The first PC was strongly associated with the focus score (q = 2.1 × 10−5) and the mean OSS (q = 5.3 × 10−4) (Supplementary Table 2, available at http://onlinelibrary.wiley.com/doi/10.1002/art.39792/abstract), suggesting that this axis captures disease‐associated processes in the gland. Indeed, results of Wilcoxon's rank sum testing showed that the first PC of DNA methylation in LSG tissue was associated with case status (P = 1.3 × 10−5). Plots of the first 2 PCs place the 2 individuals of intermediate phenotype between the cases and the controls, consistent with their phenotype (Figure 1). Tests of association between case status and self‐reported medication use (Supplementary Table 1) showed that a confounding effect of medication was unlikely in the current study.
Figure 1.
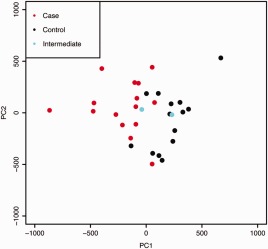
Principal components analysis of genome‐wide DNA methylation in all labial salivary gland tissue samples, including replicates. The first principal component (PC1) separates Sjögren's syndrome cases from controls, with samples from the 2 subjects with intermediate phenotype (ocular staining score ≥3 in at least 1 eye) between those of the cases and the controls. PC2 represents a spread of sample DNA methylation states orthogonal to the primary case–control contrast. This axis may represent biologic intrapatient heterogeneity. All study subjects were women of genetically confirmed European descent who were participants in the Sjögren's International Collaborative Clinical Alliance Registry.
Global hypomethylation of LSGs in SS cases
Thabet et al 8 previously reported global hypomethylation in cultured LSG epithelial cells from SS patients. We considered whether these differences could be detected in more heterogeneous LSG tissue samples. However, no significant differences in mean genome DNA methylation were observed across all CpGs (1.01‐fold hypermethylation in SS cases; P = 0.26).
Our epigenome‐wide association study identified 7,820 DMPs associated with SS case status. The median absolute β‐difference between cases and controls was 0.10 for DMPs, demonstrating that most SS‐associated DMPs identified in the current study showed modest‐to‐large differences in DNA methylation. Of the 7,820 DMPs tested, 5,699 (73%) were hypomethylated in cases. The set of DMPs contained far more hypomethylated CpGs than was expected by the distribution of non‐DMPs (P < 2.2 × 10−16 by Fisher's exact test) (Figure 2), suggesting that CpGs are generally more hypomethylated in whole LSG tissue from SS cases. Of the 7,820 DMPs tested, 338 (4%) were associated at P = 1.92 × 10−7 (q = 0.003). These top sites distinguished cases from controls in our study sample.
Figure 2.
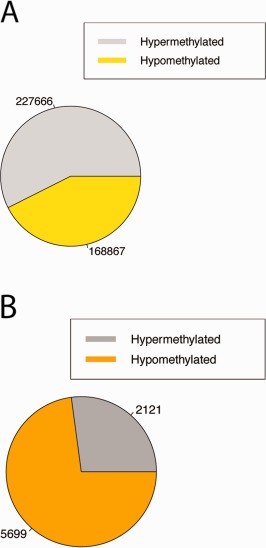
Global DNA methylation differences between labial salivary glands from the Sjögren's syndrome cases and the controls. A, Proportions of hyper‐ and hypomethylated CpGs with q values >0.01 by Wilcoxon's rank sum test. These represent a control set of CpGs, or non–differentially methylated positions (DMPs). Direction of methylation is determined by the sign of the difference in the mean β values between cases and controls. B, Proportions of hyper‐ and hypomethylated DMPs with q values <0.01 by Wilcoxon's rank sum test. DMPs are significantly enriched for hypomethylated sites as compared to non‐DMPs (P < 2.2 × 10−16 by Fisher's exact test).
Linear regression was used to model the associations between the DNA methylation level (logit transformed) for each of the 7,820 DMPs and the first PC of genetic ancestry or age at biopsy. No DMP was significantly associated with either factor at a Benjamini‐Hochberg FDR of 0.05. These 2 factors may affect DNA methylation levels of SS‐associated DMPs, but their average effects are too small to resolve in our study.
Differentially methylated promoters of various protein‐coding genes, microRNAs, and noncoding RNAs
Differentially methylated promoter analysis identified 57 genes (Table 2 and Supplementary Table 3, available at http://onlinelibrary.wiley.com/doi/10.1002/art.39792/abstract). This list includes a large number of transcription factors (e.g., RUNX3 and SPI1) and known cell‐differentiation markers (e.g., TNFRSF13B, CCR6, BST2, BTLA, and CXCR5). In addition to coding genes, the list contains a number of RNA genes, including several antisense RNA genes (e.g., PSMB8‐AS1) and microRNAs (e.g., MIR339). The results could reflect differential regulation of neighboring coding genes or primary transcripts. Interestingly, 3 of the differentially methylated promoters are located within 1 interval of the major histocompatibility complex (MHC) genomic region: PSMB8, PSMB8‐AS1, and TAP1.
Table 2.
Top 25 DMPs in labial salivary glands from Sjögren's syndrome patientsa
| Upstream region | DMP range | Total DMPs | Fold enrichment | q value for enrichment | % DMPs hypo. |
|---|---|---|---|---|---|
| PSMB8‐AS1 | Chr. 6: 32810001–32811253 | 11 | 38.3 | 1.4 × 10−11 | 100 |
| CTSZ | Chr. 20: 57582706–57583474 | 10 | 26.5 | 1.9 × 10−8 | 100 |
| PTPRCAP | Chr. 11: 67205096–67206434 | 8 | 35.3 | 1.2 × 10−7 | 100 |
| LTA | Chr. 6: 31539539–31540440 | 7 | 38.6 | 7.6 × 10−7 | 100 |
| MIR339 | Chr. 7: 1062652–1064100 | 7 | 30.9 | 4.8 × 10−6 | 100 |
| TNFRSF13B | Chr. 17: 16875129–16875596 | 5 | 55.1 | 1.8 × 10−5 | 100 |
| PSMB8 | Chr. 6: 32813084–32815091 | 7 | 22 | 5.7 × 10−5 | 100 |
| MTNR1A | Chr. 4: 187476543–187476608 | 5 | 33.1 | 0.00053 | 0 |
| MPEG1 | Chr. 11: 58980157–58981095 | 4 | 52.9 | 0.00066 | 100 |
| CCR6 | Chr. 6: 167535909–167536184 | 5 | 27.6 | 0.0013 | 100 |
| TAP1 | Chr. 6: 32822565–32823941 | 4 | 44.1 | 0.0016 | 100 |
| SSH3 | Chr. 11: 67070233–67070967 | 5 | 23.6 | 0.0027 | 0 |
| BST2 | Chr. 19: 17516282–17518018 | 4 | 37.8 | 0.0029 | 100 |
| PPFIA4 | Chr. 1: 203019107–203020617 | 4 | 37.8 | 0.0029 | 100 |
| AIM2 | Chr. 1: 159046937–159047163 | 3 | 66.1 | 0.0036 | 100 |
| BTLA | Chr. 3: 112217973–112218761 | 3 | 66.1 | 0.0036 | 100 |
| CXCR5 | Chr. 11: 118754280–118763863 | 5 | 20.7 | 0.0036 | 100 |
| FCRL3 | Chr. 1: 157670328–157670869 | 4 | 33.1 | 0.0036 | 100 |
| KCNQ1DN | Chr. 11: 2890394–2890725 | 7 | 10.8 | 0.0036 | 0 |
| LINC00926 | Chr. 15: 57592040–57592438 | 3 | 66.1 | 0.0036 | 100 |
| MIR3186 | Chr. 17: 79419796–79420279 | 4 | 33.1 | 0.0036 | 100 |
| MIR4269 | Chr. 2: 240225062–240226201 | 3 | 66.1 | 0.0036 | 100 |
| WDFY4 | Chr. 10: 49892741–49893463 | 5 | 20.7 | 0.0036 | 100 |
| RUNX3 | Chr. 1: 25291472–25292225 | 7 | 10.1 | 0.0055 | 100 |
| FERMT3 | Chr. 11: 63973846–63974153 | 4 | 26.5 | 0.0093 | 100 |
Promoter enrichment results are shown for the most‐significant regions. The genomic interval for each differentially methylated position (DMP) range is given, as well as the total number of DMPs and the fold enrichment for DMPs in the region. Hypergeometric enrichment q values and hypomethylated (hypo.) fractions are also reported. Chr. = chromosome.
DMP‐enriched promoters of candidate gene sets
The promoter enrichment results emphasized both the inflammation and tissue specificity of the observed DNA methylation differences. The set of differentially methylated promoters was found to be enriched for several gene ontology terms involving immune response and signal transduction. We also observed evidence of enrichment of genes known to contain transcription factor binding motifs for PU.1 and Ets‐2 (mouse orthologs of targets) in their promoters (Table 3), likely representing differences in cell composition and activity resulting from SS pathogenesis. Only a small number of these genes have been highlighted by SS GWAS (CXCR5 and BLK) 30 or are known to be differentially expressed at the transcription level in SS‐affected LSG tissue (ARHGAP25) 29; however, the promoter CpG sets corresponding to both of these candidate gene sets were significantly enriched for DMPs (Bonferroni‐adjusted P = 9.2 × 10−7 and 6.0 × 10−4, respectively).
Table 3.
Differentially methylated CpG sets in labial salivary glands from Sjögren's syndrome patientsa
| MSigDB gene set | Differentially methylated promoters | Adjusted P |
|---|---|---|
| Immune response (GO:0006955) | CCR6, BST2, AIM2, LCP2, CD79B, MADCAM1 | 2.9 × 10−8 |
| Intrinsic to plasma membrane (GO:0031226) | TNFRSF13B, MTNR1A, CCR6, BST2, CXCR5, NCKAP1L, CD160, CD19, CD79B, IL12RB1 | 1.7 × 10−7 |
| Genes with promoters containing Ets2 motif RYTTCCTG (M14654) | PTPRCAP, TNFRSF13B, KCNQ1DN, RUNX3, FERMT3, LCP1, SPI1, SLAMF1, CD19, ERG, PIK3CG | 3.6 × 10−7 |
| Immune system process (GO:0002376) | CCR6, BST2, AIM2, SPI1, LCP2, CD79B, MADCAM1 | 1.0 × 10−6 |
| Cell surface receptor–linked signal transduction (GO:0007166) | TNFRSF13B, MTNR1A, CXCR5, CD160, GNB3, LCP2, CD19, IL12RB1, PIK3CG | 2.9 × 10−6 |
| Genes with promoters containing PU.1 motif WGAGGAAG (M14376) | PTPRCAP, LTA, NCKAP1L, LCP2, NR1H3, PIK3CG | 4.7 × 10−5 |
| Signal transduction (GO:0007165) | LTA, TNFRSF13B, MTNR1A, CCR6, BST2, CXCR5, CD160, GNB3, BLK, LCP2, CD19, ERG, IL12RB1, KALRN, MADCAM1, PIK3CG | 2.8 × 10−4 |
These gene sets from the Molecular Signatures Database (MSigDB) were selected as candidates for CpG enrichment because they contained a significantly high fraction of differentially methylated promoters, as shown here. Bonferroni‐adjusted P values are reported for hypergeometric CpG set enrichment tests.
To further probe the meaning of the observed enrichment in differentially expressed genes, we assigned hypomethylation significance scores (score = sign[Δβ] × logP) to each CpG falling within the promoters of 42 genes reported as being highly differentially expressed in the microarray study by Hjelmervik et al 29. Regression analysis revealed that the average hypomethylation score across a promoter is positively associated with the extent of messenger RNA up‐regulation reported in SS‐affected tissue (Supplementary Figure 2, available at http://onlinelibrary.wiley.com/doi/10.1002/art.39792/abstract). The predictive power of differential methylation suggests that many DNA methylation differences in LSGs from SS cases are associated with the same upstream biologic factors driving differential transcription in SS.
Characteristic binding motifs neighboring SS‐associated DMPs
The AME tool identified 3 enriched motifs in the immediate neighborhood of DMPs (Table 4 and Supplementary Figures 3B–D, available at http://onlinelibrary.wiley.com/doi/10.1002/art.39792/abstract). The most significant motif was annotated for TCF11/MafG 34, an antioxidant response element binding complex that is reported to play a role in proteasome regulation and stability 35. A second enriched motif was annotated for the STAT1/STAT2 heterodimer, targeting interferon‐stimulated response elements 36. The final motif is the conserved binding motif of PU‐box–binding transcription factor PU.1 37.
Table 4.
Differentially methylated position–associated motifs identified by analysis of motif enrichmenta
| JASPAR ID | Annotated transcription factor complex | Targets | Adjusted P |
|---|---|---|---|
| MA0089.1 | TCF11/MAFG heterodimer | Antioxidant response elements | 5.2 × 10−5 |
| MA0517.1 | STAT2/STAT1 heterodimer | IFN‐stimulated response elements | 7.5 × 10−4 |
| MA0080.3 | PU.1 | PU box | 5.9 × 10−3 |
P values were determined by Fisher's exact test, with Bonferroni adjustment for multiple testing. IFN = interferon.
DISCUSSION
Through whole‐genome DNA methylation profiling of a clinically well‐characterized sample of European women, we identified a strong signature of disease‐associated immune processes in LSG tissue. We observed evidence of hypomethylation at the whole‐tissue level in SS cases as compared to controls. Further, our findings showed that epigenetic states of inflammatory genes and immune‐cell markers are major contributors to DNA methylation differences that distinguish SS cases. While results from this observational study cannot establish a causal role for the observed DNA methylation patterns in the risk of SS, our DMP‐based gene set, CpG set, and transcription factor motif enrichment analyses all demonstrated that DNA methylation profiling in SS cases and controls provides unique insights into tissue‐specific differences involved in disease.
The most significant DMP enrichment observed in this study was in the promoter of PSMB8‐AS1, a long noncoding RNA neighboring the PSMB8 locus (aka PSMB5i or LMP7) in the MHC region. This antisense RNA is in a head‐to‐head configuration with PSMB8 (Supplementary Figure 4, available at http://onlinelibrary.wiley.com/doi/10.1002/art.39792/abstract). PSMB8, the promoter of which we have demonstrated to be hypomethylated in SS cases, encodes a subunit of the immunoproteasome that has been reported to be up‐regulated in the salivary glands of patients with SS 38. The greater proteasome regulatory network was further implicated by the enrichment of TCF11/MAFG motifs surrounding SS‐associated DMPs. While these differences in DNA methylation may be functionally related, there is no clear evidence of immunoproteasome regulation by the TCF11/MAFG complex 35.
We have also presented evidence here for promoter hypomethylation of TAP1, neighboring both PSMB8 and PSMB9. Rare variants of TAP1 and extended HLA haplotypes are thought to confer disease risk in some SS patients 39. Given their specific roles in antigen presentation, most DMPs observed across these 3 neighboring loci are likely to be directly associated with an increased proportion of immune cells in the tissue. This “tissue‐heterogeneity interpretation” is further supported by the abundance of differentially methylated cell differentiation markers noted in our DMP enrichment analyses; this enrichment could indicate that many‐to‐most of the extended DNA methylation differences observed in this study are consequences of varying cell proportions in the gland tissue. As a deeper understanding of cell‐type–specific DNA methylation motifs in immune‐ and tissue‐specific cells becomes available, the patterns observed in target tissue may serve as clues to which cell types are driving recurring inflammation in SS patients.
The transcription factor PU.1 was highlighted multiple times in the current study. Not only was extended hypomethylation observed in the promoter region of this gene, but there also appeared to be a spatial association between differential methylation patterns and PU.1 binding motifs, both at the promoter level (CpG set enrichment analysis) and at the nucleosome level (TFBM enrichment analysis). PU.1 is a known factor involved in B cell and macrophage differentiation, binding to the enhancers of many lineage‐specific genes 40, and it may directly recruit DNA methylation machinery to repress target genes 41. As such, differential proportions of immune cell types (i.e., B lymphoid versus myeloid lineage) may drive PU.1 target enrichments in inflamed tissue. In particular, the abundance of hypomethylated B cell and lymphoid markers, including CD19, CD79B, PTPRCAP, and TNFRSF13B, further supports this interpretation.
Thabet et al 8 report that disease‐associated gland up‐regulation of ICAM1/CD54 3, a gene critically involved in the processes of intercellular adhesion and trans‐endothelial migration, was associated with global hypomethylation of salivary gland epithelial cell genomes. The investigators hypothesized that global hypomethylation could be a regulatory mechanism upstream of increased expression 8. We found no evidence of differential methylation in or around the ICAM1 promoter, suggesting that other mechanisms are directly responsible. However, due to the heterogeneous nature of gland tissue used in the current study, both direct and indirect effects may be masked by cell proportion differences in tissue.
Promoter enrichment analysis highlighted a microRNA (miR‐339) that has been demonstrated to be a potential posttranscriptional regulator of ICAM1 42. Although this mechanism is intriguing, there exists little evidence to support it within the context of SS, beyond down‐regulation of miR‐339 reported in a microarray study of SS‐affected glands 43. Any mechanistic interpretation is further complicated by the hypomethylation observed in the upstream regulatory region, which would support up‐regulation of this gene product based on a simple model of DNA methylation–associated epigenetic regulation. Despite the unknown biologic role of the striking hypomethylation we identified at this microRNA locus, the proposed regulatory potential of miR‐339 makes it an attractive candidate for functional studies.
Recently, Imgenberg‐Kreuz et al 10 reported results from their study of DNA methylation in minor salivary gland biopsies from 15 primary SS cases and 13 controls in which they used the 450K platform. In addition to a parametric analysis approach, the authors used a conservative Bonferroni‐adjusted P value reporting criterion for DMPs. While a top hit in OAS2 (cg20870559) was successfully replicated in the current study, only 2 of the remaining 44 DMP hits reported by that study were replicated here: cg12560128 and cg16596716. Both study populations were small, and differences in phenotype or age may have contributed to the lack of replication of other findings. Enrichment analyses and more comprehensive analyses of extended patterns of DNA methylation may be better approaches to characterizing profiles associated with case status than single CpG–site testing.
Previous studies have defined a gene as being differentially methylated if it contains a number of DMPs exceeding a given threshold 25. One problem with this approach is that it is biased toward reporting genes with higher CpG coverage. Assuming that false‐positive results would be randomly distributed across the 450K chip, a gene with better coverage will have more false‐positive results. Coverage is also problematically associated with biologic function 44, but enrichment tests, such as the hypergeometric test, will take this coverage into account. Given the difficulties associated with interpreting single CpG–site results, we chose to emphasize enrichment results, at both the promoter and pathway levels.
One of the strengths of our study is its restriction to European women, which minimized potential confounding by genetic ancestry or sex. Both have been shown to influence DNA methylation profiles 45, 46, and thus, our current results may not be generalizable to other studies of non‐European or male populations. Importantly, sex differences in many immunologic parameters have been observed 47. As a result, epigenetic studies comparing male cases and controls might yield a different set of SS‐associated LSG DMPs. It is also possible that SS case subgroups (e.g., cases with specific extraglandular manifestations) exhibit different DNA methylation profiles. While the current study was not large enough to test these hypotheses, larger studies will be able to probe phenotype‐specific methylation patterns.
Studies of circulating blood cells are well poised to reveal novel mechanisms in disease etiology due to ease of sample collection and access to naive cell populations. However, disease‐associated changes observed in these cells likely reflect systemic aspects of the disease, rather than tissue‐specific disease states driven by local inflammation. Labial salivary gland biopsy is a minimally invasive procedure that provides investigators access to tissue targets of SS and may help to illuminate processes specific to a disease in progress. Furthermore, as a target tissue, these samples may prove more useful in characterizing disease phenotypes in patients with early evidence of SS symptoms. Insights from this study and larger studies may soon yield new epigenetic biomarkers for this complex and heterogeneous disease and may help to inform the development of novel treatment strategies in the future.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Criswell had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Cole, Baker, Barcellos, Criswell.
Acquisition of data
H. Quach, D. Quach, Criswell.
Analysis and interpretation of data
Cole, Taylor, Barcellos, Criswell.
Supporting information
Supplementary Table 1. Self‐reported Medication by Case‐Control Status
Supplementary Table 2. Correlations between First and Second Principal Components of DNAm, Phenotype and Selected Covariates
Supplementary Table 3. Remaining (Lower Significance) Differentially Methylated Promoters in LSGs of Sjögren's Syndrome Patients
Supplementary Figure 1. Principal component analysis on genome‐wide gland DNAm, before and after non‐linear adjustment. A. Before adjustment, the first principal component separates the three batches. B. Spearman Z‐test p‐values showing significance of correlation between PC1‐5 and categorical phenotypes and covariates: disease status, treatment plate, sentrix ID (methylation chip ID), SSA and SSB seropositivity. Red dashed line indicates Bonferroni significance threshold for 25 tests. C. Following adjustment, batches are mixed. D. Significance of correlation with batch is low across first five PCs, while preserving associations with phenotype.
Supplementary Figure 2. Correlation between DNA methylation and expression disease‐associations from two studies. The x‐axis shows extent of mRNA down‐regulation from Hjelmervik et al. and the y‐axis shows the significance of DNA hyper‐methylation – from the present study ‐ for CpGs in the promoter of the corresponding gene. DMPs from the present study are highlighted in cyan.
Supplementary Figure 3. JASPAR motifs enriched in the neighborhood of SS‐associated DMPs in labial salivary gland tissue. A. Schematic of transcription factor binding motif enrichment analysis. B. MA0089.1, annotated for heterodimer of chicken orthologs of NFE2L1 and MAFG. C. MA0517.1, annotated for the heterodimer of STAT2 and STAT1. D. MA0080.3, annotated for the mouse ortholog of SPI1.
Supplementary Figure 4. Extended differential methylation in PSMB8‐AS1 promoter. A. Highlighted region shows region designated as PSB8‐AS1 promoter, sitting within the gene body of PSMB8. All SS‐associated DMPs (promoter and non‐promoter) are annotated in the top track, UCSC Genome Browser RefGene annotations in the middle, and all 450K CpG sites at the bottom. B. Evidence of an SS‐associated differentially hypo‐methylated region within the promoter of PSMB8‐AS1.
Supported by the NIH (National Institute of Dental and Craniofacial Research grants 1F31‐DE‐025176‐01, HHSN‐268201300057C, N01‐DE‐32636, and R03‐DE‐0245316) and the Sjögren's Syndrome Foundation.
REFERENCES
- 1. Ramos‐Casals M, Brito‐Zerón P, Sisó‐Almirall A, Bosch X. Primary Sjögren syndrome. BMJ 2012:344:e3821. [DOI] [PubMed] [Google Scholar]
- 2. Theander E, Manthorpe R, Jacobsson LT. Mortality and causes of death in primary Sjögren's syndrome: a prospective cohort study. Arthritis Rheum 2004;50:1262–9. [DOI] [PubMed] [Google Scholar]
- 3. Tzioufas AG, Kapsogeorgou EK, Moutsopoulos HM. Pathogenesis of Sjögren's syndrome: what we know and what we should learn. J Autoimmun 2012;39:4–8. [DOI] [PubMed] [Google Scholar]
- 4. Ice JA, Li H, Adrianto I, Lin PC, Kelly JA, Montgomery CG, et al. Genetics of Sjögren's syndrome in the genome‐wide association era. J Autoimmun 2012;39:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Selmi C, Leung PS, Sherr DH, Diaz M, Nyland JF, Monestier M, et al. Mechanisms of environmental influence on human autoimmunity: a National Institute of Environmental Health Sciences expert panel workshop. J Autoimmun 2012;39:272–84. [DOI] [PubMed] [Google Scholar]
- 6. Hewagama A, Richardson B. The genetics and epigenetics of autoimmune diseases. J Autoimmun 2009;33:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Altorok N, Coit P, Hughes T, Koelsch KA, Stone DU, Rasmussen A, et al. Genome‐wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjögren's syndrome. Arthritis Rheumatol 2014;66:731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thabet Y, le Dantec C, Ghedira I, Devauchelle V, Cornec D, Pers JO, et al. Epigenetic dysregulation in salivary glands from patients with primary Sjögren's syndrome may be ascribed to infiltrating B cells. J Autoimmun 2013;41:175–81. [DOI] [PubMed] [Google Scholar]
- 9. Miceli‐Richard C, Wang‐Renault SF, Boudaoud S, Busato F, Lallemand C, Bethune K, et al. Overlap between differentially methylated DNA regions in blood B lymphocytes and genetic at‐risk loci in primary Sjögren's syndrome. Ann Rheum Dis 2016;75:933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Imgenberg‐Kreuz J, Sandling JK, Almlöf JC, Nordlund J, Signér L, Norheim KB, et al. Genome‐wide DNA methylation analysis in multiple tissues in primary Sjögren's syndrome reveals regulatory effects at interferon‐induced genes. Ann Rheum Dis 2016. E‐pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shiboski SC, Shiboski CH, Criswell LA, Baer A, Challacombe S, Lanfranchi H, et al. American College of Rheumatology classification criteria for Sjögren's syndrome: a data‐driven, expert consensus approach in the Sjögren's International Collaborative Clinical Alliance cohort. Arthritis Care Res (Hoboken) 2012;64:475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Criswell LA. The genetic basis of Sjögren's Syndrome (SS) clinical manifestations from genome‐wide association analysis of subphenotype extremes in an international cohort [abstract]. Arthritis Rheumatol 2014;66 Suppl:S228–9. [Google Scholar]
- 14. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet 2006;38:904–9. [DOI] [PubMed] [Google Scholar]
- 15. Team RDC . R: a language and environment for statistical computing. 2015. URL: http://www.R-project.org.
- 16. Davis S, Du P, Bilke S, Triche T Jr, Bootwalla M. Methylumi: Handle Illumina methylation data. R package version 2.14.0. 2015. URL: https://www.bioconductor.org/packages/methylumi.
- 17. Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. Orchestrating high‐throughput genomic analysis with Bioconductor. Nat Methods 2015;12:115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Triche TJ Jr, Weisenberger DJ, Van den Berg D, Laird PW, Siegmund KD. Low‐level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res 2013;41:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yousefi P, Huen K, Schall RA, Decker A, Elboudwarej E, Quach H, et al. Considerations for normalization of DNA methylation data by Illumina 450K BeadChip assay in population studies. Epigenetics 2013;8:1141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross‐reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013;8:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 2015;43:D670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez‐Cabrero D, et al. A beta‐mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450k DNA methylation data. Bioinformatics 2013;29:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat 2001;29:1165–88. [Google Scholar]
- 24. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Whitaker JW, Shoemaker R, Boyle DL, Hillman J, Anderson D, Wang W, et al. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med 2013;5:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carlson M. Org.Hs.eg.db: genome wide annotation for human. R package version 3.1.2. URL: http://bioconductor.org/packages/org.Hs.eg.db/.
- 27. Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis 2013;72:110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hjelmervik TO, Petersen K, Jonassen I, Jonsson R, Bolstad AI. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjögren's syndrome patients from healthy control subjects. Arthritis Rheum 2005;52:1534–44. [DOI] [PubMed] [Google Scholar]
- 30. Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren's syndrome. Nat Genet 2013;45:1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Y, Zhang K, Chen H, Sun F, Xu J, Wu Z, et al. A genome‐wide association study in Han Chinese identifies a susceptibility locus for primary Sjögren's syndrome at 7q11.23. Nat Genet 2013;45:1361–5. [DOI] [PubMed] [Google Scholar]
- 32. McLeay RC, Bailey TL. Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 2010;11:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley‐Hunt R, Arenillas DJ, et al. JASPAR 2014: an extensively expanded and updated open‐access database of transcription factor binding profiles. Nucleic Acids Res 2014;42:D142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnsen O, Murphy P, Prydz H, Kolsto AB. Interaction of the CNC‐bZIP factor TCF11/LCR‐F1/Nrf1 with MafG: binding‐site selection and regulation of transcription. Nucleic Acids Res 1998;26:512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Steffen J, Seeger M, Koch A, Kruger E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD‐dependent feedback loop. Mol Cell 2010;40:147–58. [DOI] [PubMed] [Google Scholar]
- 36. Hartman SE, Bertone P, Nath AK, Royce TE, Gerstein M, Weissman S, et al. Global changes in STAT target selection and transcription regulation upon interferon treatments. Genes Dev 2005;19:2953–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Portales‐Casamar E, Kirov S, Lim J, Lithwick S, Swanson MI, Ticoll A, et al. PAZAR: a framework for collection and dissemination of cis‐regulatory sequence annotation. Genome Biol 2007;8:R207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Egerer T, Martinez‐Gamboa L, Dankof A, Stuhlmuller B, Dorner T, Krenn V, et al. Tissue‐specific up‐regulation of the proteasome subunit β5i (LMP7) in Sjögren's syndrome. Arthritis Rheum 2006;54:1501–8. [DOI] [PubMed] [Google Scholar]
- 39. Fox RI, Tornwall J, Michelson P. Current issues in the diagnosis and treatment of Sjögren's syndrome. Curr Opin Rheumatol 1999;11:364–71. [DOI] [PubMed] [Google Scholar]
- 40. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suzuki M, Yamada T, Kihara‐Negishi F, Sakurai T, Hara E, Tenen DG, et al. Site‐specific DNA methylation by a complex of PU.1 and Dnmt3a/b. Oncogene 2006;25:2477–88. [DOI] [PubMed] [Google Scholar]
- 42. Ueda R, Kohanbash G, Sasaki K, Fujita M, Zhu X, Kastenhuber ER, et al. Dicer‐regulated microRNAs 222 and 339 promote resistance of cancer cells to cytotoxic T‐lymphocytes by down‐regulation of ICAM‐1. Proc Natl Acad Sci U S A 2009;106:10746–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alevizos I, Alexander S, Turner RJ, Illei GG. MicroRNA expression profiles as biomarkers of minor salivary gland inflammation and dysfunction in Sjögren's syndrome. Arthritis Rheum 2011;63:535–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harper KN, Peters BA, Gamble MV. Batch effects and pathway analysis: two potential perils in cancer studies involving DNA methylation array analysis. Cancer Epidemiol Biomarkers Prev 2013;22:1052–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barfield RT, Almli LM, Kilaru V, Smith AK, Mercer KB, Duncan R, et al. Accounting for population stratification in DNA methylation studies. Genet Epidemiol 2014;38:231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yousefi P, Huen K, Dave V, Barcellos L, Eskenazi B, Holland N. Sex differences in DNA methylation assessed by 450 K BeadChip in newborns. BMC Genomics 2015;16:911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Whitacre CC, Reingold SC, O'Looney PA. A gender gap in autoimmunity. Science 1999;283:1277–8. [DOI] [PubMed] [Google Scholar]
- 48. Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of β‐value and M‐value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010;11:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Self‐reported Medication by Case‐Control Status
Supplementary Table 2. Correlations between First and Second Principal Components of DNAm, Phenotype and Selected Covariates
Supplementary Table 3. Remaining (Lower Significance) Differentially Methylated Promoters in LSGs of Sjögren's Syndrome Patients
Supplementary Figure 1. Principal component analysis on genome‐wide gland DNAm, before and after non‐linear adjustment. A. Before adjustment, the first principal component separates the three batches. B. Spearman Z‐test p‐values showing significance of correlation between PC1‐5 and categorical phenotypes and covariates: disease status, treatment plate, sentrix ID (methylation chip ID), SSA and SSB seropositivity. Red dashed line indicates Bonferroni significance threshold for 25 tests. C. Following adjustment, batches are mixed. D. Significance of correlation with batch is low across first five PCs, while preserving associations with phenotype.
Supplementary Figure 2. Correlation between DNA methylation and expression disease‐associations from two studies. The x‐axis shows extent of mRNA down‐regulation from Hjelmervik et al. and the y‐axis shows the significance of DNA hyper‐methylation – from the present study ‐ for CpGs in the promoter of the corresponding gene. DMPs from the present study are highlighted in cyan.
Supplementary Figure 3. JASPAR motifs enriched in the neighborhood of SS‐associated DMPs in labial salivary gland tissue. A. Schematic of transcription factor binding motif enrichment analysis. B. MA0089.1, annotated for heterodimer of chicken orthologs of NFE2L1 and MAFG. C. MA0517.1, annotated for the heterodimer of STAT2 and STAT1. D. MA0080.3, annotated for the mouse ortholog of SPI1.
Supplementary Figure 4. Extended differential methylation in PSMB8‐AS1 promoter. A. Highlighted region shows region designated as PSB8‐AS1 promoter, sitting within the gene body of PSMB8. All SS‐associated DMPs (promoter and non‐promoter) are annotated in the top track, UCSC Genome Browser RefGene annotations in the middle, and all 450K CpG sites at the bottom. B. Evidence of an SS‐associated differentially hypo‐methylated region within the promoter of PSMB8‐AS1.