

Abstract
The gold(I)‐catalyzed oxidative cyclization of 7‐ethynyl‐1,3,5‐cycloheptatrienes gives 1‐substituted barbaralones in a general manner, which simplifies the access to other fluxional molecules. As an example, we report the shortest syntheses of bullvalene, phenylbullvalene, and disubstituted bullvalenes, and a readily accessible route to complex cage‐type structures by further gold(I)‐catalyzed reactions.
Keywords: barbaralones, bullvalenes, cyclization reactions, gold, valence tautomerism
Fluxional molecules, such as barbaralone (1 a), bullvalone (2 a), and bullvalene (3 a) have been central to the understanding of the phenomena of valence tautomerism (see Figure 1).1, 2 These molecules undergo low energy [3.3]‐sigmatropic rearrangements, which in the case of bullvalene lead to 1 209 600 degenerate tautomers,3, 4, 5 whereas a lower number of constitutional isomers are possible for substituted bullvalenes6, 7, 8 and only two exist for barbaralone (1 a).9
Figure 1.
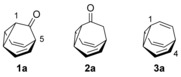
Barbaralone (1 a), bullvalone (2 a), and bullvalene (3 a).
Syntheses of these fluxional molecules requires multistep procedures that proceed with low overall yield, often using explosive and toxic diazomethane. Thus, the optimized synthesis of barbaralone (1 a), en route to bullvalene (3 a),10 starts with the Büchner reaction of ethyl diazoacetate with benzene to form 4,11, 12 which is converted into 1 a in four steps via diazomethyl ketone 5 (Scheme 1).10 Bullvalene (3 a) can be prepared from 1 a in four additional steps by two different procedures by homologation of 1 a to bullvalone (2 a) with diazomethane.2, 10 Barbaralone (1 a) has also been prepared from (cyclooctatetraene)tricarbonyliron in two steps in approximately 36 % yield.13
Scheme 1.
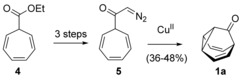
Synthesis of barbaralone (1 a) from ethyl cyclohepta‐2,4,6‐triene‐1‐carboxylate (4).
1‐Methylbarbaralone (1 w) was prepared by a procedure similar to that shown in Scheme 1 using ethyldiazomethane in the reaction with cycloheptatriene carbamoyl chloride to form the homologue of 5.9b Although some ingenious syntheses of highly substituted bullvalenes have been designed,8 most bullvalenes have been prepared from parent 3 a. Thus, for example, phenylbullvalene (3 b) was obtained in three steps (26 % yield) from 3 a by dibromination, dehydrobromination with KOtBu, and reaction of the resulting bromobullvalene with Ph2CuLi.7d
Current synthetic art does not allow preparation of substituted barbaralones in a general way,14 which limits the access to fluxional homologues and other theoretically interesting molecules.12, 15 We have recently found that 7‐aryl‐1,3,5‐cycloheptatrienes undergo a gold(I)‐catalyzed retro‐Büchner reaction to form highly reactive aryl gold(I) carbenes (a decarbenation reaction).16 However, 7‐ethynyl‐1,3,5‐cycloheptatrienes (6) react differently to form fluxional barbaralyl gold(I) intermediates 7; after a series of complex rearrangements 7 finally leads to indenes 8 and/or 9, depending on the gold catalyst (Scheme 2).17 Since the gold‐catalyzed oxidation of alkynes has been shown to take place readily with oxidants such as sulfoxides, or amine‐N‐oxides to form α‐oxo gold(I) carbenes,18, 19 we envisioned that the oxidation of intermediates 7 could lead to 1‐substituted barbaralones 1 (Scheme 2). However, if the oxidation takes place directly on 7‐ethynyl‐1,3,5‐cycloheptatrienes (6), the two regioisomeric α‐oxo gold(I) carbenes 10 a and 10 b would be formed,18 of which only 10 b would lead to barbaralones 1 by intramolecular cyclopropanation.
Scheme 2.
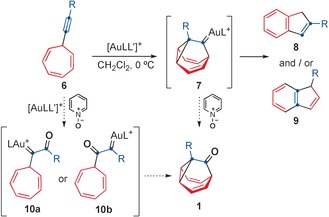
Two different pathways for the formation of barbaralones 1 by gold(I)‐catalyzed oxidative cyclization of 7‐ethynyl‐1,3,5‐cycloheptatrienes (6).
Herein, we report a general and straightforward synthesis of 1‐substituted barbaralones 1 from alkynes and commercially available tropylium tetrafluoroborate in just two steps by oxidative cyclization of 7‐ethynyl‐1,3,5‐cycloheptatrienes.
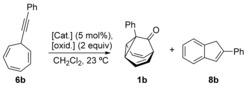
We first studied the reaction of 7‐(phenylethynyl)cyclohepta‐1,3,5‐triene (6 b) with different gold(I) catalysts in the presence of diphenylsulfoxide (Ox1), and the N‐oxides of pyridine (Ox2), 3,5‐dichloropyridine (Ox3), or 8‐methylquinoline (Ox4) as oxidants (Table 1). Using Johnphos gold(I) complex A in combination with Ox3, 1‐phenylbarbaralone (1 b) was obtained in 50 % yield, together with 2‐phenyl‐1H‐indene (8 b; Table 1, entry 3). Related gold(I) complexes B and C led to 1 b in lower yields in the presence of Ox3 (Table 1, entries 5 and 6). The best yields of 1 b were obtained using [IPrAu(MeCN)][SbF6] (D) and either Ox1 or Ox3 (Table 1, respectively entries 7 and 9). Oxidants Ox2 and Ox4 led to very poor results (Table 1, respectively entries 8 and 10). Interestingly, whereas phosphite gold(I) complex E afforded low yields (Table 1, entries 11 and 12), pycolinate gold(III) complex provided 1 b in a similar yield in the presence of Ox3 (Table 1, entry 14). Reactions of alkynes with pyridine‐N‐oxides can also be performed with Brønsted acids20 or ZnII catalysts.21 However, in our system, a complex reaction mixture was obtained in the presence of methanesulfonic acid (Table 1, entry 15) and starting material was recovered when Zn(OTf)2 was used as the catalyst (Table 1, entry 16).
Table 1.
Gold(I)‐catalyzed oxidative reaction of 6 b to give 1‐phenylbarbaralone (1 b).
| Entry | [Cat.] | Oxid. | Time [h] | 1 b Yield [%][a] | 8 b Yield [%][a] |
|---|---|---|---|---|---|
| 1 | A | Ox1 | 2.5 | 12 | 58 |
| 2 | A | Ox2 | 16 | 5 | – |
| 3 | A | Ox3 | 2.5 | 50 (50)[b] | 28 |
| 4 | A | Ox4 | 16 | 23 | – |
| 5 | B | Ox3 | 3 | 30 | 36 |
| 6 | C | Ox3 | 3 | 32 | 42 |
| 7 | D | Ox1 | 3 | (83)[b] | – |
| 8 | D | Ox2 | 24 | 2 | – |
| 9 | D | Ox3 | 3 | 64 | – |
| 10 | D | Ox4 | 3 | 7 | – |
| 11 | E | Ox1 | 2.5 | 20 | – |
| 12 | E | Ox3 | 2.5 | 30 | – |
| 13 | F | Ox1 | 5 | 14 | 61 |
| 14 | F | Ox3 | 5 | 61 | – |
| 15 | MeSO3H[c] | Ox3 | 2.5 | complex mixture | |
| 16 | Zn(OTf)2 [d] | Ox3 | 24 | starting material | |
[a] Yields determined by 1H NMR using mesitylene as an internal standard. [b] Yield of isolated products. [c] 4 equiv [d] 10 mol %. Catalyst (cat.), oxidant (oxid.). 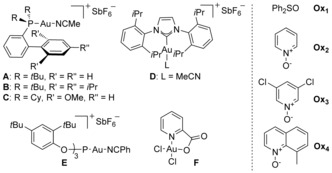
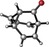
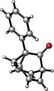
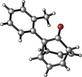
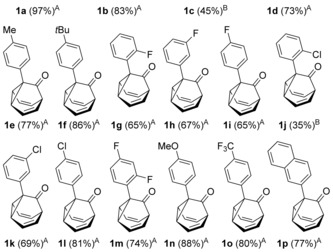
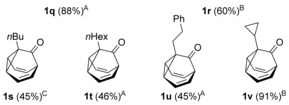
To assess the generality of this oxidative cyclization, various 7‐ethynyl‐1,3,5‐cycloheptatrienes (6 a–v) were prepared17 and the combinations of catalyst and oxidant that provided the best results in the preliminary studies (conditions: A) cat. D, Ox1; B) cat. D, Ox3; C) cat. A, Ox3) were tested on these substrates (Table 2). The most simple substrate, 7‐ethynyl‐1,3,5‐cycloheptatriene (6 a), produced barbaralone (1 a) in 97 % yield. The reaction was compatible with 7‐(arylethynyl)‐1,3,5‐cycloheptatrienes 6 c–o bearing different o‐, m‐, or p‐ substituents on the phenyl ring, such as methyl‐, tert‐butyl‐, fluorine‐, chlorine‐, methoxy‐, or trifluoromethyl‐, affording the corresponding 1‐substituted barbaralones (1 c–o) in moderate to good yields. Substrates 6 p–q, possessing a naphthyl or a thiophenyl substituent, gave barbaralones 1 p–q in yields up to 88 %. This catalytic procedure was extendible to alkynyl cycloheptatrienes with aliphatic or alkyl groups (6 s–v), including n‐butyl, n‐hexyl, 2‐phenylethyl, and cyclopropyl, giving the desired barbaralones 1 s–v in moderate to excellent yields. The reaction was also applicable to a substrate containing two alkynes (6 r), affording dibarbaralone 1 r in 60 % yield. The process was efficiently scalable, providing up to 850 mg of barbaralone (1 a) in one run in 96 % yield. Mechanistically, the formation of aryl substituted barbaralones 1 b–r strongly suggests that the oxidation takes place on barbaralyl gold(I) intermediates 7 (Scheme 2), rather than on the alkyne, which would favor formation of unproductive intermediate 10 a by benzylic oxidation.18
Table 2.
Gold(I)‐catalyzed oxidative synthesis of barbaralones 1 a–v.
|
|
|
|
| |||
|
|
||
| |||
Conditions: A) cat. D, Ox1; B) cat. D, Ox3; C) cat. A, Ox3.
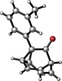
The introduction of a methyl substituent was found to shift the sigmatropic equilibrium toward the 1‐substituted isomer (Scheme 3).6c, 9b Our NMR data show that 1‐substituted barbaralones are in all cases the most stable tautomers, as confirmed by the X‐ray diffraction structures of 1 a–d, 1 q, and 1 r (Table 2).22
Scheme 3.
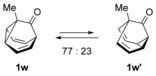
Equilibrium between 1‐methyl‐ and 5‐methylbarbaralones.9b
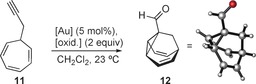
With the synthesis of barbaralones (1) in hand, and the preparation of bullvalenes (3) in mind, we examined the applicability of this oxidative cyclization for the synthesis of bullvalone (2 a) using propargyl cycloheptatriene as substrate (11). Thus, the reaction of propargyl cycloheptatriene (11) with different gold(I) catalysts in the presence of the previously employed oxidants was investigated (Table 3). However, instead of the desired bullvalone, arising from a 5‐endo‐dig oxidative cyclization, in all cases we observed the recovered starting material or formation of 1‐formylbarbaralane (12),22 the product of a 5‐exo‐dig process. While Johnphos gold(I) complex A gave poor results regardless of the oxidant used (Table 3, entries 1–4), good yields were obtained with tBuXPhos gold(I) catalyst (B′) and [IPrAu(MeCN)]SbF6 (D) with Ox1 (Table 3, entries 5 and 8). A better yield of aldehyde 12 was obtained using phosphite gold(I) complex E′ and Ox1 (Table 3, entry 10).
Table 3.
Gold(I)‐catalyzed oxidative reaction of propargyl cycloheptatriene (11) to give 1‐formylbarbaralane (12).
| Entry | [Au] | Oxid. | Time [h] | 12 Yield [%][a] |
|---|---|---|---|---|
| 1 | A | Ox1 | 18 | 27 |
| 2 | A | Ox2 | 15 | – |
| 3 | A | Ox3 | 18 | 5 |
| 4 | A | Ox4 | 15 | – |
| 5 | B′ | Ox1 | 17 | 91 (87)[b] |
| 6 | B′ | Ox3 | 17 | 7 |
| 8 | D | Ox1 | 4 | 90 (87)[b] |
| 9 | D | Ox3 | 18 | 8 |
| 10 | E′ | Ox1 | 6 | 92 |
| 11 | E′ | Ox3 | 24 | – |
[a] Yields determined by 1H NMR spectroscopy using mesitylene as an internal standard. [b] Yield of isolated products. 
At this stage we considered accessing bullvalones (2) via barbaralones (1) en route to bullvalenes (3). Homologation of 1 a with diazomethane has been reported to give bullvalone (2 a) in 24 % yield along with an isomeric aldehyde (34 %).2, 10 Reduction of 2 a followed by acetylation led to the corresponding acetate (40 %, two steps), which was pyrolyzed at 345 °C to give a 1:1 ratio of bullvalene (3 a) and cis‐9,10‐dihydronaphthalene.2, 23 An improved procedure was reported via bullvalone tosylhydrazone, providing bullvalene (3 a) in approximately 5 % yield from 2 a in four steps.10, 24
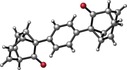
In our new approach, bullvalene (3 a) and phenylbullvalene (3 b) were prepared from barbaralones 1 a–b by a three‐step procedure. A homologation reaction of 1 a and 1 b with the lithium anion of (trimethylsilyl)diazomethane25 gave bullvalones 2 a and 2 b in 37 and 22 % yield, respectively (Scheme 4). Formation of the corresponding enol triflates using LDA and Comins’ reagent, or LiHMDS and PhNTf2 followed by immediate reduction with nBu3SnH and Pd(PPh3)4 as catalyst,26 afforded 3 a and 3 b 27 in 44 % and 60 % yield, respectively, whose structures were confirmed by X‐ray diffraction.22 This new synthesis of bullvalene (3 a) is the most efficient to date as it requires a total of five steps (10 % overall yield) from commercially available tropylium tetrafluoroborate and ethynyl magnesium bromide.
Scheme 4.
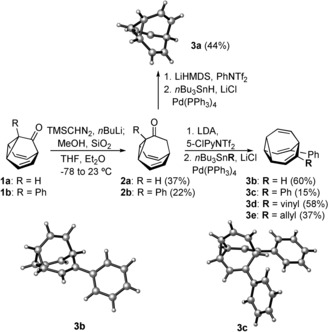
Synthesis of bullvalene (3 a), phenylbullvalene (3 b), and disubstituted bullvalenes 3 c–e.
Various disubstituted bullvalenes 3 c–e were also prepared from phenyl bullvalone (2 b) through sequential formation of the enol triflate followed by Stille couplings (Scheme 4).28 The molecular structure of diphenyl bullvalene 3 c was determined by X‐ray diffraction.22 Bullvalenes 3 c–e were in equilibrium with the 3,6‐ and 3,7‐disubstituted isomers at −40 °C; the observed ratios of the respective compounds was 7.6:5.7:1, 5.2:1.5:1, and 3.8:2.3:1.7d
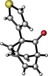
Barbaralones 1 a and 1 b were converted into 14 a and 14 b in two steps by the addition of ethynyl magnesium bromide and subsequent gold(I)‐catalyzed reaction of the corresponding alcohols 13 a and 13 b, which proceeded by a new type of cyclization/rearrangement (Scheme 5). Structures 14 a and 14 b were confirmed by X‐ray diffraction.22
Scheme 5.
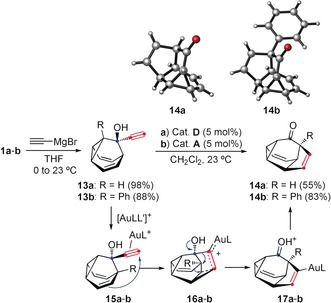
Preparation of highly fused tetracyclic molecules 14 a and 14 b.
Unprecedented tetracyclic cages 14 a and 14 b are probably formed by coordination of gold(I) of the alkyne of the minor tautomer of 13 a and 13 b to give 15 a and 15 b, followed by intramolecular attack of the alkene to form delocalized intermediates 16 a and 16 b 29, 30 and semipinacol‐type rearrangement to give 17 a and 17 b (Scheme 5). To the best of our knowledge, and despite the many different types of gold(I)‐catalyzed cycloisomerizations,30 this formation of a five‐membered ring by cyclization‐rearrangement is unprecedented.
Furthermore, alkylation of barbaralol 18 22 with propargyl bromide gives 1,7‐enyne 19, which undergoes an exo‐dig cyclization with gold(I) catalyst D to form intermediate 20,30 which then reacts with methanol as a nucleophile to form tricyclic system 21 (Scheme 6).
Scheme 6.
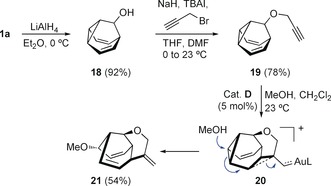
Formation of tricyclic derivative 21 from barbaralol (18).
In summary, we have developed an efficient synthesis of 1‐substituted barbaralones by gold(I)‐catalyzed oxidative cyclization of 7‐(substituted ethynyl)‐1,3,5‐cycloheptatrienes. This method has allowed accomplishment of the shortest syntheses of bullvalene and other substituted bullvalenes. Thus, parent bullvalene (3 a) is obtained in five steps from commercially available starting materials in 10 % overall yield, which compares favorably with previous procedures that require nine or more steps and proceeded with very low overall efficiency. The straightforward access to barbaralones opens a way to obtain complex cage systems with unprecedented molecular architectures.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank MINECO (Severo Ochoa Excellence Accreditation 2014–2018 (SEV‐2013‐0319) CTQ2013‐42106‐P), the MEC (FPU fellowship to SF), the European Research Council (Advanced Grant No. 321066), the AGAUR (2014 SGR 818), and the ICIQ Foundation. We also thank the ICIQ X‐ray diffraction unit and Dr. Michael E. Muratore for helpful discussions.
S. Ferrer, A. M. Echavarren, Angew. Chem. Int. Ed. 2016, 55, 11178.
References
- 1.
- 1a. Doering W. v. E., Roth W. R., Angew. Chem. Int. Ed. Engl. 1963, 2, 115–122; [Google Scholar]; Angew. Chem. 1963, 75, 27–35; [Google Scholar]
- 1b. Williams R. V., Chem. Rev. 2001, 101, 1185–1204; [DOI] [PubMed] [Google Scholar]
- 1c. Hrovat D. A., Brown E. C., Williams R. V., Quast H., Borden W. T., J. Org. Chem. 2005, 70, 2627–2632; [DOI] [PubMed] [Google Scholar]
- 1d. He M., Bode J. W., Proc. Natl. Acad. Sci. USA 2011, 108, 14752–14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Doering W. v. E., Roth W. R., Tetrahedron 1963, 19, 715–737; [Google Scholar]
- 2b. Doering W. V. E., Ferrier B. M., Fossel E. T., Hatenstein J. H., M. Jones, Jr. , Klumpp G., Rubin R. M., Saunders M., Tetrahedron 1967, 23, 3943–3963. [Google Scholar]
- 3.
- 3a. Schröder G., Angew. Chem. Int. Ed. Engl. 1963, 2, 481; [Google Scholar]; Angew. Chem. 1963, 75, 722; [Google Scholar]
- 3b. Schröder G., Chem. Ber. 1964, 97, 3140–3149; [Google Scholar]
- 3c. Merényi R., Oth J. F. M., Schröder G., Chem. Ber. 1964, 97, 3150–3161; [Google Scholar]
- 3d. Schröder G., Oth J. F. M., Merényi R., Angew. Chem. Int. Ed. Engl. 1965, 4, 752–761; [Google Scholar]; Angew. Chem. 1965, 77, 774–784; [Google Scholar]
- 3e. Oth J. F. M., Müllen K., Gilles J.-M., Schröder G., Helv. Chim. Acta 1974, 57, 1415–1433. [Google Scholar]
- 4. Saunders M., Tetrahedron Lett. 1963, 4, 1699–1702. [Google Scholar]
- 5.
- 5a. Poupko R., Zimmermann H., Luz Z., J. Am. Chem. Soc. 1984, 106, 5391–5394; [Google Scholar]
- 5b. Meier B. H., Earl W. L., J. Am. Chem. Soc. 1985, 107, 5553–5555; [Google Scholar]
- 5c. Luger P., Roth K., J. Chem. Soc. Perkin Trans. 2 1989, 649–655; [Google Scholar]
- 5d. Titman J. J., Luz Z., Spiess H. W., J. Am. Chem. Soc. 1992, 114, 3765–3771; [Google Scholar]
- 5e. Schlick S., Luz Z., Poupko R., J. Am. Chem. Soc. 1992, 114, 4315–4320. [Google Scholar]
- 6.
- 6a. Poupko R., Zimmermann H., Müller K., Luz Z., J. Am. Chem. Soc. 1996, 118, 7995–8005; [Google Scholar]
- 6b. Müller K., Zimmermann H., Krieger C., Poupko R., Luz Z., J. Am. Chem. Soc. 1996, 118, 8006–8014; [Google Scholar]
- 6c. Poupko R., Müller K., Krieger C., Zimmermann H., Luz Z., J. Am. Chem. Soc. 1996, 118, 8015–8023; [Google Scholar]
- 6d. Luz Z., Olivier L., Poupko R., Müller K., Krieger C., Zimmermann H., J. Am. Chem. Soc. 1998, 120, 5526–5538. [Google Scholar]
- 7.
- 7a. Volkmann B., Schröder G., Chem. Ber. 1984, 117, 2226–2232; [Google Scholar]
- 7b. Rebsamen K., Schröder G., Chem. Ber. 1993, 126, 1419–1423; [Google Scholar]
- 7c. Rebsamen K., Schröder G., Chem. Ber. 1993, 126, 1425–1427; [Google Scholar]
- 7d. Rebsamen K., Röttele H., Schröder G., Chem. Ber. 1993, 126, 1429–1433. [Google Scholar]
- 8.
- 8a. Lippert A. R., Kaeobamrung J., Bode J. W., J. Am. Chem. Soc. 2006, 128, 14738–14739; [DOI] [PubMed] [Google Scholar]
- 8b. Lippert A. R., Keleshian V. L., Bode J. W., Org. Biomol. Chem. 2009, 7, 1529–1532; [DOI] [PubMed] [Google Scholar]
- 8c. Lippert A. R., Naganawa A., Keleshian V. L., Bode J. W., J. Am. Chem. Soc. 2010, 132, 15790–15799; [DOI] [PubMed] [Google Scholar]
- 8d. Larson K. K., He M., Teichert J. F., Naganawa A., Bode J. W., Chem. Sci. 2012, 3, 1825–1828; [Google Scholar]
- 8e. He M., Bode J. W., Org. Biomol. Chem. 2013, 11, 1306–1317; [DOI] [PubMed] [Google Scholar]
- 8f. Teichert J. F., Mazunin D., Bode J. W., J. Am. Chem. Soc. 2013, 135, 11314–11321. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Lambert J. B., Tetrahedron Lett. 1963, 4, 1901–1906; [Google Scholar]
- 9b. Barborak J. C., Chari S., Schleyer P. v. R., J. Am. Chem. Soc. 1971, 93, 5275–5277; [Google Scholar]
- 9c. Nakanishi H., Yamamoto O., Chem. Lett. 1975, 513–516; [Google Scholar]
- 9d. Engdahl C., Ahlberg P., J. Am. Chem. Soc. 1979, 101, 3940–3946; [Google Scholar]
- 9e. Johnston E. R., Barber J. S., Jacomet M., Barborak J. C., J. Am. Chem. Soc. 1998, 120, 1489–1493. [Google Scholar]
- 10. Casas J., Serratosa F., An. Quím. 1977, 73, 300–302. [Google Scholar]
- 11. Dewar M. J. S., Pettit R., J. Chem. Soc. 1956, 2021–2025. [Google Scholar]
- 12.For two alternative procedures, see: Vedejs E., Gabel R. A., Weeks P. D., J. Am. Chem. Soc. 1972, 94, 5842–5845. [Google Scholar]
- 13. Heil V., Johnson B. F. G., Lewis J., Thompson D. J., J. Chem. Soc. Chem. Commun. 1974, 270–271. [Google Scholar]
- 14.For cyclopentannulated barbaralones by photocycloaddition of 2-(pent-4-yn-1-yl)tropones, see: Feldman K. S., Come J. H., Fegley G. J., Smith B. D., Parvez M., Tetrahedron Lett. 1987, 28, 607–610. [Google Scholar]
- 15. Kirchmeyer S., de Meijere A., Helv. Chim. Acta 1990, 73, 1182–1196. [Google Scholar]
- 16.
- 16a. Solorio-Alvarado C. R., Wang Y., Echavarren A. M., J. Am. Chem. Soc. 2011, 133, 11952–11955; [DOI] [PubMed] [Google Scholar]
- 16b. Wang Y., McGonigal P., Herlé B., Besora M., Echavarren A. M., J. Am. Chem. Soc. 2014, 136, 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGonigal P. R., de León C., Wang Y., Homs A., Solorio-Alvarado C. R., Echavarren A. M., Angew. Chem. Int. Ed. 2012, 51, 13093–13096; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 13270–13273. [Google Scholar]
- 18.
- 18a. Zhang L., Acc. Chem. Res. 2014, 47, 877–888; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b.Z. Zheng, Z. Wang, Y. Wang, L. Zhang, Chem. Soc. Rev 2016, DOI: 10.1039/c5cs00887e.
- 19.
- 19a. Vasu D., Hung H.-H., Bhunia S., Gawade S. A., Das A., Liu R.-S., Angew. Chem. Int. Ed. 2011, 50, 6911–6914; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7043–7046; [Google Scholar]
- 19b. Huple D. B., Ghorpade S., Liu R.-S., Adv. Synth. Catal. 2016, 358, 1348–1367; [Google Scholar]
- 19c.J. Schulz, J. Jasik, A. Gray, J. Roithová, Chem. Eur. J 2016, 22, DOI: 10.1002/chem.201601634, and references therein. [DOI] [PubMed]
- 20. Chen D.-F., Han Z.-Y., He Y.-P., Yu J., Gong L.-Z., Angew. Chem. Int. Ed. 2012, 51, 12307–12310; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12473–12476. [Google Scholar]
- 21. Li L., Zhou B., Wang Y.-H., Shu C., Pan Y.-F., Lu X., Ye L.-W., Angew. Chem. Int. Ed. 2015, 54, 8245–8249; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8363–8367. [Google Scholar]
- 22.CCDC 1487016 (1 a), CCDC 1487017 (1 b), CCDC 1487018 (1 c), CCDC 1487019 (1 d), CCDC 1487020 (1 q), CCDC 1487021 (1 r), CCDC 1487022 (2 b), CCDC 1487023 (3 a), CCDC 1487024 (3 b), CCDC 1487025 (3 c), CCDC 1487026 (12), CCDC 1487027 (14 a), and CCDC 1487028 (14 b) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 23.The yield for the last step was not provided in the original report.[2b]
- 24.
- 24a. Serratosa F., López F., Font J., Tetrahedron Lett. 1972, 2589–2590; [Google Scholar]
- 24b. Serratosa F., López F., Font J., An. Quím. 1974, 70, 893–899. [Google Scholar]
- 25. Liu H., Sun C., Lee N.-K., Henry R. F., Lee D., Chem. Eur. J. 2012, 18, 11889–11893. [DOI] [PubMed] [Google Scholar]
- 26. Scott W. J., Stille J. K., J. Am. Chem. Soc. 1986, 108, 3033–3040. [Google Scholar]
- 27.Phenylbullvalene 3 b has been shown to be in a 3:1 equilibrium with the 3-substituted isomer at −40 °C.[7d] We have confirmed this result and have observed that the equilibrium can be shifted to 5.6:1 at −90 °C.
- 28. Scott W. J., Crisp G. T., Stille J. K., J. Am. Chem. Soc. 1984, 106, 4630–4632. [Google Scholar]
- 29. López-Carrillo V., Huguet N., Mosquera Á., Echavarren A. M., Chem. Eur. J. 2011, 17, 10972–10978. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Jiménez-Núñez E., Echavarren A. M., Chem. Rev. 2008, 108, 3326–3350; [DOI] [PubMed] [Google Scholar]
- 30b. Dorel R., Echavarren A., Chem. Rev. 2015, 115, 9028–9072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30c. Dorel R., Echavarren A. M., J. Org. Chem. 2015, 80, 7321–7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary