

Abstract
A randomized, partial‐blind, repeat‐dose, 3‐period crossover study (NCT02027454) assessed the effect of cabotegravir on QT interval in healthy subjects. To achieve a supratherapeutic dose, each subject received cabotegravir 150 mg (30 mg × 5 tablets) every 12 hours for a total of 3 doses over 2 days, matching placebo (every 12 hours) over 2 days, or a single open‐label 400‐mg dose of the positive control moxifloxacin, with a 21‐day washout between treatments. Blood samples for pharmacokinetic analyses were collected up to 24 hours after the third dose on day 2. QT interval data were obtained by continuous Holter monitoring for approximately 24 hours at baseline (day ‐1) and from 2 hours before to 24 hours after the third dose on day 2. Plasma cabotegravir exposure was approximately 3‐fold above clinically relevant doses. After 3 doses of 150 mg of cabotegravir administered every 12 hours, all upper limits of 2‐sided 90% confidence intervals for ΔΔQTcF (difference in time‐matched change from baseline for QTcF between cabotegravir and placebo) were <10 milliseconds. There was no relationship between cabotegravir plasma concentrations and ΔΔQTcF. No subject receiving cabotegravir had a QTcF value > 450 milliseconds. There were no serious or grade 3 or 4 adverse events or clinically significant changes in laboratory values, vital signs, or electrocardiogram results. These data demonstrate that cabotegravir at a supratherapeutic dose had no effect on cardiac repolarization.
Keywords: cabotegravir, GSK1265744, QT interval, cardiac conduction, HIV‐1, integrase inhibitor
Integrase strand transfer inhibitors (INSTIs) have provided a valuable addition to treatment options for patients with HIV. This class targets the HIV integrase enzyme, thereby blocking integration of the viral genome into the host genome.1 First‐generation INSTIs (ie, raltegravir, elvitegravir) have been shown to be effective and generally well tolerated.2 However, clinical resistance to first‐generation INSTIs has been a challenge,3, 4 and a second‐generation INSTI (dolutegravir) has been developed that has demonstrated limited cross‐resistance to other members of the class.2 Although the INSTI class allows for single‐tablet regimens that significantly reduce pill burden, there remains a need for longer‐acting agents that do not require daily administration. Cabotegravir (GSK1265744) is an investigational HIV‐1 INSTI and is a structural analogue of dolutegravir that is in development as a long‐acting injectable to address this treatment need. In addition, the long‐acting nature supports investigation for prevention of HIV.
Early clinical studies have examined oral doses of cabotegravir in healthy and HIV‐1‐infected patients. These studies have shown that oral cabotegravir has potent antiretroviral activity, with an extended plasma half‐life (estimated to be 40 hours), and is generally well tolerated.5 Following oral administration of single doses (5 to 50 mg) in healthy subjects after overnight fast for ≥10 hours, cabotegravir was readily absorbed, with the maximum concentration achieved between 0.50 and 1.5 hours across doses when dosed as a solution5 and 1.5 to 3.5 hours when dosed as a tablet. Dose‐proportional increases in cabotegravir AUC(0–t), Cmax, Cτ, and Cmin were observed following repeated administration of cabotegravir for 14 days.5 Furthermore, available data support time‐invariant pharmacokinetics of cabotegravir.
Cabotegravir is metabolized via UDP‐glucuronosyltransferase (UGT) 1A1, with a secondary contribution from UGT1A9 to inactive glucuronide metabolites.6 The primary metabolite of cabotegravir (M1) is an ether glucuronide excreted predominantly in urine. In vitro, cabotegravir has not been shown to inhibit or induce UGT and cytochrome P450 (CYP) enzymes.7 In addition, CYP enzymes do not appear to be involved in cabotegravir metabolism as demonstrated in a clinical study in which etravirine, a known CYP and UGT inducer, did not affect the pharmacokinetics of cabotegravir.8 Overall, cabotegravir has a favorable drug interaction profile and is expected to have few clinically significant drug interactions.6
In accordance with the International Conference on Harmonization (ICH) recommendations for new agents,9 supratherapeutic doses of INSTIs have been examined for their potential to affect cardiac repolarization and have not been shown to have significant effects on the QTc interval.10, 11 In nonclinical studies, no effects of cabotegravir on cardiac conduction have been observed. For example, human ether‐a‐go‐go‐related gene (hERG) tail current was examined in human embryonic kidney 293 cells that had been transfected with hERG complementary DNA; the effects of cabotegravir (5.28 μM [2.14 μg/mL] and maximum soluble concentration of 17.61 μM [7.14 μg/mL]) on hERG channel tail current were no different than those observed with vehicle.
In a cardiovascular study performed in unrestrained male monkeys, a single dose of cabotegravir 1000 mg/kg was associated with a mild transient increase in mean arterial pressure (∼6%) and a transient increase in heart rate (∼20%) within the first 2 hours of dosing, but no electrocardiogram (ECG) waveform or interval changes were associated with these changes, and no effects occurred at doses of 8 and 25 mg/kg. The cabotegravir dose used in this preclinical study (1000 mg/kg) was associated with 3‐fold‐higher drug exposure (Cmax, 67.0 μg/mL) than that observed in the present study (Cmax, 22.5 μg/mL), which evaluated a supratherapeutic dose of cabotegravir (3 doses of 150 mg every 12 hours). When a repeat‐dosing study was performed in monkeys using these same doses, there were no effects on ECG tracings. In addition to evidence from nonclinical studies, the results of a meta‐analysis of safety data from 8 previous phase 1/2a clinical studies (n = 245 subjects) that included 2540 postdose ECGs also support a lack of effect of cabotegravir on cardiac repolarization.12 The overall mean difference in QTcF change from baseline between cabotegravir and placebo was –2.9 milliseconds (95% confidence interval [CI], –4.96 to –0.81 milliseconds). In addition, no subject had a QTcF ≥ 480 milliseconds or a change from baseline in QTcF of >60 milliseconds. Therefore, on the basis of nonclinical and clinical studies, cabotegravir was predicted to have a low potential to exert cardiovascular effects. Although the ICH guidelines recommend the use of therapeutic and supratherapeutic doses of investigational products, more recent clinical studies, including QTc studies of raltegravir10 and dolutegravir,11 have used single supratherapeutic doses to efficiently assess effects on cardiac repolarization. The use of supratherapeutic doses allows a thorough examination of the drug at high concentration (much higher than the anticipated efficacious dose) and thus at maximal effect. Furthermore, the use of such high dosing allows for assessment at concentrations that are in excess of those that may be observed clinically when dosed concurrently with agents that cause increases in exposure.
This study was undertaken to examine the effect of a supratherapeutic oral dose of cabotegravir (150 mg every 12 hours × 3 doses) on QTcF compared with placebo in healthy subjects. A single dose of moxifloxacin (400 mg), a fluoroquinolone known to have a consistently small effect on the QT interval,9 was used as a positive control.
Methods
Study Design and Protocol
This was a randomized, partial‐blind, repeat‐dose, 3‐period, balanced‐crossover study. Subjects were randomly assigned to 1 of 6 treatment sequences in which they received a supratherapeutic dose of cabotegravir 150 mg (30 mg × 5 tablets) every 12 hours for a total of 3 doses over 2 days, matching placebo (5 tablets), or a single 400‐mg tablet of moxifloxacin as a positive control administered using a similar dosing interval. The subjects and site staff were blinded only to the administration of cabotegravir or placebo; moxifloxacin was given open‐label. The central ECG reader was blinded to all 3 treatments. During blinded treatment with cabotegravir or placebo, subjects fasted for at least 6 hours prior to the third dose, and no food was allowed for 4 hours postdose. For doses 1 and 2, no food fast was required, but food was not permitted for 4 hours following dose 1 or dose 2. For moxifloxacin treatment (open‐label), subjects fasted for at least 6 hours, and no food was permitted for 4 hours postdose. The study design consisted of a 30‐day screening period, 3 treatment periods separated by washout periods of ≥21 days, and a follow‐up period 10 to 14 days after administration of the last dose of study medication. Subjects were admitted to the clinical unit on day ‐2 of each dosing period and remained in the unit until completion of the 24‐hour assessments on day 3 of the dosing period.
The supratherapeutic dose of cabotegravir was selected based on a simulated prediction that it would achieve a Cmax approximately 3‐fold higher than that observed with the standard clinical dose of 30 mg once daily. Previous work (unpublished data) showed that a single dose of cabotegravir 150 mg was rapidly absorbed, with a median time to Cmax (Tmax) of 2.5 hours. In the same study, the geometric mean plasma cabotegravir AUC0–∞ of 418 μg·h/mL and Cmax of 10.4 μg/mL following single‐dose administration (150 mg) were approximately half the predicted values (870 μg·h/mL and 21.2 μg/mL, respectively) based on extrapolation from 30‐mg data. Geometric mean cabotegravir Cmax was only 39% higher than the 7.49 μg/mL observed following repeat administration of 30 mg once daily in HIV‐infected subjects. Therefore, a single 150‐mg dose of cabotegravir does not represent a supratherapeutic dose, and, thus, a repeat dosing strategy was required to achieve exposures sufficiently high to evaluate effects on QTc.
The study protocol and consent forms were reviewed and approved by Midlands Independent Review Board (Overland Park, Kansas), and the study was conducted at Quintiles Phase One Services, LLC (Overland Park, Kansas) in accordance with the Declaration of Helsinki and ICH Good Clinical Practice.
Study Subjects
Healthy adults aged 18 to 55 years in general good health with no clinically relevant abnormalities as determined by medical history (including risk factors for torsades de pointes), physical examination, laboratory tests, and cardiac monitoring were eligible for study participation. Subjects with body weight ≥ 50 kg for men and ≥ 45 kg for women and body mass index between 18.5 and 31 kg/m2 were eligible for inclusion. Female subjects of reproductive potential who were pregnant, lactating, or unwilling to use highly effective contraception methods were excluded from the study. Subjects were also excluded if cardiac abnormalities were observed during the initial screening as determined by ECGs, including a corrected QT (QTc) interval value > 450 milliseconds. Additional exclusion criteria included the following: a positive test for HIV, hepatitis B surface antigen, or hepatitis C antibody; current or chronic history of liver disease; a positive prestudy drug or alcohol screen; participation in a clinical trial involving an investigational drug within 30 days of the screening visit; a history of sensitivity to the study drugs; use of prescription or nonprescription drugs, including vitamins and dietary supplements, within 7 days of dosing; or use of nicotine‐ or tobacco‐containing products within 6 months of screening.
Study Assessments
Digital ECG data were obtained for all subjects using a 24‐hour continuous 12‐lead Mortara H12+ 1000‐Hz Holter monitor (Mortara Instrument, Inc, Milwaukee, Wisconsin). Subjects underwent continuous Holter monitoring for approximately 24 hours prior to receiving the first dose of randomized treatment on day 1. Continuous Holter monitoring was repeated beginning 2 hours before subjects received the third and final dose on the morning of day 2 and continuing until 30 minutes after the last collection time on the morning of day 3 of each dosing period. Triplicate digital ECGs were extracted from the Holter monitor recordings by a central reader at 10 points: 0.5, 1, 1.5, 2, 2.5, 3, 4, 6.5, 8, 12, and 24 hours after day 2 dosing or on day ‐1 at a preset time. At each time, three 10‐second intervals were selected a few minutes apart, and the mean QTc interval from 3 separate beats was analyzed on each ECG. The 24‐hour digital ECGs collected on day ‐1 of each period prior to day 1 dosing were considered baseline ECGs for each period. Analysis of baseline and on‐treatment ECGs was based on the same lead. Analysis of lead II was conducted with V5 as a backup and V2 as an alternative when T waves were not well defined in leads II or V5. Electrocardiograms were analyzed directly from the monitor using digital on‐screen calipers. Central reading of all electrocardiograms was conducted at Quintiles ECG (QECG, Bangalore, India). All ECGs of each subject were read by the same cardiologist, who was blinded to time, treatment, and subject identifiers. Subjects remained on bed rest in a semisupine position during the period of most frequent ECG acquisition (usually the first 4–6 hours of ECG sampling) and also for 30 minutes before the later acquisition times. Dose 3 was administered following a food fast of at least 6 hours, and no food was allowed for 4 hours after the dose. Light activity such as reading and watching television was allowed during this period. Extraction of Holter monitor data did not occur within approximately 2 hours after a meal. In addition, safety ECGs (12 lead) were performed on subjects in a semisupine position collected 0, 3, 12, and 15 hours after dosing on day 1; 0 and 3 hours after dosing on day 2; and 24 hours after dosing on day 3.
The QT interval was defined as the period from the beginning of the Q wave to the end of the T wave. The end of the QT interval was defined as the visual return of the T wave to the isoelectric baseline. If a discrete U wave was present, then it was excluded from the QT measurement, and QT was measured up to the end of the T wave or the nadir between the T and U waves. Abnormality of the U wave, if any, was specifically described and reported. The QT intervals were corrected for heart rate using Fridericia's formula (QTcF =). For each treatment and time, the time‐matched change from baseline QTcF was derived, with matching based on the time after dose administration. The analysis parameter was the time‐matched change from baseline under treatment QTcF minus the time‐matched change in QTcF from baseline during placebo use at the corresponding time (ΔΔQTcF). Baseline QTcF was established with time‐matched 24‐hour QTcF taken on day ‐1 of each period.
Serial blood samples for analyses of cabotegravir pharmacokinetics (PK) were collected before dosing and 0.5, 1, 2, 3, 4, 6.5, 8, 12, and 24 hours after dosing on day 2. Plasma samples were analyzed using a validated analytic method employing liquid chromatography followed by tandem mass spectrometry. The lower limit of quantification for cabotegravir was 25 ng/mL, and the upper limit was 25 000 ng/mL. Pharmacokinetics analyses were conducted using a noncompartmental model with Phoenix WinNonlin 6.3 (Pharsight Corp., St. Louis, Missouri). The PK parameters determined from the cabotegravir plasma concentration–time data included the following: area under the plasma concentration–time curve from 0 to 24 hours after administration of dose 3 (AUC0–24), AUC0–12, the maximum observed plasma concentration (Cmax), and Tmax.
Safety assessments were collected throughout the duration of the study and included the monitoring of adverse events (AEs), 12‐lead safety ECGs, clinical laboratory tests, vital signs, and physical examinations.
Bioanalytical Methods
After extraction from plasma by protein precipitation, plasma cabotegravir concentrations were determined by high‐performance liquid chromatography using tandem mass spectrometry with TurboIonSpray and multiple‐reaction monitoring at GlaxoSmithKline (AB Sciex Analyst, version 1.4.2; Applied Biosystems, Framingham, Massachusetts; SMS2000, version 2.1; GlaxoSmithKline, Research Triangle Park, North Carolina). The validated calibration range was 25 to 25 000 ng/mL, and each analytical run included quality control (QC) samples at 75, 1200, 18 800, and 62 500 ng/mL (10× dilution). On the basis of the analysis of these QC samples, the cabotegravir bias ranged from ‐4.3% to 6.7%, with a precision of <7.4%.
Statistical Analyses
On the basis of past QT studies with 3‐replicate ECG study design, a within‐subject standard deviation of 10 milliseconds for the QTcF was assumed. To rule out an effect size of ≥10 milliseconds and assuming a true treatment difference of 1 millisecond with a 5% significance level and 90% power, 34 evaluable subjects were required. Using a potential 20% dropout rate, 42 subjects were planned for enrollment. The primary end point of change from baseline in QTcF was analyzed for comparisons between placebo and cabotegravir using a repeated‐measures analysis of covariance model, fitting subject as a random effect and period, time, treatment, and time‐by‐treatment interaction as fixed effects. Subject and period baseline QTcF were included as continuous covariates, and sex effect was added to the full mean model for exploration. Point estimates for ΔΔQTcF at each point corresponding to 90%CIs were constructed for each comparison at each time. A linear mixed‐effects PK/pharmacodynamic model with cabotegravir concentration as a fixed effect and subject as a random effect was used to examine the relationship between observed cabotegravir concentration and ΔΔQTcF. Safety data were analyzed with descriptive statistics.
Results
Subject Demographics
Forty‐two subjects participated in the study (Table 1). Of the 42 subjects, 6 withdrew early: 1 because of a traumatic tear in the Achilles tendon 5 days after the last dose of cabotegravir, an AE that was not considered drug related; 3 because of protocol violations; and 2 because of being lost to follow‐up. Overall, the majority of subjects were male (33 [79%]) and white (22 [52%]) or of black/African heritage (17 [40%]). The mean age of subjects was 33.9 years, and the mean body mass index was 26.54 kg/m2.
Table 1.
Demographic Characteristics of Healthy Subjects
| Parameter | Overall (n = 42) |
|---|---|
| Age (y), mean ± SD | 33.9 ± 11.4 |
| Sex | |
| Female, n (%) | 9 (21) |
| Male, n (%) | 33 (79) |
| BMI (kg/m2), mean ± SD | 26.54 ± 3.18 |
| Height (cm), mean ± SD | 172.49 ± 7.75 |
| Weight (kg), mean ± SD | 78.96 ± 10.93 |
| Ethnicity | |
| Hispanic or Latino, n (%) | 3 (7) |
| Not Hispanic or Latino, n (%) | 39 (93) |
| Race | |
| White, n (%) | 22 (52) |
| Black/African heritage, n (%) | 17 (40) |
| American Indian or Alaskan native, n (%) | 2 (5) |
| Native Hawaiian or other Pacific Islander, n (%) | 1 (2) |
BMI, body mass index; SD, standard deviation.
Effect on QT Interval
The effects of cabotegravir and moxifloxacin on cardiac repolarization were evaluated as ΔΔQTcF and are summarized in Table 2 and Figure 1. For the primary end point, ΔΔQTcF, all time‐matched values and the corresponding upper bounds of the 90%CI were less than 10 milliseconds for cabotegravir. In contrast, the lower limit of the 90%CI exceeded the 5‐millisecond threshold at several times during treatment with moxifloxacin, demonstrating that the positive control was valid in detecting change in QTcF. Eight hours after dosing, the maximum observed time‐matched ΔΔQTcF was observed for both cabotegravir (2.62 milliseconds; 90%CI, ‐0.02 to 5.26 milliseconds) and moxifloxacin (13.03 milliseconds; 90%CI, 10.32 to 15.74 milliseconds). Cabotegravir was not associated with any instances of QTcF > 450 milliseconds, and none of the subjects who received cabotegravir had changes from baseline in QTcF values of >30 milliseconds. Similar patterns were observed for the QT interval corrected using Bazett's formula (QTcB), and individual subject‐specific QTc and sex were included in the final model to adjust for the difference between men and women (data not shown). In addition, there were no clinically significant differences between the 3 treatments in changes from baseline in heart rate.
Table 2.
Estimated ΔΔQTcF Values of the Effect of Cabotegravir and Moxifloxacin on QTcF Interval in Healthy Subjects
| Cabotegravir 150 mg Every | ||||
|---|---|---|---|---|
| 12 Hours × 3 Doses | Moxifloxacin 400 mg | |||
| Time Point (h) | ΔΔQTcF (ms),a mean ± SE | 90%CI | ΔΔQTcF (ms),b mean ± SE | 90%CI |
| 0.5 | 0.70 ± 2.00 | −2.62, 4.02 | 5.40 ± 2.05 | 2.00–8.81 |
| 1 | −0.37 ± 1.74 | −3.26, 2.53 | 8.75 ± 1.79 | 5.78–11.71 |
| 2 | 0.44 ± 1.83 | −2.61, 3.48 | 11.67 ± 1.88 | 8.55–14.79 |
| 3 | −0.62 ± 1.69 | −3.43, 2.19 | 10.49 ± 1.73 | 7.60–13.37 |
| 4 | −0.37 ± 1.53 | −2.91, 2.17 | 11.81 ± 1.57 | 9.20–14.42 |
| 6.5 | 0.50 ± 1.63 | −2.21, 3.22 | 10.08 ± 1.68 | 7.29–12.86 |
| 8 | 2.62 ± 1.59 | −0.02, 5.26 | 13.03 ± 1.63 | 10.32–15.74 |
| 12 | −0.34 ± 1.66 | −3.10, 2.42 | 7.79 ± 1.69 | 4.98–10.60 |
| 24 | 1.13 ± 2.05 | −2.27, 4.53 | 6.53 ± 2.10 | 3.04–10.02 |
Note: Lack of effect is demonstrated if the upper limit of the 90%CI (95th percentile) for cabotegravir–placebo at each time is completely contained within 10 ms.
ΔΔQTcF, change from baseline in QTcF between each active treatment relative to placebo; CI, confidence interval; QTcF, corrected QT interval using Fridericia's formula; SE, standard error.
Change from baseline of (cabotegravir–placebo).
Change from baseline of (moxifloxacin–placebo).
Figure 1.
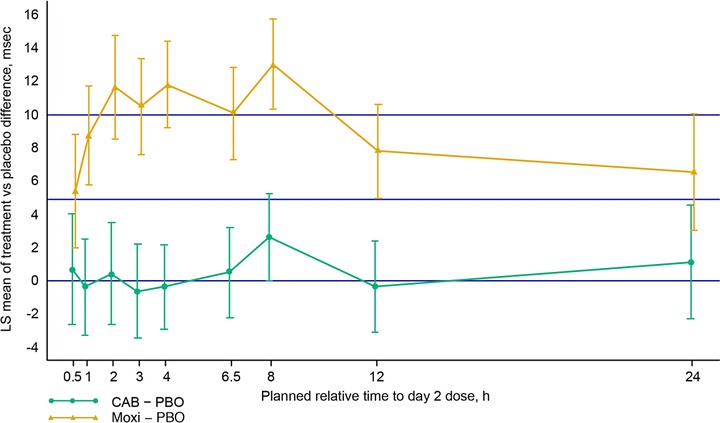
LS mean of treatment difference from placebo for QTcF change from baseline (± 90%CI). ΔΔQTcF, change from baseline in QTcF between each active treatment relative to placebo; CAB, cabotegravir; LS, least squares; Moxi, moxifloxacin; PBO, placebo.
Pharmacokinetics and Pharmacodynamics of Cabotegravir
Plasma cabotegravir exposure was similar between male and female subjects (Table 3). The geometric mean Cmax was 22.5 μg/mL following 3 doses of cabotegravir 150 mg every 12 hours; this was 3‐fold above the Cmax at steady state of 7.5 μg/mL that has been observed with a clinically therapeutic dose of cabotegravir 30 mg once daily, demonstrating that supratherapeutic concentrations were achieved.
Table 3.
Noncompartmental Pharmacokinetic Parameters After Repeat Doses of Oral Cabotegravir 150 mg in Healthy Subjects
| Cabotegravir 150 mg Every 12 Hours × 3 Dosesa | |||
|---|---|---|---|
| Parameter | Women (n = 9) | Men (n = 31) | All Subjects (n = 40) |
| Cmax (μg/mL), geometric mean (CV,%) [90%CI] | 25.1 (22) [21.2–29.6] | 21.9 (19) [20.4–23.4] | 22.5 (20) [21.1–24.0] |
| AUC0–12 (μg·h/mL), geometric mean (CV,%) [90%CI] | 234 (22) [198–277] | 213 (15) [202–225] | 217 (17) [206–229] |
| AUC0–24 (μg·h/mL), geometric mean (CV,%) [90%CI] | 409 (22) [345–484] | 380 (15) [359–402] | 386 (17) [366–408] |
| tmax (h), median (range) | 2.0 (1.0–4.0) | 2.0 (1.0–4.0) | 2.0 (1.0–4.0) |
AUC0–12, area under the plasma concentration–time curve from 0 to 12 hours; AUC0–24, AUC from 0 to 24 hours; CI, confidence interval; Cmax, maximum observed plasma concentration; CV, coefficient of variation (ratio of the standard deviation to the mean); tmax, time to Cmax.
Three doses of cabotegravir 150 mg (5 × 30‐mg tablets) every 12 hours.
The potential relationship between cabotegravir concentration and QTcF was examined using a linear mixed‐effects model, and no discernable concentration effect of cabotegravir on ΔΔQTcF was apparent (Figure 2). Parameter estimates of the relationship between ΔΔQTcF and time‐matched cabotegravir concentration were evaluated. The mean estimated intercept was 1.382 milliseconds (95%CI, ‐3.328 to 6.092 milliseconds), and the mean estimated slope for ΔΔQTcF was ‐0.00005 milliseconds (95%CI, ‐0.00029 to 0.000192 milliseconds). The P for the slope estimate was not significant, with values approaching zero, which indicated lack of a relationship between cabotegravir plasma concentration and ΔΔQTcF.
Figure 2.
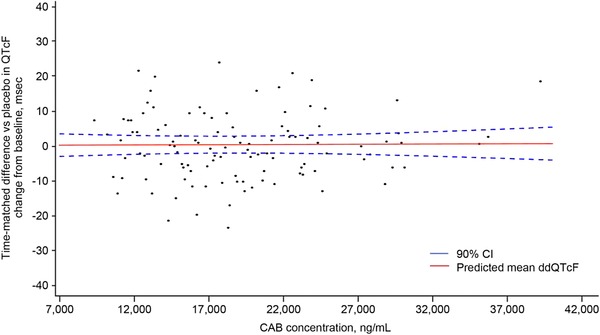
Scatterplot of individual ΔΔQTcF values versus CAB concentrations with regression line and 90%CI. ΔΔQTcF, change from baseline in QTcF between each active treatment relative to placebo; CAB, cabotegravir; CI, confidence interval.
Safety
Overall, a supratherapeutic dose of cabotegravir (150 mg every 12 hours × 3 doses) was well tolerated. The most frequently reported AE (n > 2) was contact dermatitis at the location of the ECG and Holter leads, which was reported by 6 subjects (15%) receiving cabotegravir, 9 subjects (23%) receiving placebo, and 2 subjects (6%) receiving moxifloxacin (Table 4). All other AEs were reported in 2 subjects each or less for each treatment group. All AEs were mild in intensity, with the exception of 2 grade 2 events. One subject reported a ruptured Achilles tendon following a fall that was recorded as a grade 2 AE during treatment with cabotegravir, and 1 subject had a transient elevation in creatine phosphokinase, presumably related to strenuous exercise, which was recorded as a grade 2 AE during treatment with moxifloxacin. Neither grade 2 event was considered related to study drug. There were no deaths or serious or significant AEs reported during the study. One subject withdrew because of a non‐drug‐related AE (ruptured Achilles tendon). No clinically significant trends in clinical laboratory values, vital signs, or safety ECG results were observed.
Table 4.
Adverse Events Occurring in at Least 2 Patients by Treatment (Safety Population)
| Adverse Event, n (%) | Cabotegravir 150 mg (n = 40) | Cabotegravir Placebo (n = 39) | Moxifloxacin 400 mg (n = 36) |
|---|---|---|---|
| Any adverse event | 13 (33) | 16 (41) | 12 (33) |
| Dermatitis contact | 6 (15) | 9 (23) | 2 (6) |
| Headache | 1 (3) | 2 (5) | 2 (6) |
| Nausea | 0 | 0 | 2 (6) |
| Alanine aminotransferase increased | 1 (3) | 1 (3) | 0 |
| Blood creatine phosphokinase increased | 0 | 1 (3) | 1 (3) |
Discussion
In a phase 2b dose‐ranging study in treatment‐naive HIV‐1‐infected adults (LATTE), potent activity and short time to viral suppression were observed at all doses of oral cabotegravir (10–60 mg once daily) plus dual nucleoside reverse transcriptase inhibitor therapy.13 Review of the overall data on efficacy and safety from the LATTE study led to the selection of the 30‐mg dose for future clinical studies. For the present study, 3 doses of cabotegravir given every 12 hours were administered to achieve a Cmax of cabotegravir that was approximately 3‐fold higher than that observed with the standard clinical dose of 30 mg once daily. Previous work has indicated that cabotegravir pharmacokinetics increase in a dose‐proportional manner from 10 to 30 mg, but in a less than proportional manner from 30 to 60 mg. Therefore, the dose used in the present investigation (150 mg every 12 hours x 3) achieved exposures that are likely on the plateau of the dose–response curve. Cabotegravir exposure in a long‐acting injectable regimen is expected to be lower than that observed with the 30‐mg oral tablet.
Prior studies with raltegravir10 and dolutegravir11 suggest that INSTIs do not affect cardiac repolarization. Consistent with previous preclinical and clinical12 evidence, this study in healthy subjects demonstrates that doses of cabotegravir achieving supratherapeutic concentrations compared with anticipated therapeutic doses do not prolong the QT interval and, thus, have no effect on cardiac repolarization. The point estimate for ΔΔQTcF did not exceed 5 milliseconds at any point, and the corresponding upper bounds of the 90%CI were less than 10 milliseconds at all times. In contrast, during moxifloxacin exposure, the lower limit of the 90%CI exceeded the 5‐ millisecond threshold at several times, consistent with established clinical studies,14 thus validating the sensitivity of this study to assess the effects of cabotegravir on cardiac repolarization. In a pooled analysis of 20 QTc studies involving moxifloxacin,14 the maximum effect of moxifloxacin‐induced QTc effect occurred between 1 and 4 hours after moxifloxacin dosing. In the present study, the maximum effect of ΔΔQTcF occurred at 8 hours for both cabotegravir and moxifloxacin, even though peak cabotegravir exposure occurred at approximately 2 hours. In a previous randomized, placebo‐controlled study of 20 healthy subjects, the first peak of placebo‐adjusted moxifloxacin QTcF change from baseline was observed 4 hours after the dose, followed by a second increase in QTc observed 8 hours postdose.15 This effect is likely related to eating (subjects in this study were given a meal 4 hours after the dose) and an increase in activity (subjects were not required to remain in a supine and resting position after the meal). In the present study, subjects were to remain on bed rest only during the period of most frequent ECG acquisition, the first 4 to 6 hours of sampling.
In the present study, there was no significant change from baseline in QTcF at any time in the cabotegravir‐exposed groups, as the 90%CIs included zero, compared with a positive effect observed in the moxifloxacin group, in which zero was excluded from the 90%CI at all measured times. In addition, the PK/PD model demonstrates no relationship between cabotegravir plasma concentration and ΔΔQTcF. No clinically significant changes in clinical laboratory values, vital signs, or safety ECG results were observed. These results suggest that cabotegravir is not expected to have an effect on QT prolongation at the highest Cmax that might be expected in a clinical setting. This study supports the use of standard cardiac monitoring for both HIV‐infected and HIV‐uninfected subjects enrolled in future studies of cabotegravir.
Declaration of Conflicting Interests
At the time the study was conducted, all authors were employees of GlaxoSmithKline. All authors hold stock options within GlaxoSmithKline.
Funding
Funding for this study was provided by ViiV Healthcare.
Acknowledgments
The authors thank Stephen C. Piscitelli, PharmD, for contributions to the study design and the investigator and research subjects at Quintiles Phase One Services, LLC, Overland Park, Kansas. Editorial assistance was provided under the direction of the authors by Todd Parker and Meredith MacPherson, MedThink SciCom.
References
- 1. Marchand C, Maddali K, Metifiot M, Pommier Y. HIV‐1 IN inhibitors: 2010 update and perspectives. Curr Top Med Chem. 2009;9(11):1016–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dow DE, Bartlett JA. Dolutegravir, the Second‐Generation of Integrase Strand Transfer Inhibitors (INSTIs) for the Treatment of HIV. Infect Dis Ther. 2014;3:83–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Markowitz M, Nguyen BY, Gotuzzo E, et al. Sustained antiretroviral effect of raltegravir after 96 weeks of combination therapy in treatment‐naive patients with HIV‐1 infection. J Acquir Immune Defic Syndr. 2009;52(3):350–356. [DOI] [PubMed] [Google Scholar]
- 4. Steigbigel RT, Cooper DA, Teppler H, et al. Long‐term efficacy and safety of raltegravir combined with optimized background therapy in treatment‐experienced patients with drug‐resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 phase III trials. Clin Infect Dis. 2010;50(4):605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spreen W, Min S, Ford SL, et al. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials. 2013;14(5):192–203. [DOI] [PubMed] [Google Scholar]
- 6. Bowers GD, Culp A, Reese MJ, et al. Disposition and metabolism of cabotegravir: a comparison of biotransformation and excretion between different species and routes of administration in humans. Xenobiotica. 2016;46(2):147–162. [DOI] [PubMed] [Google Scholar]
- 7. Ford SL, Gould E, Chen S, et al. Lack of pharmacokinetic interaction between rilpivirine and integrase inhibitors dolutegravir and GSK1265744. Antimicrob Agents Chemother. 2013;57(11):5472–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ford SL, Gould E, Chen S, et al. Effects of etravirine on the pharmacokinetics of the integrase inhibitor S/GSK1265744. Antimicrob Agents Chemother. 2013;57(1):277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Darpo B, Nebout T, Sager PT. Clinical evaluation of QT/QTc prolongation and proarrhythmic potential for nonantiarrhythmic drugs: the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use E14 guideline. J Clin Pharmacol. 2006;46(5):498–507. [DOI] [PubMed] [Google Scholar]
- 10. Iwamoto M, Kost JT, Mistry GC, et al. Raltegravir thorough QT/QTc study: a single supratherapeutic dose of raltegravir does not prolong the QTcF interval. J Clin Pharmacol. 2008;48(6):726–733. [DOI] [PubMed] [Google Scholar]
- 11. Chen S, Min SS, Peppercorn A, et al. Effect of a single supratherapeutic dose of dolutegravir on cardiac repolarization. Pharmacotherapy. 2012;32(4):333–339. [DOI] [PubMed] [Google Scholar]
- 12. Lou Y, Gould E, Chen S, et al. Meta‐analysis of safety data from 8 clinical studies with GSK1265744, an HIV integrase inhibitor, dosed orally or as injection of long‐acting parenteral nanosuspension (LAP). Poster presented at: 53rd Interscience Conference of Antimicrobial Agents and Chemotherapy; September 10‐13, 2013; Denver, CO.
- 13. Margolis DA, Brinson CC, Smith GH, et al. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral‐naive adults with HIV‐1 infection (LATTE): a randomised, phase 2b, dose‐ranging trial. Lancet Infect Dis. 2015;15(10):1145–1155. [DOI] [PubMed] [Google Scholar]
- 14. Yan LK, Zhang J, Ng MJ, Dang Q. Statistical characteristics of moxifloxacin‐induced QTc effect. J Biopharm Stat. 2010;20(3):497–507. [DOI] [PubMed] [Google Scholar]
- 15. Bloomfield DM, Kost JT, Ghosh K, et al. The effect of moxifloxacin on QTc and implications for the design of thorough QT studies. Clin Pharmacol Ther. 2008;84(4):475–480. [DOI] [PubMed] [Google Scholar]