

Abstract
The pharmacokinetics of apremilast and its major metabolite M12 were evaluated in subjects with varying degrees of renal impairment. Men and women with renal impairment (estimated glomerular filtration rate, 60‒89 mL/min [mild, n = 8], 30‒59 mL/min [moderate, n = 8], or <30 mL/min [severe, n = 8]) or demographically healthy matched (control) subjects (n = 24) received a single oral dose of apremilast 30 mg. Plasma apremilast and metabolite M12 concentrations were determined, and pharmacokinetic parameters were calculated from samples obtained predose and up to 72 hours postdose. In subjects with mild to moderate renal impairment, apremilast pharmacokinetic profiles were similar to healthy matched subjects. In subjects with severe renal impairment, apremilast elimination was significantly slower, and exposures based on area under the plasma concentration‐versus‐time curve from time zero extrapolated to infinity and maximum observed plasma concentration were increased versus healthy matched subjects. Metabolite M12 pharmacokinetic profiles for subjects with mild renal impairment were similar to those of the healthy matched subjects; however, they were increased in both the moderate and severe renally impaired subjects. Dose reduction of apremilast is recommended in individuals with severe renal impairment, but not in those with mild to moderate renal impairment.
Keywords: apremilast, kidney disease, phosphodiesterase 4 inhibitor, pharmacokinetics
Apremilast is an orally available small molecule that specifically inhibits the enzymatic activity of phosphodiesterase 4 (PDE4), the predominant phosphodiesterase isoform in inflammatory immune cells.1, 2, 3 PDE4A, PDE4B, PDE4C, and PDE4D isozymes constitute a diverse family of enzymes that serve as the primary means for degradation of cyclic adenosine monophosphate, an intracellular secondary messenger that helps to maintain immune homeostasis.1, 2, 4, 5 Apremilast inhibits PDE4, thereby increasing the intracellular concentration of cyclic adenosine monophosphate. This results in decreased production of proinflammatory mediators, such as inducible nitric oxide synthase, tumor necrosis factor–α, IL‐23, IL‐17A, and IL‐22 (key cytokines in the pathophysiology of psoriatic arthritis [PsA] and psoriasis),1, 6, 7, 8 and increased production of anti‐inflammatory cytokines, such as IL‐10 and IL‐1 receptor antagonists.6, 9
Apremilast is approved in several countries, including the United States, for the treatment of adult patients with active PsA and patients with moderate to severe plaque psoriasis, and it is currently in clinical development for the treatment of various other immune inflammatory conditions.1 In phase 2 and 3 studies, apremilast has demonstrated efficacy in adult patients with active PsA10, 11 and in patients with moderate to severe plaque psoriasis.12, 13, 14 In addition, apremilast 30 mg twice daily has been shown to be generally well tolerated and safe.10, 11, 12, 13, 14
In healthy subjects, apremilast has demonstrated a linear, dose‐related pharmacokinetic profile.15 After a single oral dose in healthy subjects, apremilast undergoes rapid absorption, with ≈73% absolute bioavailability and only ≈3% of a given dose excreted in urine unchanged.16, 17 Metabolism of apremilast is extensive and diverse; the chemical structures of apremilast and M12 and their metabolic pathways have been published by Hoffmann et al.16 The predominant circulating metabolite, M12 (O‐desmethyl apremilast glucuronide), is pharmacologically inactive and accounts for 39% of the circulating radioactivity after a single oral dose of [14C]apremilast.18 Because O‐demethylation of apremilast is primarily catalyzed by the hepatic enzyme CYP3A4, CYP3A4 may play a major role in the oxidative metabolism of apremilast.
The 2 studies reported here were conducted to determine whether apremilast dose adjustments are needed in subjects with varying degrees of renal impairment. These studies evaluated the pharmacokinetics of apremilast and its major metabolite, M12, in subjects with mild and moderate renal impairment as well as in subjects with severe renal impairment who did not require routine dialysis.
Methods
Study Design
The renal impairment studies were conducted at 2 study centers (Site 001CO: St. Anthony's Medical Plaza 1, Lakewood, Colorado; Site 001MN: Minneapolis, Minnesota). The mild and moderate renal impairment study protocol, informed consent, and other related documents were reviewed and approved by the Western Institutional Review Board (Olympia, Washington) before the start of the study. The severe renal impairment study protocol, informed consent, and other related documents were approved by the Crescent City Institutional Review Board (New Orleans, Louisiana) and the Independent Investigational Review Board (Plantation, Florida) before the start of the study. All subjects were required to read and sign the approved informed consent form before entry into the study and before any study‐related procedures were performed. These 2 similarly designed, 2‐center, open‐label, single‐dose studies evaluated the potential impact of mild and moderate renal impairment or severe renal impairment on the pharmacokinetics of apremilast and its metabolite M12 after oral administration. Subjects with mild, moderate, or severe renal impairment were separately matched with healthy subjects based on age (±15 years), sex, and weight (±20%).
Baseline safety and inclusion criteria assessments were conducted. Participants were confined to the study center the evening before apremilast dosing (eg, day −1, baseline) through the pharmacokinetic sampling period. Each subject received a single oral dose of apremilast on the morning of day 1 after fasting ≥ 8 hours. Blood samples for pharmacokinetic analysis were obtained at scheduled times up to 72 hours after apremilast administration. Subjects returned for follow‐up safety evaluations 11‒18 days after apremilast administration.
Subjects
The study included men and women (≥18 and ≤80 years old) with a body mass index ≥ 18 and ≤ 36 kg/m2 who were either medically stable with renal impairment or healthy (controls) and free of acute major illness for at least 1 month before dosing, as determined by medical history, physical examination findings, vital signs, electrocardiograms (ECGs), and clinical laboratory safety tests. Subjects with mild and moderate renal impairment had an estimated glomerular filtration rate (eGFR) of 60‒89 mL/min (inclusive [mild]) or 30‒59 mL/min (inclusive [moderate]), and subjects with severe renal impairment had an eGFR < 30 mL/min (not requiring dialysis). The eGFR was calculated based on the Modification of Diet in Renal Disease equation: 175 × (Scr)‐1.154 × (age)‐0.203 × (0.742, if female) × (1.212, if African American).
Excluded from the study were individuals with any serious medical condition, clinically significant laboratory abnormality (except those related to renal impairment and associated complications), psychiatric illness that would prevent study participation, or a history of any unstable, clinically significant illness within 3 months before the study. Also excluded were individuals with renal impairment who had received a renal transplant or had hemoglobin < 9 g/dL, white blood cell count < 3000/μL or > 15 000/μL, aspartate aminotransferase or alanine aminotransferase > 2 times the upper limit of normal, total bilirubin > 2.2 times the upper limit of normal, international normalized ratio > 3, platelet count < 50 000/μL, or albumin < 3 g/dL. In both studies, healthy individuals were excluded if they had any surgical or medical conditions possibly affecting drug absorption, distribution, metabolism, and excretion, including but not limited to irritable bowel syndrome, peptic ulcer, cholecystectomy, and chronic liver disease, or use of any prescribed systemic or topical medication within 30 days before administration of the study medication. Individuals were excluded if they had a history of drug or alcohol abuse within 2 years before dosing or positive screening for illicit drugs or alcohol, or were carriers of the hepatitis B surface antigen or hepatitis C antibody. Individuals who were positive for human immunodeficiency virus antibodies and women who were pregnant or breast‐feeding were not permitted to participate in either study.
Pharmacokinetic Sampling, Collection, and Analytical Methodology
Blood samples for determining apremilast and M12 plasma concentrations were collected predose (0 hour) and 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, and 72 hours postdose. Urine samples were collected predose (≤60 minutes before dosing) and at intervals of 0‒8, 8‒24, 24‒48, and 48‒72 hours postdose.
Plasma and urine apremilast and M12 concentrations were measured using a validated liquid chromatography–mass spectrometry method conducted and validated by QPS, LLC (Newark, Delaware). The lower limit of quantitation is 1 ng/mL for apremilast and 5 ng/mL for M12. An achiral assay was used to measure plasma and urine apremilast and M12 concentrations.
For the plasma assay, apremilast and its internal standard were quantitatively extracted from 100 μL of plasma sample using a liquid–liquid extraction method with methyl tert‐butyl ether and reconstituted with 200 μL of H2O:methanol:formic acid 80:20:0.1 (v/v/v). M12 and its internal standard were quantitatively extracted from 300 μL of plasma sample using a liquid–liquid extraction method with 0.04 M citric acid using ethyl acetate:isopropanol at 95:5 (v/v) and reconstituted with 200 μL of water:methanol:formic acid at 70:30:0.1 (v/v/v). The sample extract was loaded onto a Synergi Hydro‐RP 30 × 2 mm, 4 μm for separation. The mobile phase was composed of both H2O:formic acid 100:0.1 (v/v) and MeOH:formic acid 100:0.1 (v/v) for the apremilast assay, and the mobile phase was composed of both A (10 mM ammonium acetate in water [pH ∼4]) and B (methanol) for the M12 assay. The high‐performance liquid chromatography effluent was introduced into the API‐4000 tandem mass spectrometer equipped with a TurboIon Spray source (AB Sciex Pte. Ltd., Framingham, Massachusetts) for apremilast. Positive ions were detected in the multiple‐reaction monitoring mode with precursor→product ion pairs of 461.16→257.05 m/z for apremilast and 640.3→164.2 m/z for M12.
The apremilast and M12 plasma methods had an assay range of 1‒1000 ng/mL and 15‒800 ng/mL with a precision (percent coefficient of variation [CV%]) of ≤7.3% and an accuracy (percent relative error [RE%]) of ‐2.27%‒5.3%, and ≤6.7% and an accuracy (RE%) of −1.0%‒2.9%, respectively.
For the urine assay, apremilast and M12 and their internal standards were quantitatively extracted from 50 μL of the urine sample using a liquid–liquid extraction method with 3.84 mg citric acid/mL urine using ethyl acetate:isopropanol 95:5 (v/v) and reconstituted with 1000 μL of acetonitrile:water:formic acid 20:80:0.1 (v/v/v). The sample extract was loaded onto a Synergi 4 μm, MAX‐RP, 50 × 2 mm column for separation. The mobile phase was composed of both A (5 mM ammonium acetate in water:formic acid 100:0.1 [v/v]) and B (acetonitrile:water 95:5 [v/v]). The high‐performance liquid chromatography effluent was introduced into an API‐4000 tandem mass spectrometer equipped with a TurboIon Spray source for apremilast. Positive ions were detected in the multiple reaction monitoring mode with precursor→product ion pairs of 461.16→257.05 m/z for apremilast and 640.3→164.2 m/z for M12.
The apremilast and M12 urine assay used ranged from 15 to 1600 and from 150 to 16 000 ng/mL with a precision (CV%) of ≤7.3% and an accuracy (RE%) of ‐5.5%‒3.9%, and with a CV% of ≤8.1% and an RE% of ‐3.3%‒7.7%, respectively.
Pharmacokinetic Evaluation
The data analysis and data presentation were assessed using WinNonlin Professional version 5.3 software (Pharsight Corporation, Mountain View, California) and NONMEM version VI or higher (ICON Development Solutions, Ellicott City, Maryland).
Pharmacokinetic parameters were derived for apremilast and M12 by noncompartmental analysis and included area under the plasma concentration‐versus‐time curve (AUC) from time zero to the last measurable concentration (AUC0–t), AUC from time zero extrapolated to infinity (AUC0–∞), maximum observed plasma concentration (Cmax), time to Cmax (Tmax), estimated terminal elimination half‐life (t1/2) in plasma, apparent total plasma clearance (CL/F; apremilast only), calculated as (dose/AUC0–∞), and apparent total volume of distribution (VZ/F; apremilast only), calculated as ([CL/F]/λZ), where λZ is the terminal rate constant, calculated by linear regression of the terminal portion of the log concentration‐versus‐time curve in plasma.
Amount of M12 excreted in urine was calculated by multiplying the urine concentration by the volume of urine of each collection interval. Renal clearance of M12 was then obtained by dividing the cumulative amounts of M12 in urine over a 72‐hour period by its plasma AUC from 0 to 72 hours.
Safety
Safety was monitored throughout the studies based on collection of adverse events (AEs), complete physical examinations, vital signs, 12‐lead ECGs, clinical laboratory safety tests, and a record of prior and concomitant medications.
Statistical Analysis
All subjects who received apremilast and had evaluable pharmacokinetic profiles were included in the pharmacokinetic analyses. Plasma concentration was summarized descriptively by group and time postdosing; pharmacokinetic parameters also were summarized descriptively by group (mean, standard deviation [SD], CV%, geometric mean, geometric percent coefficient of variation, minimum, median, and maximum). Mean ± SD apremilast and M12 plasma concentration‐versus‐time profiles were plotted using a linear scale and semilogarithmic scale. For AUC and Cmax, an analysis of variance (ANOVA) model was used to calculate the ratio of geometric means and its 90% confidence interval (CI) between subjects with renal impairment and healthy matched subjects. For the mild and moderate renal impairment study (impaired vs healthy), the ANOVA model included group (mild and moderate), status (impaired and healthy), and group‐by‐status interaction as a fixed effect and matched pair nested within group as a random effect. For the severe renal impairment study (impaired vs healthy), the ANOVA model included status (impaired vs healthy) as a fixed effect and matched pair as a random effect. For Tmax, the Wilcoxon signed rank test was performed, and Hodges‐Lehmann estimate with its 90%CI was calculated for the median difference between subjects with renal impairment versus each respective group of healthy matched subjects.
Results
Subjects
A total of 48 subjects were enrolled in the 2 studies; of these, 8 subjects had mild, 8 had moderate, and 8 had severe renal impairment and were separately matched with 8 healthy subjects. All subjects were included in the pharmacokinetic analysis population except for 1 healthy matched subject in the severe renal impairment study who vomited ≈1 hour postdose and did not have evaluable pharmacokinetic profiles.
Subjects ranged from 33 to 72 years old in the mild and moderate renal impairment study and 50 to 74 years old in the severe renal impairment study (Table 1). The baseline demographic and clinical characteristics such as weight, height, and body mass index are presented in Table 1. The mild and moderate renal impairment study included 5 women and 3 men in each group, and the severe renal impairment study included 6 women and 2 men in each group.
Table 1.
Baseline Demographic and Clinical Characteristics of Fasting Subjects for the 2 Renal Impairment Studies
| Mild Renal Impairment | Moderate Renal Impairment | Severe Renal Impairment | ||||
|---|---|---|---|---|---|---|
| Impaired Subjects (n = 8) | Healthy Matched Subjects (n = 8) | Impaired Subjects (n = 8) | Healthy Matched Subjects (n = 8) | Impaired Subjects (n = 8) | Healthy Matched Subjects (n = 8) | |
| Age, y | ||||||
| Mean (SD) | 55.6 (4.4) | 48.0 (5.4) | 56.4 (14.6) | 51.1 (6.3) | 62.4 (7.8) | 58.3 (8.6) |
| Min–max | 51–64 | 43–57 | 33–72 | 41–59 | 52–73 | 50–74 |
| Weight, kg | ||||||
| Mean (SD) | 75.8 (14.4) | 82.2 (13.7) | 88.7 (16.9) | 86.8 (17.4) | 83.5 (20.6) | 77.2 (15.7) |
| Min–max | 50.1–100.6 | 53.5–99.7 | 64.3–107.8 | 64.2–114.0 | 62.4–118.9 | 54.6–101.1 |
| Height, cm | ||||||
| Mean (SD) | 170.9 (5.9) | 169.6 (9.2) | 176.3 (10.4) | 175.0 (10.9) | 167.6 (11.3) | 169.8 (8.1) |
| Min–max | 165.0–181.0 | 154.0–181.0 | 162.0–186.5 | 161.5–187.0 | 157.0–190.0 | 160.0–188.0 |
| BMI, kg/m2 | ||||||
| Mean (SD) | 26.0 (5.1) | 28.5 (4.1) | 28.3 (2.9) | 28.2 (4.2) | 29.6 (5.7) | 26.7 (4.5) |
| Min–max | 18.4–33.4 | 21.4–33.7 | 24.5–31.7 | 21.3–34.5 | 21.9–36.0 | 19.2–32.6 |
BMI, body mass index; Min‐max, minimum‐maximum.
Apremilast and M12 Pharmacokinetics
The plasma concentration‐versus‐time profiles for apremilast among subjects with mild renal impairment were similar in shape to those observed in healthy matched subjects (Figure 1A). Among subjects with moderate renal impairment, mean apremilast plasma profiles were slightly higher than those in the healthy matched subjects (Figure 1B), and among subjects with severe renal impairment, the overall apremilast plasma concentrations across times were higher than those in healthy matched subjects (Figure 1C). Apremilast pharmacokinetic parameters in subjects with mild or moderate renal impairment were generally comparable to those observed in healthy matched subjects; however, those with severe renal impairment differed from the healthy matched subjects (Table 2).
Figure 1.
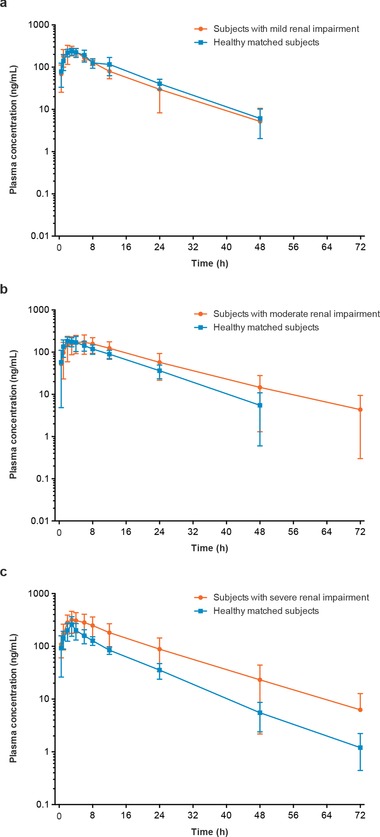
Mean (SD) apremilast plasma concentration‐versus‐time profiles (semilog scale) in subjects with (a) mild, (b) moderate, and (c) severe renal impairment versus healthy matched subjects.
Table 2.
Summary of Apremilast Plasma Pharmacokinetic Parameters and Statistical Analysis
| Mean (CV%) | |||||||
|---|---|---|---|---|---|---|---|
| AUC0–∞, ng·h/mLa | Cmax, ng/mLa | Tmax, hb | t1/2, h | CL/F, L/ha | Vz/F, La | ||
| Mild renal impairment | Impaired, apremilast 30 mg (n = 8) | 3032.24 (20.9) | 273.52 (25.4) | 3.00 (2.0–4.0) | 8.54 (19.3) | 10.28 (20.9) | 123.24 (12.1) |
| Healthy matched, apremilast 30 mg (n = 8) | 3522.84 (20.6) | 252.00 (15.5) | 3.00 (2.0–4.1) | 8.34 (23.5) | 8.79 (17.5) | 106.77 (31.1) | |
| Ratio (90%CI)c | 85.9 (65.8–112.0) | 106.2 (84.7–133.1) | 0.0 (‐1.00 to 1.00) P > .9999 | 116.5 (89.3–151.9) | 120.4 (92.2–157.2) | ||
| Moderate renal impairment | Impaired, apremilast 30 mg (n = 8) | 3993.5 (51.7) | 197.2 (41.9) | 3.50 (0.5–8.0) | 11.12 (35.6) | 10.31 (70.2) | 144.31 (51.8) |
| Healthy matched, apremilast 30 mg (n = 8) | 2905.19 (22.2) | 215.31 (25.0) | 2.00 (1.0–6.0) | 8.51 (24.8) | 10.84 (25.2) | 129.12 (21.3) | |
| Ratio (90%CI)c | 122.1 (93.6–159.3) | 87.5 (69.8–109.7) | 1.0 (‐0.50 to 2.50) P = .25 | 81.9 (62.8–106.8) | 103.2 (79.0–134.8) | ||
| Severe renal impairment | Impaired, apremilast 30 mg (n = 8) | 6050.0 (50.5) | 384.3 (32.7) | 3.0 (1.0–6.0) | 11.994 (17.2) | 6.125 (45.9) | 104.11 (47.0) |
| Healthy matched, apremilast 30 mg (n = 7) | 2917.1 (17.5) | 271.0 (36.0) | 3.0 (2.0–4.0) | 9.476 (16.9) | 10.564 (17.8) | 143.29 (21.7) | |
| Ratio (90%CI)c | 188.5 (132.5–268.0) | 141.6 (102.9–194.8) | 0.00 (‐1.5 to 1.5) P > .9999 | 2.485d (NC) | 53.10 (37.3–75.4) | 67.35 (NC) | |
ANOVA, analysis of variance; AUC0–∞, area under the concentration‐versus‐time curve from time 0 to infinity; CI, confidence interval; CL/F, apparent total plasma clearance; Cmax, maximum observed plasma concentration; CV%, percent coefficient of variation; NC, not calculated; t½, elimination half‐life; Tmax, time to Cmax; Vz/F, apparent total volume of distribution.
The ratio of geometric means (renal impaired/healthy matched) with its 90%CI was calculated from an analysis of variance (ANOVA) model, based on the natural log‐transformed pharmacokinetic values. For the mild and moderate renal impairment study, the ANOVA model included group (mild and moderate), status (impaired and healthy), and group‐by‐status interaction as fixed effects and matched pair nested within group as a random effect. For the severe renal impairment study, the ANOVA model included status (impaired and healthy) as a fixed effect and matched pair as a random effect.
The Tmax is summarized by median (range); statistical comparison based on the Wilcoxon signed rank test and Hodges‐Lehmann estimate with its 90%CI for the median difference (renal impaired/healthy matched).
The geometric mean ratio and 90%CI of the geometric mean ratio are presented as percentages.
The t1/2 statistical comparison displays geometric mean difference (severely renal impaired/healthy matched).
Statistical analyses of AUC0–∞, Cmax, and Tmax indicated comparable overall exposure to apremilast in subjects with mild renal impairment and in healthy matched subjects (Table 2). The apremilast AUC0–∞ was ≈22% higher and Cmax was ≈13% lower in the moderate renal impairment group relative to the healthy matched subjects; however, the 90%CI for the apremilast AUC0–∞ ratio (93.6%–159.3%), CL/F ratio (62.8%–106.8%), and Cmax ratio (69.8%–109.7%) contained unity or 100%, suggesting that the differences noted are not statistically significant (Table 2). Statistical analysis of AUC0–∞, Cmax, and Tmax indicated increased overall exposure to apremilast in subjects with severe renal impairment compared with healthy matched subjects (Table 2). Mean apremilast AUC0–∞ was 88.5% higher and mean Cmax was 41.6% higher in subjects with severe renal impairment compared with healthy matched subjects. The corresponding 90%CIs did not contain unity or 100%, indicating significantly greater overall apremilast exposure. Tmax was largely unchanged (Table 2). Further statistical analysis revealed that increased apremilast exposure was likely due to slower elimination. The t1/2 was prolonged by ≈27% (2.5 hours), and systemic CL/F and VZ/F were decreased by ≈47.1% and 32.7%, respectively. Based on the calculated 90%CI, the decrease in apremilast CL/F with severe renal impairment was statistically significant compared with healthy matched subjects.
The plasma concentration‐versus‐time profiles for M12 among subjects with mild or moderate renal impairment were generally higher than those observed in healthy matched subjects (Figure 2A,B). The plasma concentration‐versus‐time profiles for M12 among subjects with severe renal impairment differed in shape compared with healthy matched subjects (Figure 2C), marked by relatively greater M12 plasma concentrations throughout the postdose evaluation period. Statistical analysis of AUC0–∞ and Cmax indicated that overall exposure to M12 was higher in subjects with mild, moderate, or severe renal impairment compared with healthy matched subjects (Table 3). Subjects with mild renal impairment had AUC0–∞ and Cmax values that were 29.6% and 30.8% higher, respectively, than those in healthy matched subjects. The 90%CI for M12 AUC0–∞ and Cmax ratio contained unity or 100%, suggesting that the difference was not statistically significant between the mild renal impairment group and the healthy matched group in M12 AUC0–∞ and Cmax. In the subjects with moderate renal impairment, AUC0–∞ and Cmax were 61.4% and 16.9% higher, respectively, than those in the healthy matched subjects. The 90%CI for the M12 AUC0–∞ ratio did not contain unity or 100% (Table 3), suggesting that the difference was statistically significant between the moderate renal impairment group and the healthy matched group in M12 AUC0–∞. The 90%CI for M12 Cmax ratio contained unity or 100%, suggesting that the difference in M12 Cmax was not statistically significant between the moderate renal impairment group and the healthy matched group. Analysis of pharmacokinetic parameters indicated a greater overall M12 plasma concentration throughout the postdose evaluation period for subjects with severe renal impairment compared with healthy matched subjects. M12 pharmacokinetic parameters in subjects with severe renal impairment also differed from those observed in healthy matched subjects (Table 3). Significantly greater overall exposure to M12 was observed among subjects with severe renal impairment than among healthy matched subjects, based on increases in AUC0–∞ (191.7%) and Cmax (42.9%). Tmax was delayed ≈6.25 hours in subjects with renal impairment compared with healthy matched subjects, which was statistically significant (P ≤ .05). Renal clearance of M12 was decreased in subjects with renal impairment compared with healthy matched subjects (geometric mean renal clearance was 2.35 L/h for subjects with mild renal impairment, 3.67 L/h for healthy subjects matched with subjects with mild renal impairment, 1.84 L/h for subjects with moderate renal impairment, and 3.02 L/h for healthy subjects matched with subjects with moderate renal impairment). As with apremilast, increased M12 exposure was accompanied by elimination (t1/2) that was prolonged by 62% (10.5 hours); M12 CL/F and VZ/F were not examined.
Figure 2.
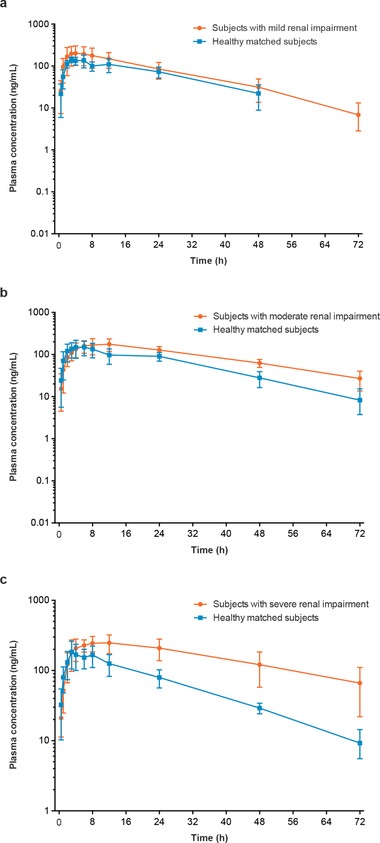
Mean (SD) M12 plasma concentration‐versus‐time profiles (semilog scale) in subjects with (a) mild, (b) moderate, and (c) severe renal impairment versus healthy matched subjects.
Table 3.
Summary of M12 Plasma Pharmacokinetic Parameters
| Geometric Mean (Geometric CV%) | |||||
|---|---|---|---|---|---|
| Study/Group | Group | AUC0–∞, ng·h/mLa | Cmax, ng/mLa | Tmax, hb | t1/2, h |
| Mild renal impairment | Impaired, apremilast 30 mg (n = 8) | 5424.52 (40.4) | 224.66 (42.7) | 5.0 (2.0–12.0) | 14.51 (22.2) |
| Healthy matched, apremilast 30 mg (n = 8) | 3973.1 (25.4) | 155.9 (23.2) | 6.0 (3.0–8.0) | 13.24 (12.6) | |
| Ratio (90%CI)c | 129.6 (99.5–168.8) | 130.8 (91.7–186.6) | NC | NC | |
| Moderate renal impairment | Impaired, apremilast 30 mg (n = 8) | 7902 (23.0) | 191.55 (33.7) | 10.0 (4.0–12.1) | 23.74 (50.4) |
| Healthy matched, apremilast 30 mg (n = 8) | 4875.3 (21.0) | 166.1 (39.7) | 5.0 (2.0–24.0) | 15.53 (28.8) | |
| Ratio (90%CI)c | 161.4 (122.8–212.3) | 116.9 (81.9–166.8) | NC | NC | |
| Severe renal impairment | Impaired, apremilast 30 mg (n = 8) | 15 042.9 (47.0) | 276.3 (26.0) | 12.0 (3.0–24.0) | 29.7 (44.4) |
| Healthy matched, apremilast 30 mg (n = 7) | 4820.0 (24.9) | 198.4 (36.0) | 4.0 (3.0–8.0) | 17.2 (22.4) | |
| Ratio (90%CI)c | 291.7 (204.3–416.4) | 142.9 (106.3–192.1) | 6.25 (2.975–11.0) | 10.498d (NC) | |
ANOVA, analysis of variance; AUC0–∞, area under the concentration‐versus‐time curve from time 0 to infinity; CI, confidence interval; Cmax, maximum observed plasma concentration; CV%, percent coefficient of variation; NC, not calculated; t½, elimination half‐life; Tmax, time to Cmax.
The ratio of geometric means (renal impaired/healthy matched) with its 90%CI was calculated from an ANOVA model based on the natural log‐transformed pharmacokinetic values. For the mild and moderate renal impairment study, the ANOVA model included group (mild and moderate), status (impaired and healthy), and group‐by‐status interaction as fixed effects and matched pair nested within group as a random effect. For the severe renal impairment study, the ANOVA model included status (impaired vs healthy) as a fixed effect and matched pair as a random effect.
The Tmax is summarized by median (range); statistical comparison based on the Wilcoxon signed rank test and Hodges‐Lehmann estimate with its 90%CI for the median difference (renal impaired/healthy matched).
The geometric mean ratio and 90%CI of the geometric mean ratio are presented as percentages.
The t1/2 statistical comparison displays geometric mean difference (severely renal impaired/healthy matched).
Safety
In the mild and moderate renal impairment groups and their healthy matched groups, a total of 9 subjects (mild renal impairment, n = 2; moderate renal impairment, n = 2, and healthy matched, n = 5) reported 22 AEs. Of these, 6 subjects (mild renal impairment, n = 2; moderate renal impairment, n = 1; and healthy matched, n = 3) had AEs suspected to be related to study medication or procedures, including headache, nausea, epigastric discomfort, and dysgeusia in subjects with mild or moderate renal impairment and cheilitis, back pain, increased blood creatinine phosphokinase, and headache in healthy matched subjects.
In the severe renal impairment group and the healthy matched group, 6 subjects (severe renal impairment, n = 4; healthy matched, n = 2) reported 9 AEs. Of these, 5 subjects had AEs considered possibly related to study medication or procedures, including headache, dizziness, and upper abdominal pain in 3 subjects with severe renal impairment and nausea, vomiting, and pain in the extremity in 2 healthy matched subjects. All AEs were each reported by 1 subject, except headache (n = 2).
The most commonly reported AEs were nervous system disorders (dizziness, headache). Most AEs were mild or moderate in severity and resolved without intervention. One serious AE, acute exacerbation of existing chronic obstructive pulmonary disease, was reported in a subject with severe renal impairment; this AE occurred several days after the subject received apremilast, was considered unrelated to study medication, and resolved. One serious AE, myocardial infarction, was reported in a subject with mild renal impairment; this AE occurred >1 week after the subject received apremilast, was considered unrelated to study medication, and resolved. No deaths or AEs leading to discontinuation occurred.
No apparent group‐related trends were observed in subjects with renal impairment (mild, moderate, or severe) or in healthy matched subjects after apremilast administration, based on physical examination findings, vital signs, 12‐lead ECGs, and clinical laboratory investigations.
Discussion
Main findings from the 2 renal impairment studies demonstrated that the pharmacokinetic exposure of apremilast, based on the AUC0–∞ and Cmax of apremilast, was largely unaffected by mild and moderate renal impairment. However, apremilast pharmacokinetic exposure was increased among subjects with severe renal impairment compared with healthy matched subjects. The pharmacokinetic profile of apremilast in these subjects with severe renal impairment indicated that the elimination was significantly slower. The plasma concentration‐versus‐time profile in the subjects with severe renal impairment can be described with a 1‐compartment population pharmacokinetic model with a first‐order absorption rate constant and lag time. A population pharmacokinetic model was used to simulate concentration‐versus‐time profiles in subjects with severe renal impairment. The disease effect of PsA (≈36% slower clearance), based on a population pharmacokinetic model built in a phase 3 study, was also taken into consideration. Modeling and simulation suggest that a reduced dose of apremilast 30 mg once daily produces apremilast exposure in subjects with severe renal impairment (eGFR < 30 mL/min/1.73 m2 or creatinine clearance < 30 mL/min) comparable to that of apremilast 30 mg twice daily in subjects without renal impairment (Figure 3). Thus, dose adjustment is not needed when administering apremilast to subjects with mild or moderate renal impairment. However, in subjects with severe renal impairment, the dose should be lowered to 30 mg administered once daily.
Figure 3.
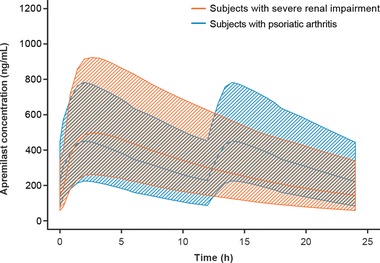
Simulated apremilast concentration‐versus‐time profiles (mean and 90%CI) in psoriatic arthritis subjects with (thick orange lines and shaded area) and without (blue lines and shaded area) severe renal impairment following oral administration of apremilast at 30 mg once daily and 30 mg twice daily.
The absolute bioavailability of apremilast is ≈73% in healthy subjects; therefore, if an increase in absorption is responsible for this increase in apremilast exposure, then it can increase by ≈37% at most. In these studies, the AUC increased by ≈89% in subjects with severe renal impairment; thus, the increase in exposure in subjects with severe renal impairment is unlikely to be a result of an increase in bioavailability. Because the apparent clearance of apremilast in subjects with severe renal impairment was nearly half (≈53%) that of the healthy matched subjects, and it is unlikely that all this change can be attributed to a change in absorption, it is likely that this change in exposure can be attributed to a decrease in the elimination of the parent apremilast compound. As discussed, apremilast is only minimally eliminated unchanged in urine, and metabolism plays a significant role in its elimination. Apremilast is extensively metabolized via multiple hepatic and nonhepatic pathways, such as nonenzymatic hydrolysis, non–cytochrome P450 (CYP)‐dependent N‐deacetylation, and oxidative metabolism followed by glucuronide conjugation, catalyzed by multiple enzymes, and generating a total of 21 known metabolites.16 The M12 metabolite is the primary circulating metabolite and is formed by glucuronide conjugation of O‐demethylated apremilast. O‐demethylation of apremilast is primarily catalyzed by the hepatic enzyme CYP3A4; therefore, CYP3A4 may play a major role in the oxidative metabolism of apremilast. Interestingly, although metabolism is a major route of elimination for apremilast, the pharmacokinetic profile of apremilast is largely unaffected by moderate and severe hepatic impairment.19 The diverse metabolism of apremilast may explain the lack of effect of moderate and severe hepatic impairment on the pharmacokinetics of apremilast observed.20, 21, 22, 23, 24 In vivo and in vitro studies have shown that uremia and various uremic by‐products, which build up in renally impaired patients, can change the drug metabolism of various compounds. Therefore, when compounds that are highly metabolized (ie, fraction of unchanged drug excreted through urine [fe] < 5%) show altered clearance (in the absence of changes in blood flow or protein binding), it can be assumed that this change in nonrenal clearance may be attributed to a change in the metabolic activity or a change in the intrinsic clearance. An example of this is repaglinide, which is a hypoglycemic agent metabolized predominantly by CYP2C8 and CYP3A4 and excreted through bile in healthy volunteers, with an fe < 0.1%. Pharmacokinetic values for repaglinide were similar between subjects with mild to moderate renal impairment and subjects with normal renal function. Mean half‐life increased nearly 4‐fold after 1 week of treatment in subjects with severe renal impairment, and AUCs were significantly greater after single and multiple dosing.25 Protein binding was similar in subjects with renal impairment and healthy matched subjects. Therefore, it is likely that with repaglinide, intrinsic clearance was decreased in subjects with severe renal impairment. These finding are supported by the expert opinions that chronic renal failure alters and decreases intestinal, renal, and hepatic drug metabolism, including CYP3A4 and transport, producing a clinically significant impact on drug disposition.26 Therefore, a decrease in metabolism in patients with severe renal impairment may explain the observation in the present study that apremilast clearance was reduced in patients with severe renal impairment.
Apremilast has not been evaluated in patients on hemodialysis. Because apremilast is a small molecule, the drug is expected to readily pass through a dialyzer. Therefore, drug exposure may be lower than the target exposure in patients on hemodialysis following apremilast therapy.
The pharmacokinetic profile of apremilast's major metabolite, M12, did not change with mild renal impairment. However, in subjects with moderate renal impairment, the overall exposure of M12, based on AUC0–∞ only, was increased compared with that in their healthy matched subjects, and in subjects with severe renal impairment, the exposure of M12 based on both AUC0–∞ and Cmax was increased. The increase in M12 AUC0–∞ was directly related to the increase in severity of renal impairment. M12 is eliminated via the renal route. Renal clearance of M12 decreased ≈40% in subjects with mild or moderate renal impairment. Similar to that of other small hydrophilic molecules such as creatinine, M12 elimination from the body is expected to slow and its plasma level to rise in patients with renal impairment. M12 is a glucuronide conjugate of O‐demethylated apremilast, and it is pharmacologically inactive. These results support the finding that no dose adjustment for apremilast is needed in patients with mild and moderate renal impairment.
In both studies, no unexpected AEs or safety signals were considered related to apremilast after a single oral 30‐mg dose. The most commonly reported AEs (nervous system disorders such as dizziness and headache) are consistent with those observed in clinical studies.10, 12, 27 A number of AEs were considered to be related to underlying renal impairment. Changes in vital signs, clinical laboratory parameters, or 12‐lead ECGs after apremilast administration demonstrated no clinically significant trends or patterns.
Although this evidence is encouraging for the potential use of apremilast in patients with renal impairment, the small, single‐dose nature of the current studies limits interpretability in clinical settings in which a repeated daily dosing regimen would be used. Patients with clinically significant renal impairment should be closely monitored when initiating any new drug regimen and routinely followed for possible treatment‐related AEs.
Conclusions
The pharmacokinetics of apremilast were unaltered in subjects with mild and moderate renal impairment, but were changed significantly in subjects with severe renal impairment who had slower clearance and increased exposure of apremilast compared with healthy matched subjects. As a result, a dose adjustment is not necessary in patients with mild and moderate renal impairment. A reduced dose of apremilast 30 mg once daily is recommended in patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2 or CLcr < 30 mL/min).
Declaration of Conflicting Interests
All authors are employees of Celgene Corporation.
Funding
These studies were sponsored by Celgene Corporation.
Acknowledgments
The authors thank Joga Gobburu for scientific insight on dose adjustment in subjects with severe renal impairment. The authors also thank colleagues Eric Laille, Xiaomin Wang, and Liangang Liu for their technical contributions to pharmacokinetic analysis, bioanalysis, and biostatistical analysis. The authors received editorial support in the preparation of this article from Amy Zannikos, PharmD, Peloton Advantage, LLC, and Prachi Wickremasingha, PharmD, funded by Celgene Corporation. The authors, however, directed and are fully responsible for all content and editorial decisions for this article.
References
- 1. Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83:1583–1590. [DOI] [PubMed] [Google Scholar]
- 2. Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase‐4 as a therapeutic target. Drug Discov Today. 2005;10:1503–1519. [DOI] [PubMed] [Google Scholar]
- 3. Salari P, Abdollahi M. Phosphodiesterase inhibitors in inflammatory bowel disease. Expert Opin Investig Drugs. 2012;21:261–264. [DOI] [PubMed] [Google Scholar]
- 4. Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. [DOI] [PubMed] [Google Scholar]
- 5. Tasken K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev. 2004;84:137–167. [DOI] [PubMed] [Google Scholar]
- 6. Schafer PH, Parton A, Gandhi AK, et al. Apremilast, a cAMP phosphodiesterase‐4 inhibitor, demonstrates anti‐inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159:842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McCann FE, Palfreeman AC, Andrews M, et al. Apremilast, a novel PDE4 inhibitor, inhibits spontaneous production of tumour necrosis factor‐alpha from human rheumatoid synovial cells and ameliorates experimental arthritis. Arthritis Res Ther. 2010;12:R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gottlieb AB, Matheson RT, Menter A, et al. Efficacy, tolerability, and pharmacodynamics of apremilast in recalcitrant plaque psoriasis: a phase II open‐label study. J Drugs Dermatol. 2013;12:888–897. [PubMed] [Google Scholar]
- 9. Schafer PH, Chen P, Fang L, Wang A, Chopra R. The pharmacodynamic impact of apremilast, an oral phosphodiesterase 4 inhibitor, on circulating levels of inflammatory biomarkers in patients with psoriatic arthritis: substudy results from a phase III, randomized, placebo‐controlled trial (PALACE 1). J Immunol Res. 2015;2015:906349. doi: 10.1155/2015/906349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schett G, Wollenhaupt J, Papp K, et al. Oral apremilast in the treatment of active psoriatic arthritis: results of a multicenter, randomized, double‐blind, placebo‐controlled study. Arthritis Rheum. 2012;64:3156–3167. [DOI] [PubMed] [Google Scholar]
- 11. Kavanaugh A, Mease PJ, Gomez‐Reino JJ, et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis: results of a phase 3, randomized, controlled trial [abstract L13]. Arthritis Rheum. 2012;64:4172–4173. [Google Scholar]
- 12. Papp K, Cather J, Rosoph L, et al. The efficacy of apremilast, a phosphodiesterase‐4 inhibitor, in the treatment of moderate to severe psoriasis: results of a phase 2 randomised study. Lancet. 2012;380:738–746. [DOI] [PubMed] [Google Scholar]
- 13. Reich K, Papp K, Leonardi C, et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate to severe psoriasis: 16‐week results of a phase 3, randomized, controlled trial (ESTEEM 1). Presented at: the Annual Meeting of the American Academy of Dermatology; March 1–5, 2013; Miami, FL.
- 14. Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM 1]). J Am Acad Dermatol. 2015;73:37–49. [DOI] [PubMed] [Google Scholar]
- 15. Wu A, Scheffler M. First‐time‐in‐man, safety/tolerability and pharmacokinetics of ascending oral doses of apremilast (APR) in healthy subjects (HS) [abstract 515]. J Invest Dermatol. 2011;131:S86. [Google Scholar]
- 16. Hoffmann M, Kumar G, Schafer P, et al. Disposition, metabolism and mass balance of [14C]apremilast following oral administration. Xenobiotica. 2011;41:1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu A, Wan Y, Laille E, et al. Absolute bioavailability of apremilast using a [14C] labeled microtracer IV solution concomitantly with an oral dose [abstract]. Presented at: the 2011 AAPS Annual Meeting and Exposition; October 23–27, 2011; Washington, DC.
- 18. Hoffmann JH, Hartmann M, Enk AH, Hadaschik EN. Autoantibodies in psoriasis as predictors for loss of response and anti‐infliximab antibody induction. Br J Dermatol. 2011;165:1355–1358. [DOI] [PubMed] [Google Scholar]
- 19. Assaf MS, Laille E, Liu L, et al. Pharmacokinetics and safety of apremilast (CC‐10004) in subjects with hepatic impairment. Int J Med Engineering Informatics. 2014;6:100–114. [Google Scholar]
- 20. Dowling TC, Briglia AE, Fink JC, et al. Characterization of hepatic cytochrome p4503A activity in patients with end‐stage renal disease. Clin Pharmacol Ther. 2003;73:427–434. [DOI] [PubMed] [Google Scholar]
- 21. Dreisbach AW, Japa S, Gebrekal AB, et al. Cytochrome P4502C9 activity in end‐stage renal disease. Clin Pharmacol Ther. 2003;73:475–477. [DOI] [PubMed] [Google Scholar]
- 22. Guevin C, Michaud J, Naud J, Leblond FA, Pichette V. Down‐regulation of hepatic cytochrome p450 in chronic renal failure: role of uremic mediators. Br J Pharmacol. 2002;137:1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leblond F, Guevin C, Demers C, Pellerin I, Gascon‐Barre M, Pichette V. Downregulation of hepatic cytochrome P450 in chronic renal failure. J Am Soc Nephrol. 2001;12:326–332. [DOI] [PubMed] [Google Scholar]
- 24. Leblond FA, Giroux L, Villeneuve JP, Pichette V. Decreased in vivo metabolism of drugs in chronic renal failure. Drug Metab Dispos. 2000;28:1317–1320. [PubMed] [Google Scholar]
- 25. Schumacher S, Abbasi I, Weise D, et al. Single‐ and multiple‐dose pharmacokinetics of repaglinide in patients with type 2 diabetes and renal impairment. Eur J Clin Pharmacol. 2001;57:147–152. [DOI] [PubMed] [Google Scholar]
- 26. Dreisbach AW, Lertora JJ. The effect of chronic renal failure on drug metabolism and transport. Expert Opin Drug Metab Toxicol. 2008;4:1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gottlieb AB, Strober B, Krueger JG, et al. An open‐label, single‐arm pilot study in patients with severe plaque‐type psoriasis treated with an oral anti‐inflammatory agent, apremilast. Curr Med Res Opin. 2008;24:1529–1538. [DOI] [PubMed] [Google Scholar]