

Veno‐occlusive disease (VOD) is a serious complication commonly occurring after stem cell transplantation (SCT). In VOD, sinusoidal endothelial cells and hepatocytes are damaged because of progressive venular occlusions, leading to hepatocellular necrosis.1, 2, 3 VOD severity ranges from mild to severe; severe VOD is defined by the presence of multiorgan failure and is associated with a greater than 85% 100‐day mortality rate.4, 5, 6 VOD currently has no approved prophylactic or curative treatment in Japan.
Defibrotide (DF) is a polydisperse mixture of 90% single‐stranded polydeoxyribonucleotides that has antithrombotic, antiischemic, and profibrinolytic functions.7, 8, 9, 10 DF, which is given 4 times a day at a dose of 6.25 mg/kg, has been approved by the European Medicines Agency for the treatment of severe hepatic VOD in SCT therapy. It is an investigational drug that has been granted orphan drug status by the Food and Drug Administration.
To support the clinical development of DF for VOD treatment in Japan, we investigated the pharmacokinetics and evaluated the safety of DF in healthy Japanese subjects.
Methods
Subjects
The protocol was approved in advance by the Human Institutional Review Board of Hamamatsu University School of Medicine, Hamamatsu, Japan. Written informed consent was obtained from each subject before participation in this study.
Twenty healthy male Japanese subjects between 20 and 43 years years old who weighted between 55.8 and 70.9 kg were enrolled in the study. Physical examination and standard biochemical, hematologic, and urinalysis screening tests were conducted to assess the suitability of the test subjects. The subjects took no other medication during the week leading up to and for the duration of the study. The consumption of alcohol and caffeinated beverages was prohibited during the study.
Study Design
This study had a randomized, single‐blind, placebo‐controlled study design. The 20 subjects were randomly divided into 2 groups. From each group, 8 subjects received either a 3.0 or 6.25 mg/kg dose of DF, and 2 subjects received placebo. Because DF has yet to be administered to many Japanese patients, we first administered the 3.0 mg/kg dose, as a 2‐hour infusion to verify dose safety. Once this dose was deemed safe, the second group received a 6.25 mg/kg dose, administered as a 2‐hour infusion.
Tolerance
Subjective and objective symptoms as well as vital signs, including blood pressure, heart rate, and body temperature, were recorded before treatment and periodically for 24 hours after DF administration. Routine laboratory tests, including hematology, blood biochemistry, and urinalysis, were also performed. Twelve‐lead electrocardiograms were recorded immediately before and 24 hours after DF administration.
Pharmacokinetic Analysis
Blood samples for pharmacokinetic analysis were collected in heparinized tubes prior to DF administration, 1 and 2 hours after the start of administration, and 5, 15, 30, and 60 minutes after administration completion. All plasma samples were stored below –80°C until assayed. Gentium S.p.A developed the analytical method for plasma DF quantification, and it was validated by Shin Nippon Biomedical Laboratories, Ltd. (Pharmacokinetics and Bioanalysis Center, Japan) for use in this study. Plasma samples (50 μL) were prepared by adding 50 mmoL/L ammonium acetate (450 μL) and filtered through a 0.45‐μm centrifuge filter (Centricut Mini, Kubota, Japan) by centrifuging at 5000g for 5 minutes. The percentage of drug recovered from the plasma was 110.7%, 85.8%, and 79.7% for 30, 140, and 225 μg/mL DF doses, respectively. Concentrations of DF in the plasma were measured using high‐performance liquid chromatography (Waters 2690 system) equipped with a C8 Column (3.5 μm, 2.1 × 100 mm, Waters X Bridge) at 50°C in a constant temperature bath and a UV detector (Waters 2487). DF was detected at a wavelength of 260 nm. The mobile phase was composed of 25 mmoL/L ammonium acetate buffer (pH 9.0, mobile phase A), 50 mmoL/L ammonium acetate buffer (pH 9.0)/acetonitrile/methanol (50/45/5 v/v/v, mobile phase B), 1% ammonia solution (mobile phase C), and acetonitrile (containing 0.1% formic acid)/ultra‐pure water (9/1 v/v, mobile phase D). The following parameters were employed: flow rate of 0.4 to 0.5 mL/min and 20‐μL injection volume. Accuracy was determined by calculating the relative error (RE %) based on the theoretical concentrations, calculated as follows: (measured concentration ‐ theoretical concentration)/theoretical concentration × 100 (the absolute value of 100 minus the average estimated concentration divided by the theoretical concentration). The lower limit of quantification (LLQ) was 10 μg/mL, and the RE was within ±15%, with the exception of the values of the LLQ. The coefficients of variation (CV%) within and between days in detecting from 10 to 300 μg/mL were less than 3.1% and 6.9%, respectively.
Pharmacokinetic parameters, including time to maximum plasma concentration (Tmax), plasma half‐life (t1/2), area under the plasma concentration–time curve (AUC), and total plasma clearance ([CL]tot), were analyzed by standard noncompartmental methods using the computer program WinNonlin 5.2.1 (Pharsight Corporation). AUC0–3 was estimated by the linear trapezoidal rule, and AUC0–∞ was determined using AUC0–3 + AUC3–∞, which was calculated by dividing the concentration at 3 hours by the elimination rate constant value of the terminal slope.
Pharmacodynamic Analysis
Blood samples were obtained prior to treatment and 2 and 24 hours after the start of the 2‐hour DF infusion to determine the fibrinolytic and coagulation activities. The parameters studied included prothrombin time, activated partial thromboplastin time, and levels of the following coagulation and fibrinolytic factors: fibrinogen, fibrinogen/fibrin degradation products, D‐dimer, plasminogen activator inhibitor‐1 antigen, protein C antigen, α2‐plasmin inhibitor, plasmin/α2‐plasmin inhibitor complex, plasminogen activity, thrombin–antithrombin complex, tissue factor pathway inhibitor (TFPI), platelet factor 4, thromboxane‐B2, and 6‐keto‐prostaglandin F1α.
Statistical Analysis
Data are presented as mean ± standard deviation (SD). Analysis of variance (ANOVA) and Dunnett's test were used for comparison of the pharmacodynamics data. The statistical analysis was performed using SAS (SAS Institute) and SPSS (IBM). Results were deemed significant at P < .05.
Results
Safety and Tolerance
One subject exhibited mild disorders in liver function when administered 3.0 mg/kg DF. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) values increased 7 days postadministration and returned to normal by the 17th postadministration day (AST, 26 IU/L prior to administration, 20 IU/L 24 hours after administration, 46 IU/L on day 7, 18 IU/L on day 17; ALT, 29, 21, 58, and 18 IU/L, respectively).
No other abnormalities in objective symptoms or in laboratory test results, including abnormalities in blood pressure, heart rate, electrocardiogram, body temperature, hematology, blood chemistry, and urinalysis, were attributable to DF.
Pharmacokinetics of DF
The changes in DF plasma concentrations after infusion with a 6.25 mg/kg DF dose are shown in Figure 1, and the associated pharmacokinetic parameters are shown in Table 1. When 3.0 mg/kg of DF was administered, plasma concentrations were below the limit of detection. On administration of 6.25 mg/kg DF, the mean values for the maximum observed plasma concentration Cmax and AUC0–∞ were 20.59 ± 4.11 ng/mL and 42.32 ± 6.95 ng·h/mL, respectively. The mean values for t1/2 and (CL)tot were 0.47 ± 0.10 hours and 9.629 ± 1.175 L/h, respectively.
Figure 1.
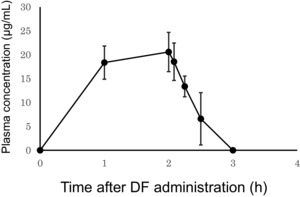
The defibrotide (DF) plasma concentration peaks immediately after the completion of a 2‐hour infusion of 6.25 mg/kg DF. Blood plasma DF levels were determined using high‐performance liquid chromatography. Data represent mean ± standard deviation (SD); n = 8. Plasma concentrations from a 3.0 mg/kg DF were below the assay's limit of detection.
Table 1.
Pharmacokinetic Parameters of a 6.25 mg/kg Dose of Defibrotide in Healthy Japanese Subjects
| Cmax (μg/mL) | Tmax (h) | kel (1/h) | T1/2 (h) | AUC0–3 (μg·h/mL) | AUC0–∞ (μg·h/mL) | (CL)tot (L/h) | Vd (L) | |
|---|---|---|---|---|---|---|---|---|
| n | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Mean | 20.59 | 2.00 | 1.55 | 0.47 | 37.09 | 42.32 | 9.27 | 7.31 |
| SD | 4.11 | 0.00 | 0.33 | 0.10 | 7.82 | 6.95 | 1.18 | 1.25 |
kel, elimination rate constant, Vd, volume of distribution.
Pharmacodynamics of DF
Compared with placebo, a 6.25 mg/kg dose of DF significantly (P < .001) increased the plasma concentration of TFPI 2 hours after the start of infusion (Figure 2). No other fibrinolytic or coagulation parameters were affected by DF administration.
Figure 2.
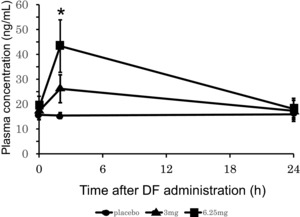
The human blood plasma tissue factor pathway inhibitor (TFPI) concentration peaked 2 hours after the start of the defibrotide (DF) treatment. Plasma TFPI levels were determined in DF‐treated (infusion over 2 hours) healthy male Japanese subjects. Data represent mean ± standard deviation (SD); n = 4–8 subjects per group. *P < .05 compared with placebo.
Discussion
Throughout the investigation period, no abnormalities attributable to the test drug were observed. Objective symptoms, vital signs, and routine laboratory test results were not altered, suggesting that DF is well tolerated in healthy subjects.
The pharmacokinetic parameters observed in the present study were approximately the same as those obtained from the clinical trial on healthy subjects performed in the United States.11 This suggests that there are no differences in the pharmacokinetic parameters of DF between Japanese and American subjects.
A previous study showed that DF reduces vascular permeability and inhibits the inflammation‐induced expression of leukocyte adhesion molecules.12 In addition, DF has been shown to have antithrombotic properties attributed to an increase in tissue plasminogen activator activity and a reduction in PAI‐1 concentration.12 Although in this study the plasma TFPI levels increased with administration of 6.25 mg/kg DF, the fibrinolytic and coagulation activities were not altered. These findings suggest that DF may not affect fibrinolytic and coagulation activity in healthy Japanese subjects.
Conclusions
From our observations, it can be concluded that DF has an acceptable pharmacokinetic profile and causes no serious adverse effects in healthy Japanese subjects.
Declaration of Conflicting Interests
Gentium provided the investigational drug and information deemed important to conduct the clinical trial. The article has been edited and proofread by a professional scientific editing company, Editage (www.editage.com).
Funding
This study was supported by Health and Labour Sciences Research Grants for Clinical Trial on Development of New Drugs and Medical Devices from the Ministry of Health, Labour and Welfare of Japan (15lk0201009h0004).
References
- 1. Copelan EA. Hematopoietic stem‐cell transplantation. N Engl J Med. 2006;354(17):1813–1826. [DOI] [PubMed] [Google Scholar]
- 2. McDonald GB, Sharma P, Matthews DE, et al. Venocclusive disease of the liver after bone marrow transplantation: diagnosis, incidence, and predisposing factors. Hepatology. 1984;4(1):116–122. [DOI] [PubMed] [Google Scholar]
- 3. Richardson P, Guinan E. The pathology, diagnosis, and treatment of hepatic veno‐occlusive disease: current status and novel approaches. Br J Haematol. 1999;107(3):485–493. [DOI] [PubMed] [Google Scholar]
- 4. McDonald GB, Hinds MS, Fisher LD, et al. Veno‐occlusive disease of the liver and multiorgan failure after bone marrow transplantation: a cohort study of 355 patients. Ann Intern Med. 1993;118(4):255–267. [DOI] [PubMed] [Google Scholar]
- 5. Coppell JA, Richardson PG, Soiffer R, et al. Hepatic veno‐occlusive disease following stem cell transplantation: incidence, clinical course, and outcome. Biol Blood Marrow Transplant. 2010;16(2):157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carreras E, Bertz H, Arcese W, et al, and the European Group for Blood and Marrow Transplantation Chronic Leukemia Working Party. Incidence and outcome of hepatic veno‐occlusive disease after blood or marrow transplantation: a prospective cohort study of the European Group for Blood and Marrow Transplantation. Blood. 1998;92(10):3599–3604. [PubMed] [Google Scholar]
- 7. Paul W, Gresele P, Momi S, et al. The effect of defibrotide on thromboembolism in the pulmonary vasculature of mice and rabbits and in the cerebral vasculature of rabbits. Br J Pharmacol. 1993;110(4):1565–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bianchi G, Barone D, Lanzarotti E, et al. Defibrotide, a single‐stranded polydeoxyribonucleotide acting as an adenosine receptor agonist. Eur J Pharmacol. 1993;238(2‐3):327–334. [DOI] [PubMed] [Google Scholar]
- 9. Zhou Q, Chu X, Ruan C. Defibrotide stimulates expression of thrombomodulin in human endothelial cells. Thromb Haemost. 1994;71(4):507–510. [PubMed] [Google Scholar]
- 10. Falanga A, Vignoli A, Marchetti M, et al. Defibrotide reduces procoagulant activity and increases fibrinolytic properties of endothelial cells. Leukemia. 2003;17(8):1636–1642. [DOI] [PubMed] [Google Scholar]
- 11. Tocchetti P, Ballabio M, Tudone E, et al. A phase 1, open‐label study of defibrotide pharmacokinetics in severe/end‐stage renal disease patients not on dialysis compared with healthy matching subjects. Presented at: 41st Annual Meeting of the European Society for Blood and Marrow Transplantation; March 22–25, 2015; Istanbul, Turkey.
- 12. Pescador R, Capuzzi L, Mantovani M, Fulgenzi A, Ferrero ME. Defibrotide: properties and clinical use of an old/new drug. Vascul Pharmacol. 2013;59(1‐2):1–10. [DOI] [PubMed] [Google Scholar]