

Abstract
Long‐term efficacy and safety of bosutinib (≥4 years follow‐up from last enrolled patient) were evaluated in an ongoing phase 1/2 study in the advanced leukemia cohort with prior treatment failure (accelerated‐phase [AP, n = 79] chronic myeloid leukemia [CML], blast‐phase [BP, n = 64] CML, acute lymphoblastic leukemia [ALL, n = 24]). Fourteen AP, 2 BP, and 1 ALL patient remained on bosutinib at 4 years (vs. 38, 8, 1 at 1 year); median (range) treatment durations: 10.2 (0.1–88.6), 2.8 (0.03–55.9), 0.97 (0.3–89.2) months. Among AP and BP patients, 57% and 28% newly attained or maintained baseline overall hematologic response (OHR); 40% and 37% attained/maintained major cytogenetic response (MCyR) by 4 years (most by 12 months). In responders at 1 versus 4 years, Kaplan‐Meier (KM) probabilities of maintaining OHR were 78% versus 49% (AP) and 28% versus 19% (BP); KM probabilities of maintaining MCyR were 65% versus 49% (AP) and 21% versus 21% (BP). Most common AEs (AP, BP) were gastrointestinal (96%; 83%), primarily diarrhea (85%; 64%), which was typically low grade (maximum grade 1/2: 81%; 59%) and transient; no patient discontinued due to diarrhea. Serious AEs occurred in 44 (56%) AP and 37 (58%) BP patients, most commonly pneumonia (n = 9) for AP and pyrexia (n = 6) for BP; 11 and 13 died within 30 days of last dose (2 considered bosutinib‐related [AP] per investigator). Responses were durable in ∼50% AP responders at 4 years (∼25% BP patients responded at year 1, suggesting possible bridge‐to‐transplant role in BP patients); toxicity was manageable.Am. J. Hematol. 90:755–768, 2015. © 2015 The Authors. American Journal of Hematology Published by Wiley Periodicals, Inc.
Introduction
Philadelphia chromosome‐positive (Ph+) chronic myeloid leukemia (CML) evolves from chronic phase (CP) to the more clinically severe accelerated phase (AP) and/or blast phase (BP) after a median duration of 3 to 4 years 1, 2. Compared with CP CML, advanced Ph+ leukemias have a more rapid disease course, are more difficult to treat 3, and have a considerably poorer prognosis 4, 5, 6, 7, 8. Ph+ acute lymphoblastic leukemia (ALL) resembles lymphoid BP in severity and is commonly associated with relapse after chemotherapy and poor long‐term outcome 9. Bcr‐Abl tyrosine kinase inhibitors (TKIs) are an effective treatment option for all phases of CML 10, 11, 12, 13, 14, 15, with survival benefit also observed in Ph+ ALL 16, 17. Notably, allogeneic hematopoietic stem cell transplantation (allo‐HSCT) remains the only curative treatment option in suitable patients with advanced leukemias 18, 19, 20, 21, 22.
Bosutinib is an oral dual Src/Abl TKI approved in the US for treatment of CP, AP, and BP Ph+ CML following resistance/intolerance to prior therapy and in Europe for treatment of CP, AP, and BP Ph+ CML patients previously treated with ≥1 TKI for whom other TKIs are not considered appropriate treatment options 23, 24. Bosutinib has demonstrated activity and manageable tolerability in a phase 1/2 study of patients with CP CML following resistance/intolerance to imatinib only or imatinib plus dasatinib and/or nilotinib 25, 26, 27, 28. The current analysis from the same study presents for the first time the durability of response and long‐term safety of bosutinib in the fully enrolled advanced leukemia cohort. The data include a long‐term follow‐up to ≥4 years after the last patients’ first visit.
Methods
Patient characteristics
Eligible patients were aged ≥18 years with diagnosis of AP or BP CML or Ph+ ALL. AP CML was defined by ≥1 of the following in peripheral blood or bone marrow: 15% to 29% blasts; ≥30% blasts plus promyelocytes; ≥20% basophils; or <100 × 109/L platelets (unrelated to therapy). BP CML and ALL were characterized by ≥30% blasts in blood or bone marrow, or extramedullary disease involvement in organs other than liver/spleen. The most advanced historical CML diagnosis for each patient was considered for cohort allocation.
Eligible patients were resistant/intolerant to prior imatinib, as defined in Supporting Information Table 1, and were allowed to have received prior dasatinib and/or nilotinib. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 to 2, adequate hepatic and renal function, no prior antiproliferative treatment (except hydroxyurea or anagrelide) within 7 days of first dose, and ≥3 months since allo‐HSCT (if applicable). Patients with a documented T315I Bcr‐Abl mutation were allowed study entry until a protocol amendment (May 28, 2008) excluded such patients; patients already receiving treatment could remain on study if enrolled before the amendment or if a baseline sample subsequently tested positive for the T315I mutation.
Table 1.
Patient Demographic and Baseline Disease Characteristics
| AP CML | BP CMLa | ||||||
|---|---|---|---|---|---|---|---|
| Characteristic | 2L (n = 49) | ≥3L (n = 30) | Total (n = 79) | 2L (n = 36) | ≥3L (n = 28) | Total (n = 64) | ALL (n = 24) |
| Median (range) age (yr) | 49 (18–73) | 57 (21–83) | 51 (18–83) | 36.5 (19–75) | 52.5 (22–82) | 47 (19–82) | 59 (24–84) |
| Men, n (%) | 26 (53) | 18 (60) | 44 (56) | 25 (69) | 17 (61) | 42 (66) | 12 (50) |
| ECOG performance status, n (%) | |||||||
| 0 | 30 (61) | 15 (50) | 45 (57) | 16 (44) | 6 (21) | 22 (34) | 9 (38) |
| 1 | 18 (37) | 14 (47) | 32 (41) | 11 (31) | 18 (64) | 29 (45) | 10 (42) |
| 2 | 1 (2) | 1 (3) | 2 (3) | 9 (25) | 4 (14) | 13 (20) | 5 (21) |
| Median (range) WBC, 109/L | 13.0 (2.2–360.1) | 11.5 (0.5–153.0) | 11.6 (0.5–360.1) | 13.0 (0.7–87.5) | 18.8 (2.4–213.7) | 14.1 (0.7–213.7) | 7.9 (0.5–115.0) |
| Median (range) platelet count, 109/L | 309.0 (5.0–1939.0) | 161.5 (8.0–2189.0) | 246.0 (5.0–2189.0) | 68.7 (6.0–455.0) | 61.0 (3.0–1060.0) | 65.5 (3.0–1060.0) | 31.5 (5.0–670.0) |
| Median (range) duration of disease (yr) | 3.9 (1.1–22.1) | 8.2 (1.5–19.2) | 5.6 (1.1–22.1) | 2.3 (0.4–9.3) | 5.8 (1.1–14.5) | 3.3 (0.4–14.5) | 1.0 (0.1–20.0) |
| No. prior therapies,b n (%) | |||||||
| 1 (imatinib) | 29 (59) | 0 | 29 (37) | 30 (83) | 0 | 30 (47) | 15 (63) |
| 2 | 20 (41) | 5 (17) | 25 (32) | 6 (17) | 11 (39) | 17 (27) | 8 (33) |
| ≥3 | 0 | 25 (83) | 25 (32) | 0 | 17 (61) | 17 (27) | 1 (4) |
| Prior therapy, n (%) | |||||||
| Interferon, n (%) | 20 (41) | 21 (70) | 41 (52) | 6 (17) | 14 (50) | 20 (31) | 1 (4) |
| Imatinib, n (%) | 49 (100) | 30 (100) | 79 (100) | 36 (100) | 28 (100) | 64 (100) | 24 (100) |
| Median (range) durationc (mo) | 35.6 (0.6–108.3) | 35.6 (4.1–66.4) | 35.6 (0.6–108.3) | 13.4 (0.9–48.6) | 29.8 (1.0–62.6) | 21.2 (0.9–62.6) | 7.2 (0.5–55.8) |
| Reason for imatinib discontinuation, n (%) | |||||||
| Adverse event | 5 (10) | 6 (20) | 11 (14) | 4 (11) | 6 (21) | 10 (16) | 2 (8) |
| Disease progression/inadequate response | 44 (90) | 24 (80) | 68 (86) | 32 (89) | 22 (79) | 54 (84) | 22 (92) |
| Dasatinib, n (%) | 0 | 25 (83) | 25 (32) | 0 | 22 (79) | 22 (34) | 8 (33) |
| Median (range) duration (mo) | NA | 6.9 (0.1–30.4) | 6.9 (0.1–30.4) | NA | 7.0 (1.4–34.6) | 7.0 (1.4–34.6) | 7.4 (2.6–32.7) |
| Nilotinib, n (%) | 0 | 15 (50) | 15 (19) | 0 | 11 (39) | 11 (17) | 1 (4) |
| Median (range) duration (mo) | NA | 4.3 (0.8–34.0) | 4.3 (0.8–34.0) | NA | 1.0 (0.1–19.3) | 1.0 (0.1–19.3) | 0.4 (0.4–0.4) |
| Stem cell transplant, n (%) | 4 (8) | 3 (10) | 7 (9) | 1 (3) | 3 (11) | 4 (6) | 3 (13) |
Of 64 patients with BP CML, 10 had lymphoid BP, 23 had myeloid BP, and 31 had unspecified BP.
Includes TKI and interferon therapy.
Excludes 1 patient with unknown imatinib start and/or stop date.
2L, second‐line (prior imatinib only); ≥3L, third‐/fourth‐line (imatinib followed by dasatinib and/or nilotinib); AP, accelerated phase; CML, chronic myeloid leukemia; BP, blast phase; ALL, acute lymphoblastic leukemia; ECOG, Eastern Cooperative Oncology Group; WBC, white blood cell count.
All patients provided written informed consent before study entry. The study protocol was approved by institutional review boards at each site and conducted in accordance with the Declaration of Helsinki. This trial was registered at www.ClinicalTrials.gov (#NCT00261846).
Study design
This phase 1/2, open‐label, two‐part study evaluated oral bosutinib in Ph+ leukemia. Part 1 was a dose‐escalation study in primarily imatinib‐resistant patients with CP CML, which determined a recommended part 2 starting dose of bosutinib 500 mg/day 25. Part 2 examined the activity and tolerability of bosutinib in patients with CP CML 25, 26, 28 and advanced leukemia (AP CML, BP CML, ALL; described herein) with resistance/intolerance to prior imatinib (second‐line) and possibly dasatinib and/or nilotinib (≥third‐line). Dose escalation to bosutinib 600 mg/day was allowed for lack of efficacy (failure to reach CHR by week 8 or complete cytogenetic response [CCyR] by week 12) if no grade 3/4 bosutinib‐related toxicity had occurred. Other concomitant antileukemia therapy was prohibited; however, intrathecal cerebrospinal fluid prophylaxis/treatment was permitted during the treatment phase as clinically indicated.
Efficacy and safety assessments
Bone marrow examinations were performed at weeks 4, 8 (unless patient returned to CP), and 12; every 3 months until 2 years; and thereafter every 6 months. Complete blood count with differential was assessed at weeks 1, 2, 3, 4, 8, and 12; every 3 months until 2 years; and thereafter every 6 months. Disease status was not typically assessed after treatment discontinuation. Central laboratory sequencing was performed to identify Bcr‐Abl mutations at baseline and treatment completion; assay sensitivity was 20% in the background of non‐mutated cells.
The key endpoint was confirmed overall hematologic response (OHR) by week 48. Other endpoints included safety/tolerability, hematologic and cytogenetic responses, time to and duration of response, response by Bcr‐Abl mutational status, time to progressive disease (PD) or death, and overall survival (OS).
Hematologic responses were to be confirmed after ≥4 weeks of duration, with peripheral blood and bone marrow differential documentation and extramedullary disease assessment; for CHR, bone marrow differential documentation was required (Supporting Information Table 2). Standard criteria for cytogenetic response were employed 29. Postbaseline assessments required ≥20 metaphases. Fluorescence in situ hybridization (FISH) was used for postbaseline assessment (≥200 cells required) only if the bone marrow sample was inadequate for cytogenetic analysis (i.e., <20 metaphases); responses based on FISH were determined as indicated above except that <1% of positive cells were required for CCyR. To be considered responders, patients must have had improvement from baseline or maintained a baseline response for ≥5 weeks for hematologic response or ≥4 weeks for cytogenetic response.
Table 2.
Reasons for Treatment Discontinuation and Adverse Events Resulting in Dose Modification Over Time
| Year 1 | Year 2 | Year 3 | Year 4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AP CML | BP CML | ALL | AP CML | BP CML | ALL | AP CML | BP CML | ALL | AP CML | BP CML | ALL | |
| Reason for treatment discontinuation,a n (%) | (n = 79) | (n = 64) | (n = 24) | (n = 38) | (n = 8) | (n = 1) | (n = 19) | (n = 3) | (n = 1) | (n = 17) | (n = 2) | (n = 1) |
| Any reason | 41 (52) | 56 (88) | 23 (96) | 19 (50) | 5 (63) | 0 | 2 (11) | 1 (33) | 0 | 3 (18) | 0 | 0 |
| AE | 16 (20) | 2 (3) | 3 (13) | 4 (11) | 0 | 0 | 0 | 1 (33) | 0 | 1 (6) | 0 | 0 |
| Disease progression | 10 (13) | 29 (45) | 10 (42) | 11 (29) | 3 (38) | 0 | 0 | 0 | 0 | 2 (12) | 0 | 0 |
| Death | 5 (6) | 6 (9) | 2 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Symptomatic deterioration | 5 (6) | 6 (9) | 3 (13) | 1 (3) | 0 | 0 | 1 (5) | 0 | 0 | 0 | 0 | 0 |
| Unsatisfactory response | 3 (4) | 5 (8) | 5 (21) | 3 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Subject request | 1 (1) | 3 (5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Other | 1 (1) | 3 (5) | 0 | 0 | 1 (13) | 0 | 1 (5) | 0 | 0 | 0 | 0 | 0 |
| Investigator request | 0 | 0 | 0 | 0 | 1 (13) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Lost to follow‐up | 0 | 1 (2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Protocol violation | 0 | 1 (2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AEs resulting in dose modification, n (%) | ||||||||||||
| Discontinuationb | ||||||||||||
| Thrombocytopenia | 6 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pericardial effusion | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (12) | 0 | 0 |
| ALT increased | 2 (3) | 0 | 0 | 1 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dose interruptionc | ||||||||||||
| Thrombocytopenia | 20 (25) | 8 (13) | 0 | 1 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Neutropenia | 11 (14) | 6 (9) | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Anemia | 7 (9) | 3 (5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ALT increased | 6 (8) | 1 (2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AST increased | 5 (6) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Leukopenia | 4 (5) | 4 (6) | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Vomiting | 4 (5) | 3 (5) | 2 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Diarrhea | 3 (4) | 2 (3) | 1 (4) | 0 | 0 | 0 | 1 (5) | 0 | 0 | 0 | 0 | 0 |
| Nausea | 3 (4) | 2 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pyrexia | 3 (4) | 1 (2) | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rash | 4 (5) | 2 (3) | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (7) | 0 | 0 |
| Dose reductionc | ||||||||||||
| Thrombocytopenia | 14 (18) | 5 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Neutropenia | 5 (6) | 2 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Anemia | 3 (4) | 1 (2) | 0 | 1 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Leukopenia | 3 (4) | 1 (2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AST increased | 3 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rash | 3 (4) | 0 | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
1 year = 48 weeks.
In ≥2 patients (any cohort).
In ≥3 patients (any cohort).
AE, adverse event; ALL, acute lymphoblastic leukemia; ALT, alanine aminotransferase; AP, accelerated phase; AST, aspartate aminotransferase; BP, blast phase.
Progressive disease was defined as any of the following while receiving bosutinib: evolving from AP to BP on 2 consecutive assessments ≥1 week apart; increasing WBC (doubling over ≥1 month, with the second WBC >20 × 109/L and confirmed ≥1 week later); loss of confirmed CHR that is confirmed with a subsequent hematologic assessment ≥2 weeks after initial loss; loss of MCyR with Ph+ metaphases increased by ≥30%; and (for ALL) loss of previous hematologic response or complete remission (blasts >5% in bone marrow).
Adverse events (AEs) were assessed from the date of informed consent to 30 days after the last bosutinib dose and were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0.
Statistical analyses
Patients evaluable for efficacy had received ≥1 bosutinib dose and had a valid baseline assessment for the respective endpoint; patients evaluated for safety had received ≥1 bosutinib dose.
Efficacy was summarized using response rates, confidence intervals, and descriptive statistics. Time‐to‐event endpoints were estimated using the Kaplan‐Meier method (duration of response; OS) or cumulative incidence adjusting for the competing risk of treatment discontinuation without the event (PD/death, transformation). Two‐sided 95% confidence intervals (CI) were calculated based on the exact binomial for response rates and the Brookmeyer‐Crowley linear transformation method for Kaplan‐Meier quartiles. Kaplan‐Meier–estimated probabilities of maintaining responses at 1 and 4 years with the associated two‐sided 95% CI were calculated using Greenwood's formula; the cumulative incidence of on‐treatment PD/death at 1 and 4 years with two‐sided 95% CIs were calculated using Gray's method. Time to response was calculated from treatment initiation to earliest date of confirmed hematologic response or unconfirmed cytogenetic response. Duration of response among responders was calculated from the first date of response to confirmed loss, treatment discontinuation due to PD/death, or death within 30 days of the last bosutinib dose.
Time from first dose to PD/death was calculated from treatment initiation to earliest documented progression (per investigator), treatment discontinuation due to death, or death within 30 days of the last dose. OS was calculated from treatment initiation to date of death, with patients followed for survival for 2 years after treatment discontinuation and patients without events censored at the last known alive date. For time‐to‐event endpoints (except OS), patients without events were censored at the last follow‐up visit for the respective endpoint.
Results
Patients and treatment summary
Among 167 patients with advanced leukemia included in the safety population, all had received prior imatinib, 55 (33%) dasatinib, 27 (16%) nilotinib, and 15 (9%) patients had prior exposure to all three TKIs (Table 1 ).
As of the data cutoff (May 23, 2014) based on an unlocked database for this interim manuscript, 14 (18%) AP CML, 2 (3%) BP CML, and 1 (4%) ALL patient was still receiving bosutinib at 4 years, compared with 38 (48%), 8 (13%), and 1 (4%) patient at 1 year; 1 year = 48 weeks). Time from last enrolled patient's first dose to the data cutoff for AP CML, BP CML, and ALL cohorts was 49.2, 54.5, and 78.5 months, respectively; median (range) duration of follow‐up from bosutinib initiation to last contact was 28.4 (0.3–88.6), 10.4 (0.4–79.9), and 3.6 (0.4–89.2) months. Most permanent treatment discontinuations (72% [n = 120]) occurred within the first year of treatment; fewer patients in the AP CML cohort permanently discontinued (52% [n = 41]) within the first year compared with patients in the BP CML (88% [n = 56]) and ALL (96% [n = 23]) cohorts (Table 2 ). Across cohorts, the most common primary reasons (≥5% of patients) for treatment discontinuation (≤1 year vs. >1 to 4 years) for those on‐treatment during those years were PD (29% vs. 34%), AEs (13% vs. 13%), unsatisfactory response (8% vs. 6%), symptom deterioration (8% vs. 4%), and death (8% vs. 0%).
The median (range) duration of bosutinib treatment for AP CML, BP CML, and ALL was 10.2 (0.1–88.6), 2.8 (0.03–55.9), and 0.97 (0.3–89.2) months; median treatment duration was longer for second‐line patients versus ≥third‐line patients (AP, 11.5 vs. 6.9 months; BP, 3.31 vs. 1.8 months). Among AP CML, BP CML, and ALL cohorts, respectively, AEs were managed by treatment interruption (47 [59%], 28 [44%], 10 [42%], respectively) and/or dose reductions (38 [48%], 17 [27%], 1 [4%]); dose escalation to 600 mg/day (due to lack of efficacy) occurred in 14 (18%), 16 (25%), and 5 (21%) patients with initial dose <600 mg/day.
Efficacy
Among 72 evaluable patients with AP CML, confirmed OHR by week 48 (key endpoint) was newly attained or maintained by 41 (57%) patients (Table 3), including 32 (44%) with a major hematologic response (MHR) and 22 (31%) with CHR. Among AP CML patients, 41 (57%), 34 (47%), and 24 (33%), respectively, attained/maintained an OHR, MHR, or CHR by 4 years. Among patients without a CHR at baseline (n = 63), 18 (29%) achieved a CHR on bosutinib, all within the first 2 years; among patients with a CHR at baseline (n = 9), 6 (67%) maintained a CHR on bosutinib. Median (95% CI) duration of OHR in responders was 119.9 (63.1 – not reached [NR]) weeks, with Kaplan‐Meier–estimated probabilities (95% CI) of 78% (59%–89%) for maintaining an OHR at 1 year versus 49% (30%–66%) at 4 years (Fig. 1A). Among evaluable patients without a CCyR at baseline (n = 69), 26 (38%) achieved MCyR and 19 (28%) achieved CCyR by 4 years; the three AP CML patients with a CCyR at baseline maintained CCyR on bosutinib. Median (95% CI) duration of MCyR among responders was 84.0 (47.3–NR) weeks, with Kaplan‐Meier–estimated probabilities (95% CI) for maintaining an MCyR of 65% (43%–80%) at 1 year versus 49% (28%–66%) at 4 years (Fig. 1B). Hematologic and cytogenetic response rates appeared higher in the second‐line versus ≥third‐line AP CML cohort (Table 3).
Table 3.
Response to Therapy by Disease State and Prior Treatment With TKIs
| AP CML | BP CML | ||||||
|---|---|---|---|---|---|---|---|
| Response, n (%) | 2L (n = 49) | ≥3L (n = 30) | Total (n = 79) | 2L (n = 36) | ≥3L (n = 28) | Total (n = 64) | ALL (n = 24) |
| Week 48 hematologic responsea | |||||||
| Evaluable patientsb | 43 | 29 | 72 | 34 | 26 | 60 | 22 |
| OHR [95% CI] | 29 (67) [52–81] | 12 (41) [24–61] | 41 (57) [45–69] | 13 (38) [22–56] | 4 (15) 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 17 (28) 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41 | 1 (5) [<1–23] |
| Cumulative hematologic response by 4 yrs | |||||||
| Evaluable patientsa ,b | 43 | 29 | 72 | 34 | 26 | 60 | 22 |
| OHR [95% CI] | 29 (67) [52%–81%] | 12 (41) [24%–61%] | 41 (57) [45%–69%] | 13 (38) [22%–56%] | 4 (15) [4%–35%] | 17 (28) [18%–41%] | 2 (9) [1%–29%] |
| MHR | 23 (54) | 11 (38) | 34 (47) | 9 (27) | 2 (8) | 11 (18) | 2 (9) |
| CHR | 17 (40) | 7 (24) | 24 (33) | 9 (27) | 1 (4) | 10 (17) | 2 (9) |
| Median [range] time to OHRc (wk) | 12.0 [3.7–49.0] | 12.1 [4.0–24.0] | 12.0 [3.7–49.0] | 8.0 [4.0–12.0] | 11.6 [4.1–12.1] | 8.9 [4.0–12.1] | 43.6 [4.0–83.1] |
| Evaluable patients without a CHR at baseline | 39 | 24 | 63 | 32 | 23 | 55 | 21 |
| OHR | 22 (57) | 7 (29) | 29 (46) | 9 (28) | 1 (4) | 10 (18) | 1 (5) |
| Cumulative cytogenetic response by 4 years | |||||||
| Evaluable patientsa ,d | 46 | 26 | 72 | 30 | 24 | 54 | 20 |
| MiCyR | 3 (7) | 0 | 3 (4) | 1 (3) | 1 (4) | 2 (4) | 0 |
| MCyR [95% CI] | 22 (48)d [33%–63%] | 7 (27) [12%–48%] | 29 (40)d [29%–53%] | 15 (50) [31%–69%] | 5 (21)e [7%–42%] | 20 (37)e [24%–51%] | 4 (20) [6%–44%] |
| CCyR | 16 (35) | 6 (23) | 22 (31) | 11 (37) | 4 (17) | 15 (28) | 4 (20) |
| Median [range] time to MCyRc (wk) | 12.3 [3.9–42.0] | 144.7 [4.0–144.7] | 24.0 [3.9–144.7] | 12.1 [3.9–25.1] | NR [4.1–12.3] | 12.1 [3.9–25.1] | 9.0 [4.0–12.0] |
| Evaluable patients without a CCyR at baseline | 46 | 23 | 69 | 28 | 22 | 50 | 18 |
| MCyR | 22 (48) | 4 (17) | 26 (38) | 13 (46) | 3 (14) | 16 (32) | 3 (17) |
To be considered a responder, the patient must have improved from their baseline assessment or maintained their baseline response.
Evaluable patients included those who received ≥1 dose of bosutinib and had a valid baseline hematologic measurement.
Time to response was calculated among responders from first study dosing to the earliest date of response (confirmed response for OHR and unconfirmed response for MCyR).
Evaluable patients included those who received ≥1 dose of bosutinib and had a valid baseline cytogenetic measurement.
Included four patients with a partial cytogenetic response determined by using FISH instead of cytogenetic analysis.
Included one patient with CCyR determined by using FISH instead of cytogenetic analysis.
2L, second‐line (prior imatinib only); ≥3L, third‐/fourth‐line (imatinib followed by dasatinib and/or nilotinib); AP, accelerated phase; CML, chronic myeloid leukemia; BP, blast phase; ALL, acute lymphoblastic leukemia; OHR, overall hematologic response; CI, confidence interval; CHR, complete hematologic response; MiCyR, minor cytogenetic response; MCyR, major cytogenetic response; CCyR, complete cytogenetic response.
Figure 1.
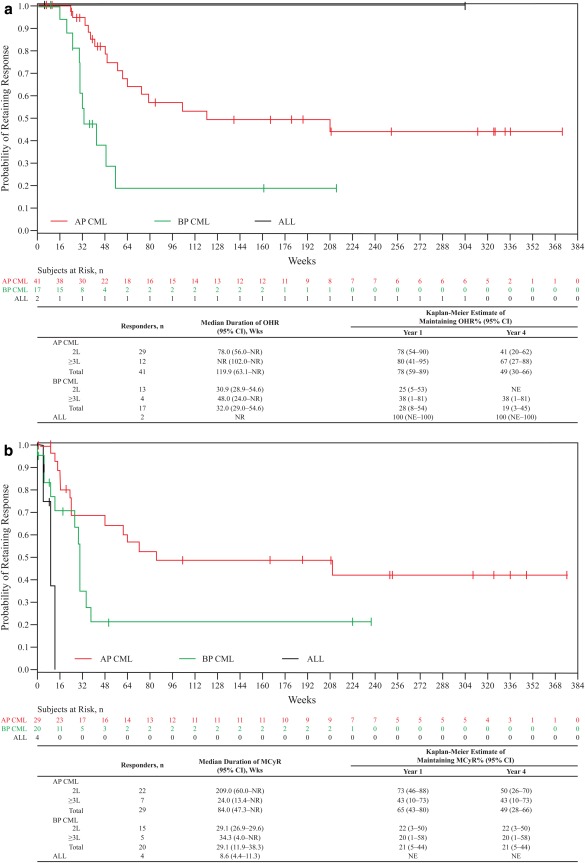
Duration of OHR (A) and MCyR (B). Duration of response was calculated from the first date of response to confirmed loss, treatment discontinuation due to progressive disease or death, or death within 30 days of the last dose of study drug; responders without events were censored at the last follow‐up visit. 2L, Second‐line (prior imatinib only); ≥3L, third‐/fourth‐line (imatinib followed by dasatinib and/or nilotinib); ALL, acute lymphoblastic leukemia; AP, accelerated phase; BP, blast phase; CI, confidence interval; CML, chronic myeloid leukemia; MCyR, major cytogenetic response; NR, not reached; NE, not evaluable; OHR, overall hematologic response.
Among 60 evaluable patients with BP CML, a confirmed OHR by week 48 was newly attained/maintained by 17 (28%) patients (Table 3), including 11 (18%) with an MHR and 10 (17%) with a CHR; no patient had an initial response after week 48. Among patients without a CHR at baseline (n = 55), 7 (13%) achieved a CHR on bosutinib, all approximately within the first 12 months; among patients with a CHR at baseline (n = 5), 3 (60%) maintained a CHR on bosutinib. Median (95% CI) duration of OHR among responders was 32 (29.0–54.6) weeks, with Kaplan‐Meier–estimated probabilities (95% CI) of maintaining response of 28% (8%–54%) at 1 year versus 19% (3%–45%) at 4 years (Fig. 1A). Among patients without a CCyR at baseline (n = 50), 16 (32%) achieved an MCyR and 11 (22%) achieved a CCyR by 4 years; all four patients with a CCyR at baseline maintained a CCyR on bosutinib. Median (95% CI) duration of MCyR among responders was 29.1 (11.9–38.3) weeks for patients with BP CML, with similar Kaplan‐Meier–estimated probabilities (95% CI) of maintaining an MCyR at 1 year and at 4 years (both 21% [5%–44%]; Fig. 1B). Hematologic and cytogenetic response rates appeared higher for the second‐line versus ≥third‐line BP CML cohort (Table 3).
Among 22 evaluable patients with ALL, 2 (9%) newly attained or maintained a confirmed OHR (both CHR) by 4 years (Table 3). One of the patients had a CHR at baseline and maintained CHR on bosutinib, with a duration of response of 304.3+ weeks (patient discontinued the study to enroll in bosutinib extension study, NCT01903733). This patient also had a CCyR for 327 weeks, but was not considered evaluable for cytogenetic response due to an invalid baseline cytogenetic assessment. The other patient did not have a CHR at baseline and newly achieved a CHR on bosutinib (duration of response, 3.9 weeks [discontinued treatment due to symptomatic deterioration]). Among patients without a CCyR at baseline (n = 18), three (17%) achieved MCyR (all CCyR); between the two patients who had a CCyR at baseline, one had retained a CCyR on bosutinib. The median (95% CI) duration of MCyR among responders was 8.6 (4.4–11.3) weeks for patients with ALL.
BCR‐ABL mutations
Sixty‐two (78%) AP, 53 (83%) BP, and 13 (54%) ALL patients had mutation assessment at baseline. There were 16, 13, and 5 unique mutations in 32 (52%) AP, 28 (53%) BP, and 7 (54%) ALL patients, including 2 (3%) AP and 6 (11%) BP patients who had ≥2 mutations. Mutations occurring in more than five patients (all cohorts) included T315I (AP, n = 3; BP, n = 10; ALL, n = 3), F317L (AP, n = 4; BP, n = 4; ALL, n = 1), G250E (AP, n = 4; BP, n = 2; ALL, n = 1), and Y253H (AP, n = 3; BP, n = 4; ALL, n = 0). In the AP CML cohort, OHR and MCyR rates were 57% (n = 16/28) and 39% (n = 11/28) in patients with ≥1 mutation (62% [n = 16/26] and 44% [n = 11/25], respectively, excluding T315I), versus 62% (n = 18/29) and 43% (n = 12/28) without a mutation. Among the 11 AP patients with ≥1 baseline mutation who had an MCyR, 10 newly attained an MCyR whereas only 1 (with a G321R mutation) maintained an MCyR from baseline while on bosutinib treatment. In the BP CML cohort, OHR and MCyR rates were 27% (n = 7/26) and 17% (n = 4/23) in patients with ≥1 mutation (35% [n = 6/17] and 20% [n = 3/15], respectively, excluding T315I), versus 25% (n = 6/24) and 45% (n = 9/20) without a mutation. None of the seven ALL patients with ≥1 mutation (including the three patients with T315I) achieved an OHR or MCyR, compared with 40% (n = 2/5) and 50% (n = 2/4) of ALL patients without a mutation. Responses were broadly achieved across baseline Bcr‐Abl kinase domain mutations for AP and BP CML patients, except for patients with T315I for whom only one response was achieved (Fig. 2).
Figure 2.
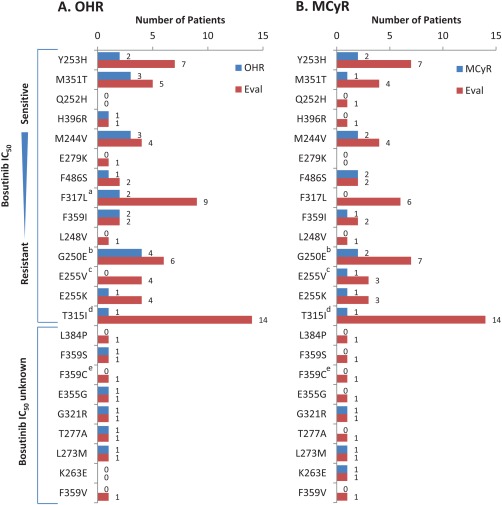
OHR (A) and MCyR (B) by individual baseline Bcr‐Abl mutations in AP, BP, and ALL cohorts. Individual patients may have had more than one detected mutation. Bosutinib IC50 concentrations were based on data from 34. aIncludes one evaluable ALL patient with a F317L mutation who did not achieve a response; bincludes one evaluable ALL patient with a G250E mutation who did not achieve a response; cincludes one evaluable ALL patient with a E255V mutation who did not achieve a response; dincludes three evaluable ALL patients with a T315I mutation who did not achieve a response; eincludes one evaluable ALL patient with a F359C mutation who did not achieve a response. Eval, number of patients with each baseline mutation who had a valid baseline efficacy assessment for the respective endpoint; IC50, half‐maximal inhibitory concentration; MCyR, major cytogenetic response; OHR, overall hematologic response.
Among 48 patients who were assessed for mutations at baseline and on‐treatment, 14 had an emergent mutation at treatment discontinuation (AP CML, 4/24 [17%]; BP CML, 9/21 [43%]; ALL, 1/3 [33%]). The most frequently detected emergent mutations were T315I (n = 6) and V299L (n = 5); reasons for discontinuation among these patients were PD (n = 7), unsatisfactory response (n = 1 [T315I]), AE (n = 1 [T315I]), and patient request to switch treatment (n = 1 [T315I]). One BP CML patient with an emergent V299L mutation also had a F311L mutation; this patient discontinued at the request of the investigator. F317L emerged in two patients, one of whom also had a G250E mutation and discontinued treatment due to an AE; the other patient discontinued due to PD. An A365G mutation was detected in one patient who discontinued treatment due to PD.
Long‐term outcomes
The cumulative incidence of on‐treatment PD/death (95% CI) by 1 year versus 4 years in the AP CML, BP CML, and ALL cohorts, respectively, were 24% (16%–36%) versus 38% (29%–50%), 58% (47%–71%) versus 61% (50%–74%), and 54% (38%–78%) versus 54% (38%–78%), respectively; 44%, 36%, and 42% discontinued without on‐treatment PD/death before year 4 (Fig. 3A). The cumulative incidence (95% CI) of on‐treatment transformation to BP CML in the AP CML cohort was 4% (1%–12%); 79% discontinued without on‐treatment transformation before year 4. Three AP CML patients had on‐treatment transformation to BP CML (two patients with second‐line and one with ≥third‐line bosutinib), which occurred 164, 315, and 744 days after treatment initiation.
Figure 3.
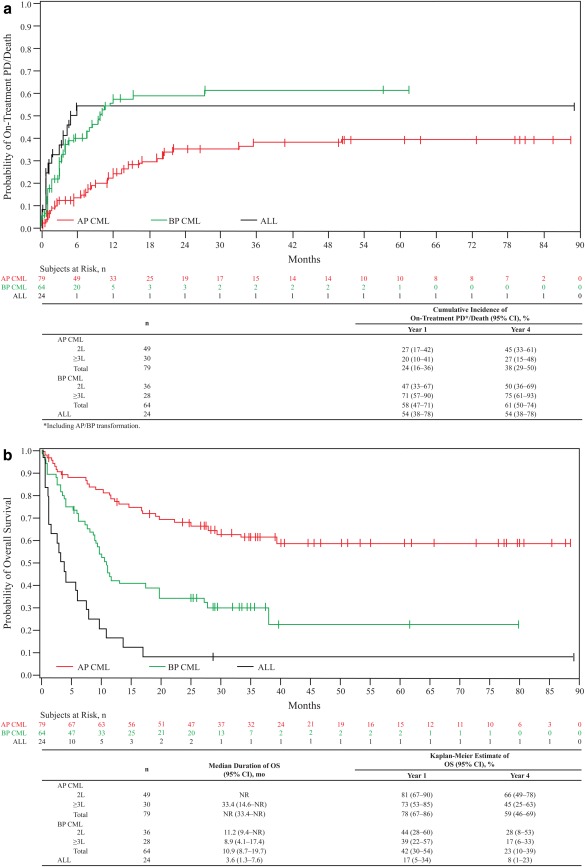
Cumulative incidence of PD/death adjusting for the competing risk of treatment discontinuation without PD/death (A) and overall survival (B). Criteria for PD included transformation to AP/BP CML, increasing white blood cell count (doubling over ≥1 month with second count >20 × 109/L and confirmed ≥1 week later, or loss of confirmed CHR or unconfirmed MCyR. OS was calculated from the date of first study dosing to the date of death, with patients without events censored at the last contact, and was evaluated throughout the 2‐year follow‐up period after treatment discontinuation. The median follow‐up was 28.4 (0.3–88.6), 10.4 (0.4–79.9), and 3.6 (0.4–89.2) months for AP, BP, and ALL patients, respectively. 2L, Second‐line (prior imatinib only); ≥3L, third‐/fourth‐line (imatinib followed by dasatinib and/or nilotinib); ALL, acute lymphoblastic leukemia; AP, accelerated phase; BP, blast phase; CI, confidence interval; CML, chronic myeloid leukemia; NR, not reached; OS, overall survival; Ph+, Philadelphia chromosome‐positive; PD, progressive disease.
As of the data cutoff, a total of 30 (38%) treated patients with AP CML had died (11 within 30 days of the last bosutinib dose); median OS had not yet been reached; Kaplan‐Meier–estimated OS (95% CI) was 78% (67%–86%) at 1 year and 59% (46%–69%) at 4 years with 38% censored before year 4 (Fig. 3B). Among treated patients with BP CML, 44 (69%) had died (13 within 30 days of the last dose), with a median OS (95% CI) of 10.9 (8.7–19.7) months; Kaplan‐Meier–estimated OS (95% CI) was 42% (30%–54%) at 1 year and 23% (10%–39%) at 4 years with 28% censored before year 4. Among treated patients with ALL, 22 (92%) had died (8 within 30 days of the last dose), with a median OS (95% CI) of 3.6 (1.3–7.6) months; Kaplan‐Meier–estimated OS (95% CI) at 4 years was 8.3% (1%–23%) with one patient censored before year 4. Across cohorts, 32 (19%) patients died within 30 days of their last bosutinib dose; reasons included disease progression (n = 19), AE unrelated to treatment (n = 11), and AE considered to be related to bosutinib (n = 2; myocardial infarction and acidosis, both occurring in year 1). Among the 11 patients who died within 30 days of their last bosutinib dose due to AEs considered unrelated to treatment, 6 deaths were due to cardiac or vascular AEs, including left intraventricular hemorrhage (BP CML), posterior cerebral artery distribution infarct (ALL), congestive heart failure (BP CML), cerebral hemorrhage (ALL), coronary artery disease (AP CML), and brain vascular accident (BP CML).
Safety and tolerability
The most common treatment‐emergent adverse events (TEAEs) occurring in ≥30% of all advanced leukemia patients were gastrointestinal TEAEs, which were primarily of low grade (diarrhea [any grade, 74%; maximum grade 1/2, 69%], nausea [48%; 46%], and vomiting [44%; 40%]; Table 4). Other frequently occurring TEAEs (any grade; grade 3/4) were thrombocytopenia (45%; 40%), anemia (39%; 26%), pyrexia (38%; 2%), and rash (31%; 4%). No notable differences in TEAE incidence were observed between cohorts (Table 4), and there was no clear pattern of differences in the incidences of TEAEs according to prior treatment (data not shown).
Table 4.
Incidence of TEAEs, any Causality
| AP CML (n = 79) | BP CML (n = 64) | ALL (n = 24) | Total (n = 167) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Event,a n (%) | All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | |||
| Nonhematologic TEAEs occurring in ≥10% (any grade) of patients (any cohort) | |||||||||||
| Diarrhea | 67 (85) | 3 (4) | 41 (64) | 3 (5) | 15 (63) | 1 (4) | 123 (74) | 7 (4) | |||
| Nausea | 36 (46) | 2 (3) | 32 (50) | 1 (2) | 12 (50) | 0 | 80 (48) | 3 (2) | |||
| Vomiting | 35 (44) | 3 (4) | 27 (42) | 2 (3) | 11 (46) | 1 (4) | 73 (44) | 6 (4) | |||
| Pyrexia | 28 (35) | 1 (1) | 25 (39) | 2 (3) | 11 (46) | 1 (4) | 64 (38) | 4 (2) | |||
| Rash | 27 (34) | 3 (4) | 20 (31) | 2 (3) | 4 (17) | 1 (4) | 51 (31) | 6 (4) | |||
| Abdominal pain | 21 (27) | 3 (4) | 12 (19) | 2 (3) | 3 (13) | 0 | 36 (22) | 5 (3) | |||
| Fatigue | 17 (22) | 4 (5) | 12 (19) | 3 (5) | 5 (21) | 0 | 34 (20) | 7 (4) | |||
| Headache | 12 (15) | 2 (3) | 13 (20) | 4 (6) | 6 (25) | 1 (4) | 31 (19) | 7 (4) | |||
| Cough | 24 (30) | 0 | 8 (13) | 0 | 1 (4) | 0 | 33 (20) | 0 | |||
| Dyspnea | 15 (19) | 7 (9) | 12 (19) | 2 (3) | 4 (17) | 0 | 31 (19) | 9 (5) | |||
| Constipation | 14 (18) | 0 | 9 (14) | 1 (2) | 4 (17) | 0 | 27 (16) | 1 (1) | |||
| Arthralgia | 12 (15) | 0 | 8 (13) | 0 | 3 (13) | 1 (4) | 23 (14) | 1 (1) | |||
| Pneumonia | 10 (13) | 9 (11) | 9 (14) | 5 (8) | 3 (13) | 3 (13) | 22 (13) | 17 (10) | |||
| Dizziness | 11 (14) | 1 (1) | 8 (13) | 0 | 2 (8) | 0 | 21 (13) | 1 (1) | |||
| Decreased appetite | 7 (9) | 0 | 12 (19) | 0 | 1 (4) | 0 | 20 (12) | 0 | |||
| Asthenia | 10 (13) | 1 (1) | 4 (6) | 0 | 5 (21) | 0 | 19 (11) | 1 (1) | |||
| Pain in extremity | 9 (11) | 1 (1) | 6 (9) | 0 | 3 (13) | 0 | 18 (11) | 1 (1) | |||
| Elevated ALT | 12 (15) | 6 (8) | 4 (6) | 1 (2) | 2 (8) | 0 | 18 (11) | 7 (4) | |||
| Elevated AST | 12 (15) | 4 (5) | 4 (6) | 0 | 1 (4) | 1 (4) | 17 (10) | 5 (3) | |||
| Abdominal pain upper | 10 (13) | 1 (1) | 7 (11) | 2 (3) | 0 | 0 | 17 (10) | 3 (2) | |||
| Back pain | 8 (10) | 1 (1) | 4 (6) | 1 (2) | 4 (17) | 1 (4) | 16 (10) | 3 (2) | |||
| Insomnia | 8 (10) | 0 | 5 (8) | 0 | 2 (8) | 0 | 15 (9) | 0 | |||
| Pleural effusion | 10 (13) | 4 (5) | 3 (5) | 2 (3) | 2 (8) | 1 (4) | 15 (9) | 7 (4) | |||
| Peripheral edema | 6 (8) | 0 | 5 (8) | 1 (2) | 4 (17) | 0 | 15 (9) | 1 (1) | |||
| Chest pain | 8 (10) | 2 (3) | 4 (6) | 0 | 2 (8) | 0 | 14 (8) | 2 (1) | |||
| Oropharyngeal pain | 8 (10) | 0 | 3 (5) | 0 | 2 (8) | 0 | 13 (8) | 0 | |||
| Anxiety | 8 (10) | 0 | 3 (5) | 0 | 0 | 0 | 11 (7) | 0 | |||
| Bone pain | 1 (1) | 0 | 7 (11) | 2 (3) | 3 (13) | 1 (4) | 11 (7) | 3 (2) | |||
| Dyspepsia | 9 (11) | 0 | 1 (2) | 0 | 1 (4) | 0 | 11 (7) | 0 | |||
| Upper respiratory tract infection | 8 (11) | 0 | 2 (3) | 0 | 0 | 0 | 10 (6) | 0 | |||
| Edema | 5 (6) | 0 | 2 (3) | 0 | 3 (13) | 0 | 10 (6) | 0 | |||
| Chills | 3 (4) | 0 | 2 (3) | 0 | 4 (17) | 1 (4) | 9 (5) | 1 (1) | |||
| Petechiae | 1 (1) | 0 | 3 (5) | 0 | 3 (13) | 0 | 7 (4) | 0 | |||
| Hematoma | 1 (1) | 0 | 0 | 0 | 3 (13) | 0 | 4 (2) | 0 | |||
| Hematologic TEAEsb occurring in ≥10% (any grade) of patients (any cohort) | |||||||||||
| Thrombocytopenia | 42 (53) | 35 (44) | 24 (38) | 23 (36) | 9 (38) | 8 (33) | 75 (45) | 66 (40) | |||
| Anemia | 36 (46) | 26 (33) | 19 (30) | 13 (20) | 10 (42) | 4 (17) | 65 (39) | 43 (26) | |||
| Neutropenia | 15 (19) | 14 (18) | 17 (27) | 16 (25) | 5 (21) | 4 (17) | 37 (22) | 34 (20) | |||
| Leukopenia | 10 (13) | 5 (6) | 12 (19) | 12 (19) | 3 (13) | 3 (13) | 25 (15) | 20 (12) | |||
| Leukocytosis | 5 (6) | 3 (4) | 6 (9) | 2 (3) | 4 (17) | 2 (8) | 15 (9) | 7 (4) | |||
| Febrile neutropenia | 1 (1) | 1 (1) | 3 (5) | 2 (3) | 4 (17) | 4 (17) | 8 (5) | 7 (4) | |||
| Vascular TEAEs occurring in ≥1 patient (any cohort)c | |||||||||||
| Hypertension | 7 (9) | 4 (5) | 2 (3) | 1 (2) | 2 (8) | 0 | 11 (7) | 5 (3) | |||
| Cerebral hemorrhage | 0 | 0 | 1 (2) | 0 | 1 (4) | 0 | 2 (1) | 0 | |||
| Subarachnoid hemorrhage | 1 (1) | 1 (1) | 1 (2) | 0 | 0 | 0 | 2 (1) | 1 (1) | |||
| Blood pressure increased | 1 (1) | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 | |||
| Cerebral artery occlusion | 1 (1) | 1 (1) | 0 | 0 | 0 | 0 | 1 (1) | 1 (1) | |||
| Cerebral infarction | 0 | 0 | 0 | 0 | 1 (4) | 1 (4) | 1 (1) | 1 (1) | |||
| Cerebrovascular accident | 0 | 0 | 1 (2) | 0 | 0 | 0 | 1 (1) | 0 | |||
| Intraventricular hemorrhage | 0 | 0 | 1 (2) | 1 (2) | 0 | 0 | 1 (1) | 1 (1) | |||
| Ischemic stroke | 1 (1) | 1 (1) | 0 | 0 | 0 | 0 | 1 (1) | 1 (1) | |||
| Raynaud's phenomenon | 0 | 0 | 1 (2) | 0 | 0 | 0 | 1 (1) | 0 | |||
| Thrombosis | 0 | 0 | 1 (2) | 0 | 0 | 0 | 1 (1) | 0 | |||
National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0.
Combined with similar MeDRA Preferred Terms from the Investigations System Organ Class (Platelet count decreased, Hemoglobin decreased, Neutrophil count decreased, White blood cell count decreased).
See Supporting Information Table 3 for definition of vascular TEAEs.
AP, accelerated phase; CML, chronic myeloid leukemia; BP, blast phase; ALL, acute lymphoblastic leukemia; TEAE, treatment‐emergent adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Overall, serious AEs (SAEs) occurred in 59% (99/167) of patients; the most frequently occurring individual SAEs (≥5% of patients overall) included pneumonia (10%), pyrexia (7%), febrile neutropenia (6%), thrombocytopenia (6%), disease progression (5%), headache (5%), and pleural effusion (5%). Most newly occurring SAEs (i.e., those not experienced by the same patient in previous years for patients on‐treatment during that specific year) were most common in year 1 (91/167 [55%]); incidences in years 2, 3, and 4 were 11/40 (28%), 4/23 (17%), and 5/18 (28%), respectively (Table 5 ). The only newly occurring individual SAE reported in more than two patients within year 2, 3, or 4 was pneumonia (three patients with events in year 2).
Table 5.
Incidence of Newly Occurring Serious AEs, any Causality Over Timea
| Year 1 | Year 2 | Year 3 | Year 4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AP CML (n = 79) | BP CML (n = 64) | ALL (n = 24) | AP CML (n = 34) | BP CML (n = 5) | ALL (n = 1) | AP CML (n = 19) | BP CML (n = 3) | ALL (n = 1) | AP CML (n = 15) | BP CML (n = 2) | ALL (n = 1) | |
| Any AE,b n (%) | 36 (46) | 37 (58) | 18 (75) | 10 (29) | 1 (20) | 0 | 3 (16) | 1 (33) | 0 | 5 (33) | 0 | 0 |
| Thrombocytopenia | 6 (8) | 1 (2) | 3 (13) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| General physical health deterioration | 1 (1) | 3 (5) | 2 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Anemia | 5 (6) | 1 (2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Febrile neutropenia | 1 (1) | 4 (6) | 4 (17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Leukocytosis | 1 (1) | 2 (3) | 2 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nausea | 0 | 5 (8) | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Vomiting | 0 | 4 (6) | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Disease progression | 5 (6) | 3 (5) | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pyrexia | 4 (5) | 6 (9) | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pneumonia | 6 (8) | 5 (8) | 3 (13) | 3 (9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Headache | 4 (5) | 2 (3) | 2 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pleural effusion | 3 (4) | 2 (3) | 1 (4) | 0 | 0 | 0 | 1 (5) | 0 | 0 | 1 (7) | 0 | 0 |
| Respiratory failure | 1 (1) | 2 (3) | 2 (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Newly occurring serious AEs refers to those not experienced by the same patient in previous years for patients on‐treatment during that specific year (1 year = 365.25 days).
In ≥5 patients in the safety population (n = 167) in year 1, or in ≥2 patients in years 2, 3, or 4.
AE, adverse event; AP, accelerated phase; CML, chronic myeloid leukemia; BP, blast phase; ALL, acute lymphoblastic leukemia.
Across the cohorts (n = 167), patients with bosutinib toxicities were frequently managed with dose interruption (51% [n = 85]) and/or dose reduction (34% [n = 56]); some variability was observed across cohorts. In general, most dose interruptions and reductions due to AEs occurred during the first year of treatment (Table 2 ). Thrombocytopenia was the most common TEAE leading to dose interruption (n = 29 [17%] in total; n = 28/167 [17%] in year 1) or dose reduction (n = 19 [11%] in total; n = 19 [11%] in year 1). Adverse events as the primary reason for discontinuation occurred more frequently in AP CML (30% [n = 24]) versus the BP CML (6% [n = 4]) and ALL (13% [n = 3]) patients including discontinuations after year 4. Across the cohorts, the most common AE leading to treatment discontinuation was thrombocytopenia (n = 6); in general, most discontinuations due to AEs occurred during the first year of treatment (Table 2 ).
Diarrhea AEs (defined in Supporting Information Table 3) were typically of mild severity (maximum grade 1/2 severity in 69% of patients [AP: 81%; BP: 59%; ALL: 58%]), with first events occurring early (median [range] time to first event, 2 [1–209] days), and transient (median [range] any grade event duration, 2 [1–910] days). Diarrhea was primarily managed with concurrent medication (62% of affected patients used loperamide or similar medication) and less frequently with dose interruption (n = 7 [6% of patients with events]) or dose reduction (n = 3 [2% of patients with events]). Most nausea cases were mild (maximum grade 1/2 severity in 46% of patients), with first event occurring early (median [range] time to first event, 4.0 [1–1113] days), and had limited duration (median [range] any grade event duration, 7.0 [1–274] days). No newly occurring AEs of diarrhea were reported after year 1; newly occurring nausea was reported in only two patients after year 1 (1 BP patient in year 2 and 1 AP patient in year 4). No patient discontinued bosutinib due to either of these AEs, underscoring the manageability of these common toxicities.
Pleural effusions of any grade were observed in 15 (9%) patients (grade 3/4 in seven [4%] patients; five considered treatment‐related by the investigator). Ten of the 15 patients experienced pleural effusion in year 1; newly occurring AEs of pleural effusion after year 1 were reported in five AP CML patients (1 in year 2, 3 in year 3, and 1 in year 4). One AP CML patient discontinued bosutinib due to pleural and pericardial effusion TEAEs. Of the 15 patients with a pleural effusion on bosutinib, 6 had received prior dasatinib (representing 11% of all patients with prior dasatinib exposure); all 6 patients had a history of pleural effusion, and for 3 of these patients, pleural effusion was indicated as the reason for dasatinib intolerance. Pneumonia was reported as a TEAE by 22 (13%) patients (n = 19/167 [11%] in year 1; n = 3/40 [8%] newly occurring in year 2) and was the most common nonhematologic grade 3/4 TEAE (n = 17 [10%]; 3 [2%] treatment‐related pneumonia); three patients experienced concurrent pleural effusion and grade 3/4 pneumonia.
Liver‐related TEAEs (defined in Supporting Information Table 3) occurred in 35 (21%) patients, with similar incidences across cohorts (AP, 23% [n = 18]; BP, 20% [n = 13]; ALL, 17% [n = 4]); most newly occurring liver‐related AEs were reported in year 1 (n = 34/167 [20%] in year 1; n = 3/40 [8%] in year 2). Liver‐related TEAEs most commonly included alanine aminotransferase elevation (ALT, n = 18 [11%]; all initially reported in year 1) and aspartate aminotransferase elevation (AST, n = 17 [10%] in total; n = 15 [9%] in year 1); 11 of these patients experienced both ALT and AST elevations. Seventeen (22%) AP CML, 8 (13%) BP CML, and 1 (4%) ALL patients experienced treatment‐related liver TEAEs. Across cohorts, maximum grade 1/2 liver TEAEs were experienced by 12% of patients, while 9% experienced a maximum grade 3 event; there were no grade 4 events. Only three patients discontinued treatment due to a liver‐related TEAE (all AP CML). Among AP, BP, and ALL cohorts, respectively, the median (range) time to first liver TEAE was 21.5 (3–168), 33.0 (2–421), and 9.5 (1–15) days; the median (range) duration of an individual liver TEAE (any grade) was 17.0 (1–252), 9.0 (1–441), and 5.0 (1–9) days. Of nine patients with a treatment interruption due to a liver TEAE, six were successfully rechallenged with bosutinib and two were rechallenged but subsequently discontinued due to elevated alkaline phosphatase/ALT and ALT/AST. One patient with AP CML discontinued treatment without rechallenge due to recurrent treatment‐related grade 3 elevated ALT.
Overall, cardiac TEAEs (defined in Supporting Information Table 3) were experienced by 25 (15%) patients across cohorts. Cardiac disorders (in more than two patients in any cohort) included pericardial effusion (AP, n = 5 [four also had pleural effusion]; BP, n = 1 [no pleural effusion]) and tachycardia (AP, n = 2; BP, n = 4). Among AP and BP patients, newly occurring cardiac TEAEs reported in year 1 (more than two patients in either cohort) were pericardial effusion (AP, n = 4; BP, n = 1), and tachycardia (AP, n = 2; BP, n = 4). Cardiac TEAEs arising after year 1 included coronary artery disease (AP, n = 1) and acute myocardial infarction (BP, n = 1) in year 2; congestive cardiac failure (AP, n = 1; BP, n = 1) in year 3; and pericardial effusion (AP, n = 1), and sinus bradycardia and first degree atrioventricular block (AP, n = 1 [same patient]) in year 4. No on‐treatment grade 3/4 events of Fridericia's corrected QT (QTcF) interval prolongation or left ventricular ejection fractions (LVEF) were reported. Vascular TEAEs (defined in Supporting Information Table 3) occurred in 21 (13%) patients (Table 4). Among AP and BP patients, hypertension was only vascular TEAE to occur in more than two patients in year 1 (AP, n = 3; BP, n = 2); newly occurring vascular AEs arising after year 1 included cerebral artery occlusion, ischemic stroke (AP, n = 1 [same patient]) and hypertension (AP, n = 3) in year 2; blood pressure increased (AP, n = 1) in year 3; and hypertension (AP, n = 1) in year 4. No patient experienced peripheral arterial occlusive disease.
Most common on‐treatment, grade 3/4 laboratory abnormalities were hematologic (Table 6). Of note, grade 3/4 myelosuppression (defined in Supporting Information Table 3) was common at baseline (thrombocytopenia, 34%; neutropenia, 13%; anemia, 9%); shifts in severity from baseline are shown in Table 7. Myelosuppression TEAEs typically occurred early (median [range] time to onset, 12.0 [1–899]), with a median (range) any grade event duration of 8.0 (1–889) days. Despite its frequency and severity, only seven patients (all AP CML) discontinued treatment due to myelosuppression.
Table 6.
Incidence of Laboratory Abnormalities, any Causality
| Event,a n (%) | AP CML (n = 79) | BP CML (n = 64) | ALL (n = 24) | Total (n = 167) | ||||
|---|---|---|---|---|---|---|---|---|
| All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | |
| Hematologic laboratory abnormalitiesa | ||||||||
| Anemia | 78 (99) | 28 (35) | 60 (94) | 25 (39) | 22 (92) | 7 (29) | 160 (96) | 60 (36) |
| Thrombocytopenia | 64 (81) | 39 (49) | 50 (78) | 42 (66) | 20 (83) | 18 (75) | 134 (80) | 99 (59) |
| Neutropenia | 50 (63) | 18 (23) | 44 (69) | 37 (58) | 20 (83) | 12 (50) | 114 (68) | 67 (40) |
| Leukopenia | 39 (49) | 11 (14) | 42 (66) | 28 (44) | 16 (67) | 9 (38) | 97 (58) | 48 (29) |
| Lymphopenia | 38 (48) | 12 (15) | 39 (61) | 18 (28) | 12 (50) | 11 (46) | 89 (53) | 41 (25) |
| Other laboratory abnormalities occurring in ≥30% of patients, any cohort | ||||||||
| Hypocalcemia | 32 (41) | 4 (5) | 35 (55) | 3 (5) | 9 (38) | 1 (4) | 76 (46) | 8 (5) |
| Elevated alkaline phosphatase | 30 (38) | 1 (1) | 25 (39) | 1 (2) | 15 (63) | 0 | 70 (42) | 2 (1) |
| Hyperglycemia | 30 (38) | 7 (9) | 26 (41) | 2 (3) | 13 (54) | 1 (4) | 69 (41) | 10 (6) |
| Elevated INR | 31 (39) | 2 (3) | 30 (47) | 0 | 10 (42) | 1 (4) | 71 (43) | 3 (2) |
| Elevated ALT | 36 (46) | 7 (9) | 20 (31) | 1 (2) | 11 (46) | 0 | 67 (40) | 8 (5) |
| Elevated AST | 34 (43) | 4 (5) | 19 (30) | 1 (2) | 5 (21) | 0 | 58 (35) | 5 (3) |
| Elevated creatinine | 25 (32) | 0 | 22 (34) | 1 (2) | 9 (38) | 0 | 56 (34) | 1 (1) |
| Hypophosphatemia | 30 (38) | 6 (8) | 17 (27) | 4 (6) | 9 (38) | 3 (13) | 56 (34) | 13 (8) |
| Elevated PTT | 26 (33) | 2 (3) | 21 (33) | 2 (3) | 5 (21) | 0 | 52 (31) | 4 (2) |
| Hypokalemia | 18 (22) | 3 (4) | 22 (34) | 4 (6) | 12 (50) | 0 | 52 (31) | 7 (4) |
| Elevated prothrombin time | 23 (29) | 2 (3) | 20 (31) | 0 | 3 (13) | 0 | 46 (28) | 2 (1) |
| Hyponatremia | 16 (20) | 3 (4) | 22 (34) | 6 (9) | 5 (21) | 1 (4) | 43 (26) | 10 (6) |
| Hypoalbuminemia | 18 (23) | 0 | 24 (38) | 2 (3) | 5 (21) | 1 (4) | 47 (28) | 3 (2) |
Many patients had cytopenias when they started the study.
AP, accelerated phase; CML, chronic myeloid leukemia; BP, blast phase; ALL, acute lymphoblastic leukemia; ALT, alanine aminotransferase; INR, international normalized ratio; AST, aspartate aminotransferase; PTT, partial thromboplastin time.
Table 7.
Shifts From Baseline in Myelosuppression Laboratory Abnormalities
| Baseline severity | n | Maximum on‐treatment severity | ||||
|---|---|---|---|---|---|---|
| Normal | Grade 1 | Grade 2 | Grade 3 | Grade 4 | ||
| Anemia (n) | ||||||
| Normal | 31 | 5 | 12 | 10 | 4 | 0 |
| Grade 1 | 60 | 0 | 21 | 24 | 12 | 3 |
| Grade 2 | 61 | 0 | 4 | 27 | 22 | 6 |
| Grade 3 | 12 | 0 | 1 | 1 | 4 | 6 |
| Grade 4 | 3 | 0 | 0 | 0 | 0 | 3 |
| Total | 167 | 5 | 38 | 62 | 42 | 18 |
| Neutropeniaa (n) | ||||||
| Normal | 83 | 43 | 16 | 10 | 10 | 4 |
| Grade 1 | 44 | 4 | 10 | 4 | 10 | 14 |
| Grade 2 | 17 | 2 | 3 | 1 | 3 | 6 |
| Grade 3 | 9 | 0 | 0 | 3 | 1 | 5 |
| Grade 4 | 13 | 0 | 0 | 0 | 3 | 10 |
| Total | 167 | 49 | 29 | 18 | 27 | 40 |
| Thrombocytopenia (n) | ||||||
| Normal | 72 | 28 | 17 | 6 | 12 | 8 |
| Grade 1 | 29 | 3 | 8 | 1 | 8 | 8 |
| Grade 2 | 10 | 0 | 0 | 0 | 6 | 4 |
| Grade 3 | 25 | 0 | 3 | 0 | 4 | 18 |
| Grade 4 | 31 | 0 | 0 | 0 | 1 | 30 |
| Total | 167 | 31 | 28 | 7 | 31 | 68 |
Severity based on National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0.
One patient had missing baseline absolute neutrophil count data.
A retrospective evaluation of cross‐intolerance (defined as discontinuing bosutinib and a prior TKI due to the same AE) between bosutinib and imatinib found that among 22 patients in the AP CML, BP CML, and ALL cohorts intolerant to prior imatinib treatment, 8 (36%) experienced the same grade 3/4 AE with bosutinib and 4 (18%) discontinued due to the same AE. Among 21 patients intolerant to prior dasatinib treatment, 10 (48%) experienced the same grade 3/4 AE with bosutinib and 1 (5%) discontinued bosutinib due to the same AE. Among four patients intolerant to prior nilotinib treatment, two (50%) experienced the same grade 3/4 AE with bosutinib and none discontinued bosutinib due to the same AE.
Discussion
Patients with advanced Ph+ leukemia have a more rapid disease course, worse prognosis, and increased likelihood of developing imatinib resistance than those with CP disease. This 4‐year follow‐up of an ongoing phase 1/2 study demonstrates durable responses to bosutinib among patients with AP CML and high response rates among patients with BP CML after prior TKI treatment, with some responses also observed among patients with ALL. The higher observed rate of MCyR versus CHR (AP CML, 40% vs. 33%; BP CML, 37% vs. 17%; ALL, 20% vs. 9%) may be due to the requirement for bone marrow differential documentation to achieve a confirmed CHR, potentially excluding some patients who would have achieved a CHR based on peripheral blood documentation. It is also possible that the relatively lower CHR rate might have reflected residual cytopenia and an inadequate level of Ph‐negative hematopoiesis, which has been reported previously with dasatinib treatment in BP CML patients 30.
Although differences in designs, patient populations, response definitions, and follow‐up durations limit direct comparison with other studies, the MHR rate by 4 years in AP CML patients receiving second‐line bosutinib observed here (54%) is comparable with that reported for dasatinib (66% [median follow‐up, 15 months] 14) or nilotinib (55% by 2 years 31). The MCyR rate in AP CML patients was 39% with dasatinib 14 and 32% with nilotinib 31 after imatinib failure compared with 48% with bosutinib in this study. Kaplan‐Meier estimated 2‐year OS rates observed previously among patients with AP CML receiving second‐line dasatinib (63%) 14 or nilotinib (70%) 31 were each comparable to the 2‐year rate observed here for second‐line bosutinib (73%); the OS rate for second‐line bosutinib continued to be relatively high at 4 years (66%).
The MHR rate reported here among patients with BP CML receiving second‐line bosutinib (27%) was lower than that previously reported among patients receiving second‐line dasatinib (34% [myeloid BP] and 35% [lymphoid BP] after median follow‐up of 3.4 months 32), whereas the MCyR rate reported here (50%) was comparable to that reported with dasatinib (33% [myeloid BP] and 52% [lymphoid BP] 32). Median OS also appeared comparable between bosutinib as second‐line therapy (11.2 months) and dasatinib (11.8 months [myeloid BP] and 5.3 months [lymphoid BP] 32). The lack of analyses by myeloid versus lymphoid BP in the current study prevents closer comparison.
Responses to bosutinib were observed in AP and BP CML patients across a wide range of Bcr‐Abl mutations, except T315I, supporting preclinical investigations 33, 34. The more frequent emergence of T315I and V299L at treatment discontinuation in patients with disease progression or unsatisfactory response is also consistent with preclinical investigations 33, 34. Response to bosutinib among patients with F317L (associated with dasatinib resistance 35) at baseline was lower among patients with advanced CML (two of eight evaluable AP and BP CML patients had an OHR; zero of six had MCyR) compared with CP CML patients receiving third‐line bosutinib (four of eight evaluable patients had CHR; one of seven had MCyR) 28. This observation reflects the higher degree of tumor heterogeneity that emerges with increasing disease duration and exposure to several treatments, and is intricately driven by the multiple resistance mechanisms underlying the molecular pathogenesis of advanced leukemia.
The observed safety profile of bosutinib in patients with advanced disease is consistent with that reported for patients with CP CML receiving second‐line bosutinib 25, 26, 27. In the retrospective evaluation of cross‐intolerance (i.e., discontinuation of bosutinib and prior TKI due to the same AE), 4 (18%), 1 (5%), and 0/4 patients experienced cross‐intolerance to bosutinib and prior imatinib, dasatinib, or nilotinib, respectively. The incidence of newly occurring AEs generally decreased after the first year of treatment, as did discontinuations due to AEs. These observations emphasize the importance of early AE management and monitoring. Despite the frequent occurrence of diarrhea, these events were of low grade, of short duration, and none led to discontinuation. Similarly, although liver‐related AEs were common, few patients discontinued due to these AEs. Bosutinib was also associated with no grade 3/4 QTcF interval prolongation or LVEF across advanced leukemia cohorts. Myelosuppression is part of the natural history of leukemia, especially in advanced disease 6, 36, 37 and appears common among TKI therapies 6, 11, 13, 14, 15, 32, 37, 38; consistent with this, the incidence of cytopenias was high across all cohorts at baseline and during treatment. The report of two deaths due to SAEs considered to be related to bosutinib treatment should be interpreted in the context of advanced leukemia patients, for whom AEs can occur due to different causes, and for whom the assessment of a causal relationship between AE and treatment is always more problematic than for patients with chronic disease.
Although findings from this analysis and other studies show benefit of TKI treatment in patients with advanced CML, allo‐HSCT remains the only curative treatment option for some patients with advanced phase CML, albeit with a lower cure potential compared with those transplanted in chronic phase 18. Certain factors have been shown to increase the risk of poor treatment outcomes with allo‐HSCT, including increased age and increased disease burden at the time of allo‐HSCT 39, 40. The promising clinical activity of bosutinib reported herein suggests that bosutinib treatment may facilitate allo‐HSCT by in reducing disease burden in these patients. Similar observations have been made with other TKIs, including dasatinib and nilotinib 41.
In summary, bosutinib demonstrates a durable response, with ∼50% of AP responders maintaining a response at 4 years. Moreover, ∼25% of BP responders maintained response to treatment at 1 year, for whom bosutinib may be considered to be a bridge to stem cell transplantation. Responses were also observed among some ALL patients; however, the analysis of these data was limited by the small number of enrolled ALL patients. Bosutinib toxicities were manageable with long‐term treatment. Based on the current treatment landscape, bosutinib represents a long‐term treatment option for patients with advanced Ph+ leukemias following failure of prior TKI therapy.
Author Contributions
The study was created/designed by C.G.P., H.J.K., and J.E.C. C.G.P., H.M.K., D.W.K., H.J.K., A.G.T., T.H.B., and J.E.C. collected and assembled the data. C.G.P., H.M.K., D.W.K., H.J.K., A.G.T., T.H.B., E.M., N.B.B., M.S., K.T., E.L., and J.E.C. provided analysis and/or interpretation of the data and provided study materials and/or enrolled patients in the study. E.L. performed statistical analyses. All authors assisted in the writing and/or critical review of the manuscript, and all authors approved the final version of the manuscript for submission.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
The authors acknowledge all of the participating patients and their families as well as the global network of investigators, research nurses, study coordinators, and operations staff. The authors are also very grateful to Virginia Kelly, MD, Nadine Besson, MSc, and Athena Countouriotis, MD, for their contributions to this study. Editorial and medical writing support was provided by Simon Slater, PhD, and Cynthia Gobbel, PhD, CMPP, of Complete Healthcare Communications, Inc., and was funded by Pfizer.
Conflict of interest: C.G.P. has received research funding from Pfizer and consultancy or other fees from Pfizer and Bristol‐Myers Squibb; H.M.K. has received research funding from Pfizer; D.W.K. has received research funding, consultancy and other fees, and served as a member on the board of directors or advisory committees for Bristol‐Myers Squibb, Ilyang Co, Novartis, and Pfizer; H.J.K. has received research funding, consultancy and other fees, and served as a member on the board of directors or advisory committees for Pfizer; A.G.T. has served as a consultant/advisor, participated on the Speakers’ bureau, and received honoraria from Bristol‐Myers Squibb and Novartis; T.H.B. has received research funding from Novartis and Pfizer, consultancy and other fees from Ariad, Bristol‐Myers Squibb, Novartis, and Pfizer, and has received patent royalties (use of imatinib and hypusination inhibitors); E.M., N.B.B., M.S., K.T., and E.L. are employees of Pfizer, and N.B. and V.K. are former employees of Pfizer; J.E.C. has received research funding and consultancy and other fees from Ariad, Bristol‐Myers Squibb, Novartis, and Pfizer, and research funding from Teva.
This study was sponsored by Wyeth Research, which was acquired by Pfizer in October 2009. Programming support was provided by ICON plc.
References
- 1. Faderl S, Talpaz M, Estrov Z, et al. Chronic myelogenous leukemia: Biology and therapy. Ann Intern Med 1999;131:207–219. [DOI] [PubMed] [Google Scholar]
- 2. Kantarjian HM, Dixon D, Keating MJ, et al. Characteristics of accelerated disease in chronic myelogenous leukemia. Cancer 1988;61:1441–1446. [DOI] [PubMed] [Google Scholar]
- 3. Cortes J, Kantarjian H. Advanced‐phase chronic myeloid leukemia. Semin Hematol 2003;40:79–86. [DOI] [PubMed] [Google Scholar]
- 4. Kantarjian HM, Talpaz M, O'Brien S, et al. Survival benefit with imatinib mesylate versus interferon‐alpha‐based regimens in newly diagnosed chronic‐phase chronic myelogenous leukemia. Blood 2006;108:1835–1840. [DOI] [PubMed] [Google Scholar]
- 5. Kantarjian H, Talpaz M, O'Brien S, et al. Survival benefit with imatinib mesylate therapy in patients with accelerated‐phase chronic myelogenous leukemia–comparison with historic experience. Cancer 2005;103:2099–2108. [DOI] [PubMed] [Google Scholar]
- 6. Sawyers CL, Hochhaus A, Feldman E, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: Results of a phase II study. Blood 2002;99:3530–3539. [DOI] [PubMed] [Google Scholar]
- 7. Kantarjian HM, Cortes J, O'Brien S, et al. Imatinib mesylate (sti571) therapy for Philadelphia chromosome‐positive chronic myelogenous leukemia in blast phase. Blood 2002;99:3547–3553. [DOI] [PubMed] [Google Scholar]
- 8. Saglio G, Hochhaus A, Goh YT, et al. Dasatinib in imatinib‐resistant or imatinib‐intolerant chronic myeloid leukemia in blast phase after 2 years of follow‐up in a phase 3 study: Efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer 2010;116:3852–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kurzrock R, Kantarjian HM, Druker BJ, et al. Philadelphia chromosome‐positive leukemias: From basic mechanisms to molecular therapeutics. Ann Intern Med 2003;138:819–830. [DOI] [PubMed] [Google Scholar]
- 10. Kantarjian HM, Giles F, Gattermann N, et al. Nilotinib (formerly amn107), a highly selective BCR‐ABL tyrosine kinase inhibitor, is effective in patients with philadelphia chromosome‐positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood 2007;110:3540–3546. [DOI] [PubMed] [Google Scholar]
- 11. Hochhaus A, Baccarani M, Deininger M, et al. Dasatinib induces durable cytogenetic responses in patients with chronic myelogenous leukemia in chronic phase with resistance or intolerance to imatinib. Leukemia 2008;22:1200–1206. [DOI] [PubMed] [Google Scholar]
- 12. de Lavallade H, Apperley JF, Khorashad JS, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: Incidence of sustained responses in an intention‐to‐treat analysis. J Clin Oncol 2008;26:3358–3363. [DOI] [PubMed] [Google Scholar]
- 13. Apperley JF, Cortes JE, Kim DW, et al. Dasatinib in the treatment of chronic myeloid leukemia in accelerated phase after imatinib failure: The START a trial. J Clin Oncol 2009;27:3472–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kantarjian H, Cortes J, Kim DW, et al. Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice daily in patients with chronic myeloid leukemia in accelerated phase resistant or intolerant to imatinib: 15‐Month median follow‐up. Blood 2009;113:6322–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. le Coutre P, Ottmann OG, Giles F, et al. Nilotinib (formerly amn107), a highly selective BCR‐ABL tyrosine kinase inhibitor, is active in patients with imatinib‐resistant or ‐intolerant accelerated‐phase chronic myelogenous leukemia. Blood 2008;111:1834–1839. [DOI] [PubMed] [Google Scholar]
- 16. Lee S, Kim YJ, Min CK, et al. The effect of first‐line imatinib interim therapy on the outcome of allogeneic stem cell transplantation in adults with newly diagnosed philadelphia chromosome‐positive acute lymphoblastic leukemia. Blood 2005;105:3449–3457. [DOI] [PubMed] [Google Scholar]
- 17. Ravandi F, O'Brien S, Thomas D, et al. First report of phase 2 study of dasatinib with hyper‐CVAD for the frontline treatment of patients with philadelphia chromosome‐positive (ph+) acute lymphoblastic leukemia. Blood 2010;116:2070–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013;122:872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jain P, Kantarjian H, Cortes J. Chronic myeloid leukemia: Overview of new agents and comparative analysis. Curr Treat Options Oncol 2013;14:127–143. [DOI] [PubMed] [Google Scholar]
- 20. Liu‐Dumlao T, Kantarjian H, Thomas DA, et al. Philadelphia‐positive acute lymphoblastic leukemia: Current treatment options. Curr Oncol Rep 2012;14:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.NCCN Clinical Practice Guidelines in Oncology. Acute Lymphoblastic Leukemia. Version 3. 2013. Fort Washington, PA: National Comprehensive Cancer Network. [Google Scholar]
- 22.NCCN Clinical Practice Guidelines in Oncology: Chronic Myelogenous Leukemia. Version 2.2014. Fort Washington, PA: National Comprehensive Cancer Network. [DOI] [PubMed] [Google Scholar]
- 23.Bosulif: EPAR Product Information. London, United Kingdom: European Medicines Agency. [Google Scholar]
- 24.Pfizer Inc. BOSULIF® New York, NY: Pfizer Inc.; 2014.
- 25. Cortes JE, Kantarjian HM, Brummendorf TH, et al. Safety and efficacy of bosutinib (SKI‐606) in chronic phase philadelphia chromosome‐positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 2011;118:4567–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gambacorti‐Passerini C, Brummendorf TH, Kim DW, et al. Bosutinib efficacy and safety in chronic phase chronic myeloid leukemia after imatinib resistance or intolerance: Minimum 24‐month follow‐up. Am J Hematol 2014;89:732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kantarjian HM, Cortes JE, Kim DW, et al. Bosutinib safety and management of toxicity in leukemia patients with resistance or intolerance to imatinib and other tyrosine kinase inhibitors. Blood 2014;123:1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khoury HJ, Cortes JE, Kantarjian HM, et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood 2012;119:3403–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baccarani M, Saglio G, Goldman J, et al. Evolving concepts in the management of chronic myeloid leukemia: Recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2006;108:1809–1820. [DOI] [PubMed] [Google Scholar]
- 30. Cortes J, Rousselot P, Kim DW, et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib‐resistant or ‐intolerant chronic myeloid leukemia in blast crisis. Blood 2007;109:3207–3213. [DOI] [PubMed] [Google Scholar]
- 31. le Coutre PD, Giles FJ, Hochhaus A, et al. Nilotinib in patients with ph+ chronic myeloid leukemia in accelerated phase following imatinib resistance or intolerance: 24‐month follow‐up results. Leukemia 2012;26:1189–1194. [DOI] [PubMed] [Google Scholar]
- 32. Cortes J, Kim DW, Raffoux E, et al. Efficacy and safety of dasatinib in imatinib‐resistant or ‐intolerant patients with chronic myeloid leukemia in blast phase. Leukemia 2008;22:2176–2183. [DOI] [PubMed] [Google Scholar]
- 33. Redaelli S, Piazza R, Rostagno R, et al. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib‐resistant BCR/ABL mutants. J Clin Oncol 2009;27:469–471. [DOI] [PubMed] [Google Scholar]
- 34. Redaelli S, Mologni L, Rostagno R, et al. Three novel patient‐derived BCR/ABL mutants show different sensitivity to second and third generation tyrosine kinase inhibitors. Am J Hematol 2012;87:E125–E128. [DOI] [PubMed] [Google Scholar]
- 35. Soverini S, Colarossi S, Gnani A, et al. Resistance to dasatinib in Philadelphia‐positive leukemia patients and the presence or the selection of mutations at residues 315 and 317 in the BCR‐ABL kinase domain. Haematologica 2007;92:401–404. [DOI] [PubMed] [Google Scholar]
- 36. Deininger MW, O'Brien SG, Ford JM, et al. Practical management of patients with chronic myeloid leukemia receiving imatinib. J Clin Oncol 2003;21:1637–1647. [DOI] [PubMed] [Google Scholar]
- 37. Sneed TB, Kantarjian HM, Talpaz M, et al. The significance of myelosuppression during therapy with imatinib mesylate in patients with chronic myelogenous leukemia in chronic phase. Cancer 2004;100:116–121. [DOI] [PubMed] [Google Scholar]
- 38. Singer JB, Shou Y, Giles F, et al. UGT1A1 promoter polymorphism increases risk of nilotinib‐induced hyperbilirubinemia. Leukemia 2007;21:2311–2315. [DOI] [PubMed] [Google Scholar]
- 39. Szczepanski T. Why and how to quantify minimal residual disease in acute lymphoblastic leukemia? Leukemia 2007;21:622–626. [DOI] [PubMed] [Google Scholar]
- 40. Liu‐Dumlao T, Kantarjian H, Thomas DA, et al. Philadelphia‐positive acute lymphoblastic leukemia: Current treatment options. Curr Oncol Rep 2012;14:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shimoni A, Leiba M, Schleuning M, et al. Prior treatment with the tyrosine kinase inhibitors dasatinib and nilotinib allows stem cell transplantation (SCT) in a less advanced disease phase and does not increase SCT toxicity in patients with chronic myelogenous leukemia and philadelphia positive acute lymphoblastic leukemia. Leukemia 2009;23:190–194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information