

Abstract
The peroxisome proliferator activated receptor gamma (PPARγ) is a ligand‐activated transcription factor that regulates growth and differentiation within normal prostate and prostate cancers. However the factors that control PPARγ within the prostate cancers have not been characterized. The goal of this study was to examine whether the androgen receptor (AR) regulates PPARγ expression and function within human prostate cancer cells. qRT‐PCR and Western blot analyses revealed nanomolar concentrations of the AR agonist dihydrotestosterone (DHT) decrease PPARγ mRNA and protein within the castration‐resistant, AR‐positive C4‐2 and VCaP human prostate cancer cell lines. The AR antagonists bicalutamide and enzalutamide blocked the ability of DHT to reduce PPARγ levels. In addition, siRNA mediated knockdown of AR increased PPARγ protein levels and ligand‐induced PPARγ transcriptional activity within the C4‐2 cell line. Furthermore, proteasome inhibitors that interfere with AR function increased the level of basal PPARγ and prevented the DHT‐mediated suppression of PPARγ. These data suggest that AR normally functions to suppress PPARγ expression within AR‐positive prostate cancer cells. To determine whether increases in AR protein would influence PPARγ expression and activity, we used lipofectamine‐based transfections to overexpress AR within the AR‐null PC‐3 cells. The addition of AR to PC‐3 cells did not significantly alter PPARγ protein levels. However, the ability of the PPARγ ligand rosiglitazone to induce activation of a PPARγ‐driven luciferase reporter and induce expression of FABP4 was suppressed in AR‐positive PC‐3 cells. Together, these data indicate AR serves as a key modulator of PPARγ expression and function within prostate tumors. J. Cell. Physiol. 231: 2664–2672, 2016. © 2016 The Authors. Journal of Cellular Physiology Published by Wiley Periodicals, Inc.
The peroxisome proliferator activated receptor gamma (PPARγ) is a member of the nuclear receptor superfamily that is activated by prostaglandins and several synthetic compounds. Upon binding ligand, PPARγ associates with regions of genomic DNA known as PPAR response elements (PPREs) as part of a heterodimer with the retinoid X receptor (RXR). This association results in the recruitment of coactivators, such as PPARγ coactivator 1 (PGC1), steroid receptor coactivator‐1 (SRC‐1) and CBP/p300, to DNA and alterations in gene expression. While high levels of PPARγ are expressed within adipose tissue, PPARγ is also present within the normal prostate. Within the prostate epithelium PPARγ functions as a tumor suppressor, for conditional knockout of PPARγ within mouse epithelial cells results in the development of prostatic intraepithelial neoplasia (PIN), a precursor of prostate cancer (Jiang et al., 2010a). Loss of PPARγ also increases the level of autophagy within the mouse prostate (Jiang et al., 2010a,2010b). Furthermore, studies by DW Strand et al. revealed knockdown of two PPARγ isoforms (PPARγ1 and PPARγ2) within the BHPrE normal human prostate cell line results in low expression of prostate differentiation markers (Strand et al., 2013). Taken together these data suggest PPARγ is a key regulator of prostatic differentiation and cell survival in normal prostatic tissue. PPARγ protein and mRNA have been detected within human prostate cancer cell lines and prostate tumors (Butler et al., 2000; Segawa et al., 2002; Sabichi et al., 2004; Subbarayan et al., 2004; Lyles et al., 2009; Moss et al., 2010). However, the significance of PPARγ expression within prostate cancers is not fully understood. In addition, the factors that control PPARγ levels and function within human prostate cancer cells have not been characterized.
The androgen receptor (AR) is also a member of the nuclear receptor superfamily that plays a critical role in the development and differentiation of normal prostate and the progression of prostate cancer. Activation of AR via the androgens testosterone and dihydrotestosterone (DHT) promotes growth of early stage prostate cancers. For this reason the reduction of circulating androgens via castration and other types of androgen deprivation therapy (ADT) is the standard treatment for patients with advanced, metastatic prostate cancer. Unfortunately, castration‐resistant forms of the prostate tumor develop approximately 18–24 months after the start of ADT (Santen, 1992). Although castration‐resistant tumors don't require androgens for tumor growth, they continue to express active forms of AR. Multiple factors appear to contribute to the increased level of AR activation within castration‐resistant prostate cancers. These include amplifications and mutations of the AR gene, the expression of constitutively active N‐terminal AR variants, ligand‐independent activation of AR by growth factors and cytokines, and local production of androgens within prostate tumors (Knudsen and Penning, 2010). Furthermore, AR is still a major driver of tumor growth within these recurrent castration resistant prostate cancers. Data from ChIP‐seq and expression profiling studies indicate AR regulates proteins that are involved in cell cycle progression, biosynthetic pathways and cellular metabolism within human prostate cancer cells (Wang et al., 2009; Massie et al., 2011). However, the extent to which alterations in these gene products contribute to the promotion of tumor growth by AR is still unclear.
Interactions between the AR and PPARγ signaling pathways occur within adipose tissue and influence the process of adipogenesis. Data from R. Singh and colleagues revealed activation of AR by testosterone and DHT not only suppresses adipocyte differentiation but also decreases PPARγ mRNA and protein levels in mouse 3T3‐L1 preadipocytes. Furthermore, DHT produced a similar reduction in PPARγ2 mRNA and protein levels within mouse pluripotent C3H10T1/2 cells (Singh et al., 2003). It is not known if PPARγ and AR signaling pathways interact in human prostate, and whether this interaction influences the biology of normal or diseased prostate. The goal of the present study was to determine if AR might influence PPARγ function within human prostate cancer cells. Our data reveal that AR suppresses PPARγ transcriptional activity in prostate cancer cells, and that in AR‐positive prostate cancer cells this suppression is due in part to AR‐mediated reductions in PPARγ expression.
Materials and Methods
Materials
DMEM low glucose media, DMEM high glucose media, Hams’ F‐12 media, DMEM/F‐12 media (1:1), penicillin/streptomycin solution and phosphate buffered saline (PBS) were purchased from Invitrogen (Carlsbad, CA). The media additives d‐biotin, adenine hemisulfate, insulin solution, apo‐transferrin, and Nuclei EZ Prep kit were purchased from Sigma–Aldrich (St. Louis, MO). Fetal bovine serum (FBS) was obtained from HyClone (Logan, UT). Charcoal stripped FBS (CSS) was prepared within our laboratory or purchased from Invitrogen (Carlsbad, CA). Zapoglobin and Isoton II were purchased from Beckman Coulter Inc. (Fullerton, CA). Rabbit anti‐mouse IgG secondary antibody was obtained from Zymed Laboratories, Inc. Both horseradish peroxidase‐conjugated donkey anti‐rabbit and sheep anti‐mouse antibodies were purchased from GE Healthcare Biosciences (Pittsburg, PA). All tissue culture plasticware and additional chemicals were purchased from Fisher Scientific (Suwanee, GA).
Drugs
The PPARγ agonist rosiglitazone was purchased from Cayman Chemical (Ann Arbor, MI). Stock solutions of rosiglitazone were prepared by diluting the compound in 100% DMSO and stored at −20°C. The proteasome inhibitor MG132 was purchased from Sigma–Aldrich. Stock solutions of MG132 were diluted in DMSO and stored at −20°C. The AR antagonist bicalutamide was purchased from Tocris Bioscience (Minneapolis, MN) and stored at −20°C as a stock solution in 100% DMSO. The more potent AR antagonist enzalutamide, which was purchased from Selleck Chemicals (Houston, TX), was diluted in 100% ethanol (EtOH) and stored at −20°C.
Cell lines
The C4‐2 cell line was purchased from ViroMed Laboratories (Burlington, NC) and grown in T medium (80% DMEM low glucose medium, 20% Hams' F12 medium, 5% heat inactivated FBS, 1% penicillin/streptomycin, 0.244 µg/ml d‐biotin, 25 µg/ml adenine hemisulfate, 5 µg/ml insulin and 5 µg/ml apotransferrin). The PC‐3 cell line, which was purchased from ATCC (Manassas, VA), was grown in DMEM‐F12 medium supplemented with 10% FBS and 1% penicillin/streptomycin. The VCaP cell line was purchased from ATCC and grown in DMEM high glucose medium supplemented with 10% FBS and 1% penicillin/streptomycin. Each cell line was maintained in an incubator with a 5% CO2 atmosphere at 37°C.
Western blot analysis
To measure the effect of DHT on PPARγ protein levels, C4‐2 and VCaP cells were plated at a density of 600,000–750,000 cells per 10 cm dish in either T media supplemented with 5% CSS (C4‐2 cells) or DMEM high glucose media supplemented with 10% CSS (VCaP cells). The cells were then treated with ethanol vehicle (EtOH) or the indicated concentrations of dihydrotestosterone (DHT) for up to 24 h. For experiments involving proteasome inhibitors or AR antagonists, the cells were pretreated with DMSO vehicle, MG132, enzalutamide or bicalutamide prior to the addition of EtOH or DHT. Following drug exposure, cells were lysed using the Sigma–Aldrich Nuclei EZ Prep Nuclei Isolation Kit to prepare nuclear extracts or RIPA buffer (Thermo Scientific, Pittsburg, PA) to prepare whole cell extracts. Protein concentrations for each sample were calculated using the Bradford protein assay (BioRad, Hercules, CA). Equal amounts of protein from each extract were separated on SDS‐PAGE gels and transferred to a nitrocellulose membrane. Membrane blots were then blocked in TBST (1× TBS, 0.1% Tween 20) containing 1% non‐fat powdered milk and incubated with primary antibody diluted in the blocking solution overnight at 4°C. The primary antibodies used were the PPARγ rabbit polyclonal antibody (clone H‐100, Santa Cruz Biotechnology, Santa Cruz, CA; 1:200) and the AR mouse monoclonal antibody (clone AR 441, Lab Vision Corporation, Fremont, CA; 1:400). Following exposure to primary antibody, the blots were washed in blocking buffer and then incubated with either a donkey anti‐rabbit or sheep anti‐mouse secondary antibody conjugated to horseradish peroxidase. Proteins were then visualized using the Pierce Enzyme‐Linked Chemiluminescence (ECL) Western Blotting Substrate (Thermo Scientific). ECL images were captured using either X‐ray film or the Carestream Gel Logic 4000 imaging system. Blots containing nuclear extracts were stripped and reprobed with a rabbit polyclonal topoisomerase I antibody (clone H‐300, Santa Cruz Biotechnology; 1:400). Blots containing whole cell lysates were reprobed with an actin mouse monoclonal antibody (Chemicon International, Temecula, CA; 1:10,000) or alpha tubulin antibody (Santa Cruz, Biotechnology, 1:200) to confirm equal loading of the gel.
qRT‐PCR analysis
To measure basal levels of PPARγ mRNA, untreated cells were incubated in FBS‐containing media for 72 h. Total RNA was then isolated from each cell line with the Qiagen RNeasy Kit or Trizol reagent according to the manufacturer's protocol. For each sample the iScript cDNA Synthesis Kit (BioRad) was used to synthesize cDNA from 1 µg of total RNA. The cDNA was then amplified by quantitative PCR using a reaction involving iQ SYBR Green Supermix reagent (BioRad). This PCR reaction consisted of an initial denaturation step (3 min at 95°C) and 40 cycles of PCR (95°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec). The Qiagen PPARγ primer set (HsPPARG_1_SG Quantitect Primer Cat. #ATT00029841), PPARγ2 Forward (GACCACTCCCACTCCTTTGA) and Reverse (5′‐TCCATGCTGTTATGGGTGAA) primers, as well as the 18S Forward (5′‐ATC AAC TTT CGA TGG TAG TCG‐3′) and 18S Reverse (5′‐TCC TTG GAT GTG GTA GCG‐3′) primers were used to detect the presence of total PPARγ mRNA, PPARγ2 mRNA and 18S rRNA. The ΔΔCt algorithm was used to calculate the relative amounts of PPARγ mRNA and 18S rRNA in each sample. The level of PPARγ mRNA (total PPARγ or PPARγ2) was then normalized to 18S rRNA levels.
To examine the effect of DHT on PPARγ mRNA levels, C4‐2 and VCaP cells were plated in media supplemented with either 5% CSS (C4‐2 cells) or 10% CSS (VCaP). The cells were then treated with EtOH or the indicated concentrations of DHT for 0–24 h. Total RNA was isolated using the Qiagen RNeasy Kit or Trizol reagent according to the manufacturer's instructions. The amount of total PPARγ and 18S rRNA in each total RNA samples was then measured as described above.
To measure mRNA levels of the PPARγ target gene adipose fatty acid binding protein (FABP4), total RNA was extracted from treated cells using the Trizol reagent. The iScript cDNA Synthesis Kit was then used to synthesize cDNA from 1 µg of total RNA. qPCR was performed using the 18S primers described above and FABP4‐specific Forward (5′‐TCAACGTCCCTTGGCTTATGC‐3′) and reverse (5′‐TCAGTGTGAATGGGGATGTGA‐3′) primers. The ΔΔCt algorithm was used to calculate the relative amounts of FABP4 mRNA and 18S rRNA in each sample.
siRNA studies
To determine how loss of AR affects PPARγ expression, C4‐2 cells were first transfected with an AR SMARTpool siRNA or a nonspecific SMARTpool siRNA (GE Dharmacon, Lafayette, CO) via electroporation. Following transfection, the cells were placed in RPMI 1640 media containing 5% FBS at a density of 260,000 cells/well of a 6 well plate and allowed to recover for 48 h. Nuclear extracts were then isolated from transfected cells. Western blot analysis was then performed as described above to detect the level of AR and PPARγ in each cell extract. Blots were stripped and reprobed with a rabbit polyclonal topoisomerase I antibody (clone H‐300, Santa Cruz Biotechnology; 1:400) to confirm equal loading of the gel.
To determine whether AR loss affects PPARγ‐driven luciferase activity, C4‐2 cells were transfected with 20 μg PPRE3‐ luciferase, 2 μg CMV β‐galactosidase plasmid, and 20 μM of either non‐specific control SMARTpool siRNA or AR SMARTpool siRNA via electroporation (∼5 million cells per transfection). Following transfection, the cells were placed in RPMI 1640 media containing 5% FBS and allowed to recover for 24 h. After the recovery period the cells were treated for 24 h with either DMSO vehicle or 40 μM of the PPARγ agonist rosiglitazone. The luciferase activity in treated cells was measured using the Luciferase Assay System kit from Promega (Madison, WI) and normalized to β‐galactosidase activity. In parallel wells, transfected cells were lysed using RIPA buffer. The level of AR and tubulin protein in each whole cell lysate was then measured by Western blot analysis.
To assess how loss of p65 NFκB affects androgen‐induced suppression of PPARγ, C4‐2 cells were first transfected with a p65 siRNA SMARTpool siRNA (Dharmacon) or a non‐targeting control SMARTpool siRNA via electroporation and allowed to recover for 48 h. The cells were then placed in media containing 5% CSS and treated for 24 h with either EtOH or 1 nM DHT. Following treatment, the level of p65 NFκB, PPARγ, AR and actin protein in whole cell extracts from treated cells was measured by Western blot analysis.
[3H]‐thymidine incorporation assays
Transfected C4‐2 cells were treated for 48 h with either DMSO vehicle or 40 μM rosiglitazone. During the last 3 h of treatment, the cells were pulsed with 2 µCi/ml [3H]‐thymidine (MP Biomedicals, Irvine, CA). The cells were then fixed with methanol: acetic acid (3:1) for 5 min and washed with 100% methanol for 5 min. They were next incubated with 5% tricholoracetic acid for 5 min and washed three times with 100% methanol. To extract the incorporated [3H]‐thymidine, the cells were incubated with 0.1 N NaOH and neutralized with an equal amount of 0.1 N HCl. The amount of [3H]‐thymidine in each sample was then measured using a scintillation counter.
AR overexpression studies
To study the effect of AR overexpression on PPARγ transcriptional activity, PC‐3 cells were plated at a density of 75,000 cells/well in a 6 well plate. They were next transfected with 500 ng PPRE3‐ luciferase reporter plasmid, 500 ng CMV β‐galactosidase plasmid, and 1 μg of either PCR‐3.1 AR or PCR3.1 expression vector using Lipofectamine (Invitrogen). Following transfection, the cells were placed in DMEM/F12 media containing 10% FBS and allowed to recover for 24 h. After the recovery period the cells were treated with either DMSO or different concentrations of rosiglitazone (10–40 μM) for 24 h. The luciferase activity in treated cells was measured using the Luciferase Assay System and normalized to β‐galactosidase activity. Western blot analysis was performed as previously described to measure the level of AR, PPARγ, and actin protein in transfected cells.
To analyze the effect of AR overexpression on the PPARγ target gene FABP4, PC‐3 cells were plated at a density of 75,000 cells/well in a 6 well plate. Lipofectamine was then used to transfect the cells with 1 μg of either PCR‐3.1 AR or PCR3.1 expression vector. The cells were allowed to recover overnight and then treated with DMSO vehicle or 40 μM rosiglitazone for 24 h. Total RNA was extracted from treated cells using the Trizol reagent. qRT‐PCR was then performed as described above to measure the level of FABP4 mRNA and 18S rRNA in each RNA sample.
Statistical analysis
Each experiment was performed at least three times and representative data are shown. For transient transfections and qRT‐PCR experiments, One Way Analysis of Variance (ANOVA) was used to detect differences between control and treatment groups. These analyses were performed using the Sigma Stat 3.1 program (Systat Software Inc.). The standard for statistical significance was P < 0.05.
Results
Androgens decrease PPARγ protein levels in AR‐positive cell lines
Our laboratory has previously shown that PPARγ protein levels vary across castration‐resistant human prostate cancer cells. In these studies, we noted that the PC‐3 cell line, which expresses very little if any AR, contained high levels of PPARγ protein while low levels of PPARγ were present in the AR positive C4‐2 cells (Moss et al., 2010). To determine whether the presence of AR influences PPARγ expression, we first tested the ability of the AR agonist dihydrotestosterone (DHT) to modulate PPARγ protein levels within the AR‐positive C4‐2 cells. DHT produced a concentration‐dependent decrease in not only nuclear PPARγ but also the total amount of PPARγ protein within C4‐2 cells (Fig. 1A). The greatest reduction in PPARγ levels was noted at DHT concentrations ≥1 nM (Fig. 1A). This reduction was also time‐dependent. Over the time frame examined, a reduction in PPARγ protein levels was detected in C4‐2 cells after 6 h of DHT treatment. Furthermore, PPARγ levels remained low after 24 h of DHT exposure (Fig. 1B). This reduction in PPARγ protein levels was not unique to the C4‐2 cell line. Nanomolar concentrations of DHT produced a similar decrease in PPARγ protein in the AR‐positive VCaP cells (Fig. 1C).
Figure 1.
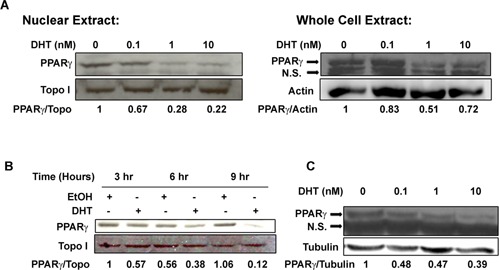
Dihydrotestosterone (DHT) down‐regulates PPARγ protein in a time‐ and concentration‐dependent manner in AR‐positive prostate cancer cells. (A) C4‐2 cells were treated with ethanol vehicle or the indicated concentrations of DHT for 24 h. Western blot analysis was then performed to detect PPARγ, topoisomerase I, or actin protein in nuclear or whole cell extracts prepared from the treated cells. (B) C4‐2 cells were treated with ethanol vehicle (EtOH) or 10 nM DHT for the indicated times. Nuclear extracts were prepared from treated cells, and the level of PPARγ and topoisomerase I protein measured by Western blot analysis. (C) VCaP cells were treated with EtOH or the indicated concentrations of DHT for 24 h. Western blot analysis was then performed to detect PPARγ and α tubulin protein in whole cell extracts from treated cells. In Western blots of whole cell lysates, the image includes a 53 kD band representing PPARγ as well as a lower nonspecific (N.S.) band.
Androgens reduce PPARγ mRNA levels
One mechanism by which androgens could suppress PPARγ protein levels is by changing the amount of PPARγ mRNA present in the cell. To explore this possibility, we measured the effect of DHT on PPARγ mRNA levels. We began these studies by defining the PPARγ isoforms expressed within C4‐2 and other prostate cancer cell lines. In mammals two isoforms of PPARγ have been identified, PPARγ1 and PPARγ2. The isoforms differ in that PPARγ2 contains an additional 30 amino acids at its N‐terminus (Desvergne and Wahli, 1999). Therefore, in these studies we used primers that could detect both PPARγ isoforms (PPARγ1 and PPARγ2; total PPARγ) as well as PPARγ2‐specific primers. In qRT‐PCR experiments that involved primers against total PPARγ, PPARγ mRNA was detected in the C4‐2, PC‐3 and VCaP cells. However, very little if any PPARγ was detected in experiments involving the PPARγ2 specific primers (Fig. 2A). These data suggest that PPARγ1 is the dominant isoform expressed in C4‐2, PC‐3, and VCaP cells. Since the total PPARγ primers were effective in detecting PPARγ within our cell lines, we used those primers in subsequent qRT‐PCR experiments.
Figure 2.
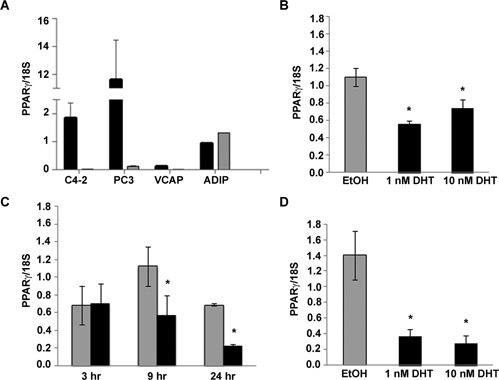
The androgen DHT reduces PPARγ mRNA in AR‐positive cells. (A) qRT‐PCR was used to detect basal levels of total PPARγ and PPARγ2 mRNA in total RNA samples from human prostate cancer cell lines and adipose tissue RNA. The amount of PPARγ mRNA in each sample was normalized to 18S rRNA. Black bars represent the normalized amount of total PPARγ (PPARγ1 and PPARγ2) while the gray bars represent PPARγ2 mRNA. (B) C4‐2 cells were treated with EtOH or different concentrations of DHT for 24 h. Total RNA was then isolated from treated cells. The amount of PPARγ mRNA and 18S rRNA in each RNA sample was measured using qRT‐PCR. (C) C4‐2 cells were treated with EtOH (gray bars) or 1 nM DHT (black bars) for 3–24 h. PPARγ mRNA and 18S rRNA levels in each sample were then measured using qRT‐PCR. (D) VCaP cells were treated for 24 h with either EtOH or increasing concentrations of DHT. The level of PPARγ and 18S RNA in treated cells was then measured by qRT‐PCR. In parts A–D, each bar represents the mean ± SEM of three independent samples. *P < 0.05 compared to EtOH group.
We next explored the ability of DHT to alter PPARγ mRNA levels within AR‐positive cell lines. qRT‐PCR revealed DHT produces a time‐ and concentration‐dependent decrease in PPARγ mRNA in C4‐2 cells (Fig. 2B and C). The nanomolar concentrations of DHT that reduced PPARγ protein levels were also effective at suppressing PPARγ mRNA levels. DHT at concentrations ≥1 nM lowered PPARγ mRNA levels by approximately 40–50%. At very early time points (i.e., ≤3 h) DHT did not produce a dramatic change in PPARγ mRNA levels. However, we did see a significant reduction in PPARγ mRNA in C4‐2 cells exposed to DHT for ≥9 h (Fig. 2C). Nanomolar concentrations of DHT were also effective at reducing PPARγ mRNA in the AR‐positive VCaP prostate cancer cell line (Fig. 2D). It therefore appears that the ability of DHT to suppress PPARγ mRNA was not limited to C4‐2 cells, but also occurs in other AR‐containing human prostate cancer cell lines.
AR regulates PPARγ expression and activity in C4‐2 cells
To determine the importance of AR in DHT‐mediated suppression of PPARγ mRNA and protein, we performed a series of experiments involving the first generation AR antagonist bicalutamide and the second generation AR antagonist enzalutamide. In these experiments, C4‐2 cells were pretreated with AR antagonists prior to the addition of 1 nM DHT. Both bicalutamide and enzalutamide blocked DHT‐induced reductions in PPARγ mRNA (Fig. 3A). Furthermore, the ability of DHT to reduce PPARγ protein was suppressed in C4‐2 cells pretreated with either bicalutamide or enzalutamide (Fig. 3B). These data suggest AR is required for DHT‐ stimulated reductions in PPARγ mRNA and protein.
Figure 3.
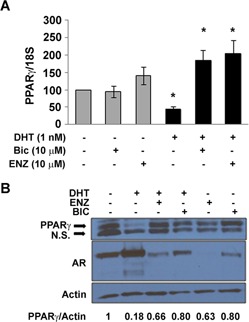
Anti‐androgens inhibit DHT‐induced reductions in PPARγ. (A) C4‐2 cells were first treated with DMSO vehicle, bicalutamide (Bic, 10μM) or enzalutamide (ENZ, 10 μM). The cells were then exposed to EtOH or 1 nM DHT for 24 h. Total RNA was extracted from treated cells, and the level of PPARγ and 18S RNA measured by qRT‐PCR. Each bar represents the mean ± SEM for three experiments. *P < 0.05 compared to the control (DHT‐, Bic‐, ENZ‐) group. (B) C4‐2 cells were first pretreated with either DMSO vehicle, 10 μM bicalutamide, or 10 μM enzalutamide. The cells were next exposed to EtOH or 1 nM DHT for 24 h. Whole cell extracts were prepared from treated cells. Western blot analysis was then performed to measure the level of AR, PPARγ and actin protein in each extract.
To further characterize the role of AR in the regulation of PPARγ, we examined how loss of AR influences PPARγ protein levels and activity. In these studies we used an AR siRNA SMARTpool reagent to reduce wild type AR levels within the C4‐2 cell line. siRNA‐ mediated knockdown of AR produced a two‐fold increase in PPARγ protein in C4‐2 cells (Fig. 4A). A PPRE3‐luciferase reporter construct was then used to determine whether the function of PPARγ might be influenced by AR levels. Luciferase‐based reporter assays revealed knockdown of wild type AR protein in C4‐2 cells increases basal PPARγ transcriptional activity. In addition, the ability of the PPARγ agonist rosiglitazone to activate PPARγ was enhanced in C4‐2 cells transfected with AR siRNA (Fig. 4B). We next used [3H]‐thymidine incorporation assays to determine whether the presence of AR modulates the anti‐proliferative effects of rosiglitazone. Exposure to rosiglitazone did not alter the level of [3H]‐thymidine incorporation in C4‐2 cells transfected with control siRNA. However, rosiglitazone did significantly reduce [3H]‐thymidine incorporation in C4‐2 cells that had been transfected with AR siRNA (Fig. 4C). These data suggest that reductions in AR expression enhance the ability of PPARγ agonists to decrease cell proliferation.
Figure 4.
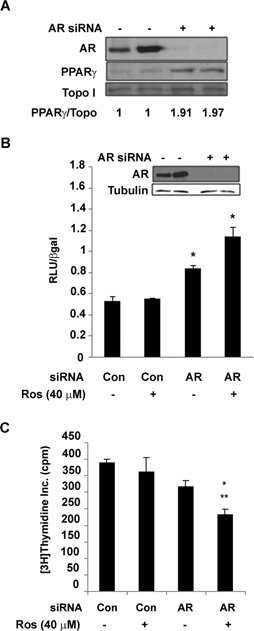
Knockdown of AR protein increases PPARγ protein expression and transcriptional activity. (A) C4‐2 cells were transfected with an AR SMARTpool siRNA (+) or a nonspecific SMARTpool siRNA (−). Forty‐eight hours following transfection, Western blot analysis was performed to detect the level of AR, PPARγ and topoisomerase I protein in nuclear extracts isolated from the transfected cells. (B) C4‐2 cells were first transfected with the PPRE3‐luciferase reporter plasmid, CMV‐β galactosidase reporter, and either the AR SMARTpool siRNA or a nonspecific control SMARTpool siRNA. The cells were then treated with DMSO vehicle (−) or 40 μM Rosiglitazone (+) for 24 h. Luciferase activity was measured in cell lysates and normalized to β‐galactosidase activity. Each bar represents the mean ± SD for three wells. *P < 0.05 compared to Control siRNA, Ros + group. (C) C4‐2 cells were first transfected with an AR SMARTpool siRNA or a nonspecific control SMARTpool siRNA. After a 24 h recovery period the cells were exposed to either DMSO vehicle (−) or 40 μM rosiglitazone (+) for 48 h. The cells were then pulsed with 2 µCi/mL [3H]‐thymidine. The amount of [3H]‐thymidine incorporated into the treated cells was measured using a scintillation counter. Each bar represents the mean ± SEM for three wells. *P < 0.05 compared to Control siRNA, Ros − group; **P < 0.05 compared to Control siRNA, Ros + group.
Proteasome inhibitors reduce AR and increase PPARγ levels
In our studies, reductions in AR activity and expression increased both PPARγ expression and activity. This observation led us to predict that other factors that lower AR expression and/or function would also alter PPARγ in human prostate cancer cells. Previous studies have shown that MG132 and other proteasome inhibitors decrease AR transcriptional activity within human prostate cancer cells by interfering with AR nuclear translocation (Lin et al., 2002; Hu et al., 2015). We therefore tested the effects of two proteasome inhibitors, MG132 and bortezomib, on PPARγ protein in C4‐2 cells. In CSS‐containing media MG132 alone lowered the amount of AR protein present in whole cell lysates and increased basal PPARγ levels. DHT increased the total amount of AR protein present within C4‐2 cells both in the absence and presence of MG132. However, MG132 pretreatment blocked the ability of DHT to reduce PPARγ levels in the C4‐2 cell line (Fig. 5A). Since MG132 reduces translocation of AR into the nucleus and increases cytoplasmic AR levels ((Lin et al., 2002) and data not shown), we believe the decrease in intracellular PPARγ levels produced by MG132 is due to MG132‐mediated reductions in AR nuclear translocation and function. Similar changes in AR and PPARγ levels were produced by the proteasome inhibitor bortezomib. In androgen‐containing media, micromolar concentrations of bortezomib increased PPARγ protein levels (Fig. 5B). At these concentrations bortezomib also reduced nuclear AR protein levels. Bortezomib not only functions as a proteasome inhibitor but also inhibits activation of the NFκB signaling pathway. However, the NFκB inhibitor BMS 345541 did not increase PPARγ levels within C4‐2 cells (Fig. 5B). Furthermore, siRNA‐mediated knockdown of p65 NFκB did not alter the ability of DHT to suppress PPARγ in C4‐2 cells (Fig. 5C). Therefore, our data suggest that bortezomib‐induced increases in PPARγ protein are primarily due to proteasome‐mediated alterations in AR expression and/or activity.
Figure 5.
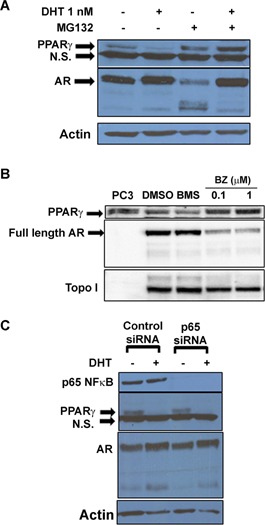
Proteasome inhibitors prevent DHT‐induced alterations in PPARγ protein. (A) C4‐2 cells were first treated with DMSO vehicle (−) or 10 μM MG132 (+). The cells were then exposed to ethanol vehicle (−) or 1 nM DHT for 24 h. Whole cell lysates were isolated from treated cells, and Western blot analysis was performed to determine the amount of AR, PPARγ and actin protein in each cell extract. (B) C4‐2 cells plated in DHT‐containing media were treated with DMSO, BMS 345541 (10 nM), or bortezomib (0.1 or 1 μM) for 24 h. Western blot analysis was performed on nuclear extracts to measure the level of AR, PPARγ and topoisomerase I protein in treated cells. (C) C4‐2 cells were first transfected with a p65 siRNA SMARTpool siRNA or a non‐targeting control SMARTpool siRNA via electroporation. The cells were then placed in CSS media and treated for 24 h with either EtOH (−) or 1 nM DHT (+). Whole cell lysates were prepared from treated cells. The level of p65 NFκB, PPARγ, AR and actin protein was then measured by Western blot analysis. A representative experiment is shown.
Overexpression of AR suppresses PPARγ transcriptional activity in PC‐3 cells
PC‐3 cells express high amounts of PPARγ protein and low, non‐detectable levels of AR protein (Moss et al., 2010). To determine whether an increase in wild type AR levels would alter PPARγ within AR‐null prostate cancer cells, we transfected PC‐3 cells with the pCR3.1‐AR expression construct. The amount of PPARγ present in AR‐positive PC‐3 cells was comparable to that found in cells transfected with the empty vector pCR3.1 (Fig. 6A). However, the addition of AR did alter PPARγ function. The basal level of PPARγ luciferase activity in PC‐3 cells transfected with the empty vector pCR3.1 was significantly higher than that found in PC‐3 cells that express wild type AR. In addition, the ability of rosiglitazone to activate the PPRE‐luciferase reporter was reduced in AR‐positive PC‐3 cells (Fig. 6B). The presence of wild type AR also decreased basal levels of the PPARγ target gene adipocyte FABP (FABP4) and reduced rosiglitazone‐induced increases in FABP4 mRNA (Fig. 6C).
Figure 6.
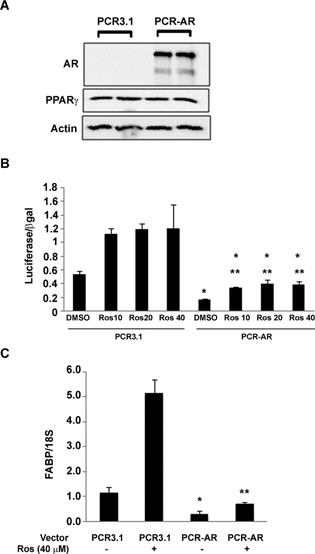
Overexpression of AR protein in PC‐3 cells decreases PPARγ function. (A) PC‐3 cells were transfected with either the PCR3.1 or PCR‐AR expression vector. Western blot analysis was then used to measure the level of AR, PPARγ, and actin protein in transfected cells. (B) PC‐3 cells were transfected with the PPRE3‐ luciferase reporter plasmid, CMV β‐galactosidase plasmid, and either PCR‐AR or PCR3.1 expression vector. The transfected cells were next treated with DMSO vehicle or different concentrations of rosiglitazone (10–40 μM) for 24 h. Luciferase activity in treated cells was then measured and normalized to β‐galactosidase activity. (C) PC‐3 cells transfected with either the PCR3.1 or PCR‐AR expression vector were treated for 24 h with DMSO vehicle (−) or 40 μM rosiglitazone (+). qRT‐PCR was used to measure the level of FABP mRNA and 18S rRNA in treated cells. In parts B and C, each bar represents the mean ±SEM of three wells. *P < 0.05 compared to the PCR3.1, DMSO group. **P < 0.05 compared to the PCR‐AR, DMSO group.
Discussion
Our laboratory and others have previously shown that ligand‐mediated activation of PPARγ can regulate AR activity in human prostate cancer cells (Hisatake et al., 2000; Yang et al., 2007; Moss et al., 2010). In this study we demonstrate the expression of PPARγ can be suppressed by activation of AR. Physiological concentrations of the AR agonist DHT reduced PPARγ mRNA and protein levels within the castration‐resistant C4‐2 and VCaP cell lines. Furthermore, inhibition or knockdown of AR increases PPARγ expression and activity within the AR‐positive C4‐2 cells. Taken together, these data indicate there is a bidirectional crosstalk between the PPARγ and AR signaling pathways. Of the two isoforms of PPARγ protein that exist in mammalian cells, PPARγ2 is primarily expressed within adipose tissue while PPARγ1 is present in multiple tissues including the prostate. While work by R. Singh et al. has shown that similar androgen concentrations reduce PPARγ2 expression in mouse adipocytes (Singh et al., 2006), ours is the first report to show androgens via AR also control PPARγ activity and expression in human prostate cancer cells that predominantly express PPARγ1. Our data suggest that androgens reduce expression of both PPARγ isoforms and, as a result, have the potential to influence PPARγ expression in the prostate and several other organ systems.
This study has primarily focused on interactions between the AR and PPARγ signaling pathways in prostate cancer cells. It is also possible that crosstalk between these two pathways occurs within the normal prostate. Within the normal prostate AR is expressed in the stroma and luminal epithelial cells (Nieto et al., 2014). PPARγ has also been detected within normal prostatic epithelial cells, although multiple reports suggest the amount of PPARγ present in normal and benign prostate cells and tissues is lower than that found in prostate cancers (Nwankwo and Robbins, 2001; Subbarayan et al., 2004; Nakamura et al., 2009; Rogenhofer et al., 2012). To our knowledge, there are no studies that have directly examined the regulation of PPARγ by AR within normal prostatic tissues. However D. Strand et al. have explored the regulation of AR signaling by PPARγ. Their studies revealed that the addition of PPARγ1 to mouse prostatic epithelial cells lacking PPARγ (mPrE‐γKO cells) resulted in a decrease in AR transcriptional activity, while restoration of PPARγ2 increased DHT‐induced AR activation (Strand et al., 2012). Therefore PPARγ may influence the function of AR in normal prostatic epithelial cells in an isoform‐specific manner.
We believe that in the AR‐positive C4‐2 cells, AR‐induced reductions in PPARγ activity are due in part to reductions in PPARγ protein. Increasing AR levels in the AR‐null PC‐3 cells was not enough to stimulate a decrease in PPARγ, as AR overexpression in the AR‐null PC‐3 cells produced a minimal effect on PPARγ protein levels. However, this elevation reduced the ability of PPARγ ligands to induce transcription in the PC‐3 cell line. These data would suggest that AR may be able to suppress PPARγ transcriptional activity via a mechanism that does not require reductions in PPARγ protein. In addition to receptor protein levels, the transcriptional activity of nuclear receptors is influenced by the recruitment of coactivators or corepressors. Coactivators such as SRC‐ 1, TIF‐2, and CBP have been shown to enhance the activity of both PPARγ and AR (DiRenzo et al., 1997; Hong et al., 1997; Ding et al., 1998; Fronsdal et al., 1998; Chen et al., 2000; Picard et al., 2002). Furthermore, the corepressor NCoR reduces the transcriptional activity of each receptor. NCoR has been shown to inhibit AR activity in human prostate cancer cells and other cell types (Cheng et al., 2002; Hodgson et al., 2005; Yoon and Wong, 2006; Godoy et al., 2012), while it promotes phosphorylation of PPARγ at Ser 273 and suppresses PPARγ activation within adipocytes (Yu et al., 2005; Li et al., 2011). The elevated level of NCoR in PC‐3 cells has also been suggested to inhibit PPARγ activity and reduce responsiveness to PPARγ agonists (Battaglia et al., 2010). It is possible that AR activation alters the availability of coactivators and/or corepressors, and ultimately reduces the pool of coregulators needed for efficient PPARγ‐mediated transcription. As a result, any increase in the amount of active AR within the cell produces a net decrease in PPARγ function. However, to confirm that AR can alter PPARγ signaling without significant alterations in PPARγ protein additional experiments need to be performed in other AR negative prostate cancer cells that express functional PPARγ.
While our data demonstrate AR suppresses the expression and activity of PPARγ in human prostate cancer cells, the consequences of this decrease in PPARγ activity are not fully understood. Data from [3H]‐thymidine incorporation studies suggest that the presence of AR interferes with the ability of PPARγ agonists to inhibit prostate cancer proliferation. Our data also indicate that AR‐driven reductions in PPARγ function influence the expression of gene products within human prostate cancer cells. In our study, the presence of AR blocked the ability of PPARγ to stimulate expression of adipocyte FABP/FABP4. FABP4 is a protein present within the cytoplasm and circulation that regulates fatty acid transport. Intracellular FABP has also been linked to alterations in prostate cell survival and proliferation. De Santis et al. showed that overexpression of FABP4 induced apoptosis within the DU‐145 prostate cancer cell line (De Santis et al., 2004). Furthermore, concentrations of bisphenol A that stimulate proliferation within the ventral prostate also decreased expression of FABP4 (Hotamisligil and Bernlohr, 2015). It is therefore possible that AR promotes growth and survival of human prostate cancer cells in part by controlling PPARγ‐mediated increases in FABP4. PPARγ activation has also been shown to induce expression of lipoprotein lipase (Lefebvre et al., 1997) and GLUT4 (Dana et al., 2001) and decrease leptin and TNF‐α levels(Spiegelman, 1998). By controlling the expression of these and other gene products, PPARγ functions as a key regulator of glucose metabolism, lipid metabolism and insulin sensitivity (Picard and Auwerx, 2002; Tontonoz and Spiegelman, 2008). A recent ChIP‐seq study by CE Massie et al. has shown that within human prostate cancer cell lines AR also regulates metabolic gene products. The AR target genes identified within their study include CAMKK2, GLUT1, hexokinase I and II, as well as other genes that regulate metabolism of glucose, lipids and amino acids (Massie et al., 2011). Our work suggests that along with above listed direct gene targets, AR may indirectly control expression of genes that regulate prostate cancer metabolism by suppressing PPARγ. However, additional studies are required to better understand how AR‐driven reductions in PPARγ function influence growth, proliferation and metabolism of prostate cancer cells.
In this study, we have primarily focused on the effect of the full length, 110 kDa form of the AR on PPARγ expression and function. However in addition to the full length AR, constitutively active N‐terminal AR variants that lack the C‐terminal ligand binding domain have been detected in human prostate cancer cell lines and tumor samples (van der Steen et al., 2013). Data from transgenic mouse studies indicate the presence of AR variants such as AR3/ARv‐7 and ARv567es is linked to the development of prostate cancer as well as the progression to castration‐resistant prostate cancer (Liu et al., 2013; Sun et al., 2014). The development of resistance to newer AR antagonists such as enzalutamide has also been associated with elevated expression of AR variants in castration‐resistant prostate cancer cells (Li et al., 2013; Nadiminty et al., 2013). Like the wild type AR, the AR variants regulate expression of several classic AR target genes such as PSA, TMPRSS2, and Nkx3.1 (Hu et al., 2009; Chan et al., 2012). However, some studies suggest AR variants may also regulate expression of unique gene targets within human tissues independent of full length AR (Guo et al., 2009; Hu et al., 2009). Studies are currently underway in our laboratory to assess whether ARv7 and other AR variants influence PPARγ expression and function in human prostate cancers.
In conclusion, AR normally functions to inhibit PPARγ expression and transcriptional activity within human prostate cancer cells. AR continues to be a primary therapeutic target for both castration‐sensitive as well as castration‐resistant prostate cancer. ADT is commonly used to reduce AR signaling in patients with advanced, metastatic prostate cancer. Furthermore, newer drugs that inhibit the AR signaling pathway have been approved by the Federal Drug Administration to treat metastatic, castration‐resistant prostate cancer. Abiraterone acetate, which blocks intratumoral and extratumoral androgen synthesis, and the more potent AR antagonist enzalutamide have been shown to enhance survival of prostate cancer patients that have developed castration‐resistant forms of prostate cancer. Our study would suggest that these and other therapeutic strategies that interfere with AR activity, whether they are competitive inhibitors of AR or other compounds that block androgen synthesis or AR nuclear localization, would ultimately result in increased PPARγ levels within prostate tumor cells. Consequently, strategies that reduce AR function could be used to increase the net amount of PPARγ and anti‐tumor effects of PPARγ agonists in prostate cancer cells. Furthermore, PPARγ expression and/or activity could serve as useful measure of AR function within human prostate cancers.
Acknowledgments
We thank Dr. Stephen Safe (Texas A&M University) for the PPRE3luciferase construct, Dr. Nancy Weigel (Baylor College of Medicine) for the CMV‐β‐galactosidase reporter construct, and Drs. Robert Matusik and RenJie Jin (Vanderbilt University School of Medicine) for the bortezomib and BMS compound. This work was supported by a NCI K01 Career Development Award (K01 CA114253), the Vanderbilt CTSA grant UL1 RR024975‐01 from NCRR/NIH, the Meharry RISE Initiative (R25 GM059994), and the NHLB1 T32 training grant (T32HL007735).
Literature Cited
- Battaglia S, Maguire O, Thorne JL, Hornung LB, Doig CL, Liu S, Sucheston LE, Bianchi A, Khanim FL, Gommersall LM, Coulter HS, Rakha S, Giddings I, O'Neill LP, Cooper CS, McCabe CJ, Bunce CM, Campbell MJ. 2010. Elevated NCOR1 disrupts PPARalpha/gamma signaling in prostate cancer and forms a targetable epigenetic lesion. Carcinogenesis 31:1650–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler R, Mitchell SH, Tindall DJ, Young CY. 2000. Nonapoptotic cell death associated with S‐phase arrest of prostate cancer cells via the peroxisome proliferator‐activated receptor gamma ligand, 15‐deoxy‐delta12, 14‐prostaglandin J2. Cell Growth Differ 11:49–61. [PubMed] [Google Scholar]
- Chan SC, Li Y, Dehm SM. 2012. Androgen receptor splice variants activate AR target genes and support aberrant prostate cancer cell growth independent of the canonical AR nuclear localization signal. J Biol Chem 287:19736–19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Johnson BA, Li Y, Aster S, McKeever B, Mosley R, Moller DE, Zhou G. 2000. Both coactivator LXXLL motif‐dependent and ‐independent interactions are required for peroxisome proliferator‐activated receptor gamma (PPARgamma) function. J Biol Chem 275:3733–3736. [DOI] [PubMed] [Google Scholar]
- Cheng S, Brzostek S, Lee SR, Hollenberg AN, Balk SP. 2002. Inhibition of the dihydrotestosterone‐activated androgen receptor by nuclear receptor corepressor. Mol Endocrinol 16:1492–1501. [DOI] [PubMed] [Google Scholar]
- Dana SL, Hoener PA, Bilakovics JM, Crombie DL, Ogilvie KM, Kauffman RF, Mukherjee R, Paterniti JR, Jr . 2001. Peroxisome proliferator‐activated receptor subtype‐specific regulation of hepatic and peripheral gene expression in the Zucker diabetic fatty rat. Metabolism 50:963–971. [DOI] [PubMed] [Google Scholar]
- De Santis ML, Hammamieh R, Das R, Jett M. 2004. Adipocyte‐fatty acid binding protein induces apoptosis in DU145 prostate cancer cells. J Exp Ther Oncol 4:91–100. [PubMed] [Google Scholar]
- Desvergne B, Wahli W. 1999. Peroxisome proliferator‐activated receptors: Nuclear control of metabolism. Endocr Rev 20:649–688. [DOI] [PubMed] [Google Scholar]
- Ding XF, Anderson CM, Ma H, Hong H, Uht RM, Kushner PJ, Stallcup MR. 1998. Nuclear receptor‐binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC‐1): Multiple motifs with different binding specificities. Mol Endocrinol 12:302–313. [DOI] [PubMed] [Google Scholar]
- DiRenzo J, Soderstrom M, Kurokawa R, Ogliastro MH, Ricote M, Ingrey S, Horlein A, Rosenfeld MG, Glass CK. 1997. Peroxisome proliferator‐activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Mol Cell Biol 17:2166–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronsdal K, Engedal N, Slagsvold T, Saatcioglu F. 1998. CREB binding protein is a coactivator for the androgen receptor and mediates cross‐talk with AP‐1. J Biol Chem 273:31853–31859. [DOI] [PubMed] [Google Scholar]
- Godoy AS, Sotomayor PC, Villagran M, Yacoub R, Montecinos VP, McNerney EM, Moser M, Foster BA, Onate SA. 2012. Altered corepressor SMRT expression and recruitment to target genes as a mechanism that change the response to androgens in prostate cancer progression. Biochem Biophys Res Commun 423:564–570. [DOI] [PubMed] [Google Scholar]
- Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y. 2009. A novel androgen receptor splice variant is up‐regulated during prostate cancer progression and promotes androgen depletion‐resistant growth. Cancer Res 69:2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisatake JI, Ikezoe T, Carey M, Holden S, Tomoyasu S, Koeffler HP. 2000. Down‐regulation of prostate‐specific antigen expression by ligands for peroxisome proliferator‐activated receptor gamma in human prostate cancer. Cancer Res 60:5494–5498. [PubMed] [Google Scholar]
- Hodgson MC, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E, Balk SP, Hollenberg AN. 2005. The androgen receptor recruits nuclear receptor CoRepressor (N‐CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J Biol Chem 280:6511–6519. [DOI] [PubMed] [Google Scholar]
- Hong H, Kohli K, Garabedian MJ, Stallcup MR. 1997. GRIP1, a transcriptional coactivator for the AF‐2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol Cell Biol 17:2735–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Bernlohr DA. 2015. Metabolic functions of FABPs‐mechanisms and therapeutic implications. Nat Rev Endocrinol 11:592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. 2009. Ligand‐independent androgen receptor variants derived from splicing of cryptic exons signify hormone‐refractory prostate cancer. Cancer Res 69:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Zhang D, Wang D, Sun B, Safoor A, Young CY, Lou H, Yuan H. 2015. Bisbibenzyls, novel proteasome inhibitors, suppress androgen receptor transcriptional activity and expression accompanied by activation of autophagy in prostate cancer LNCaP cells. Pharm Biol 54:1–11. [DOI] [PubMed] [Google Scholar]
- Jiang M, Fernandez S, Jerome WG, He Y, Yu X, Cai H, Boone B, Yi Y, Magnuson MA, Roy‐Burman P, Matusik RJ, Shappell SB, Hayward SW. 2010a. Disruption of PPARgamma signaling results in mouse prostatic intraepithelial neoplasia involving active autophagy. Cell Death Differ 17:469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Jerome WG, Hayward SW. 2010b. Autophagy in nuclear receptor PPARgamma‐deficient mouse prostatic carcinogenesis. Autophagy 6:175–176. [DOI] [PubMed] [Google Scholar]
- Knudsen KE, Penning TM. 2010. Partners in crime: Deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab 21:315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre AM, Peinado‐Onsurbe J, Leitersdorf I, Briggs MR, Paterniti JR, Fruchart JC, Fievet C, Auwerx J, Staels B. 1997. Regulation of lipoprotein metabolism by thiazolidinediones occurs through a distinct but complementary mechanism relative to fibrates. Arterioscler Thromb Vasc Biol 17:1756–1764. [DOI] [PubMed] [Google Scholar]
- Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, Sears DD, Talukdar S, Oh D, Chen A, Bandyopadhyay G, Scadeng M, Ofrecio JM, Nalbandian S, Olefsky JM. 2011. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 147:815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. 2013. Androgen receptor splice variants mediate enzalutamide resistance in castration‐resistant prostate cancer cell lines. Cancer Res 73:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Altuwaijri S, Lin WJ, Kan PY, Collins LL, Chang C. 2002. Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. J Biol Chem 277:36570–36576. [DOI] [PubMed] [Google Scholar]
- Liu G, Sprenger C, Sun S, Epilepsia KS, Haugk K, Zhang X, Coleman I, Nelson PS, Plymate S. 2013. AR variant ARv567es induces carcinogenesis in a novel transgenic mouse model of prostate cancer. Neoplasia 15:1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles BE, Akinyeke TO, Moss PE, Stewart LV. 2009. Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARgamma‐dependent and PPARgamma‐independent pathways. Cell Cycle 8:268–277. [DOI] [PubMed] [Google Scholar]
- Massie CE, Lynch A, Ramos‐Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, Bon H, Zecchini V, Smith DM, Denicola GM, Mathews N, Osborne M, Hadfield J, Macarthur S, Adryan B, Lyons SK, Brindle KM, Griffiths J, Gleave ME, Rennie PS, Neal DE, Mills IG. 2011. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 30:2719–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss PE, Lyles BE, Stewart LV. 2010. The PPARgamma ligand ciglitazone regulates androgen receptor activation differently in androgen‐dependent versus androgen‐independent human prostate cancer cells. Exp Cell Res 316:3478–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadiminty N, Tummala R, Liu C, Yang J, Lou W, Evans CP, Gao AC. 2013. NF‐kappaB2/p52 induces resistance to enzalutamide in prostate cancer: Role of androgen receptor and its variants. Mol Cancer Ther 12:1629–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Suzuki T, Sugawara A, Arai Y, Sasano H. 2009. Peroxisome proliferator‐activated receptor gamma in human prostate carcinoma. Pathol Int 59:288–293. [DOI] [PubMed] [Google Scholar]
- Nieto CM, Rider LC, Cramer SD. 2014. Influence of stromal‐epithelial interactions on androgen action. Endocr Relat Cancer 21:T147–T160. [DOI] [PubMed] [Google Scholar]
- Nwankwo JO, Robbins ME. 2001. Peroxisome proliferator‐activated receptor‐ gamma expression in human malignant and normal brain, breast and prostate‐derived cells. Prostaglandins Leukot Essent Fatty Acids 64:241–245. [DOI] [PubMed] [Google Scholar]
- Picard F, Auwerx J. 2002. PPAR(gamma) and glucose homeostasis. Annu Rev Nutr 22:167–197. [DOI] [PubMed] [Google Scholar]
- Picard F, Gehin M, Annicotte J, Rocchi S, Champy MF, O'Malley BW, Chambon P, Auwerx J. 2002. SRC‐1 and TIF2 control energy balance between white and brown adipose tissues. Cell 111:931–941. [DOI] [PubMed] [Google Scholar]
- Rogenhofer S, Ellinger J, Kahl P, Stoehr C, Hartmann A, Engehausen D, Wieland WF, Muller SC, Hofstadter F, Walter B. 2012. Enhanced expression of peroxisome proliferate‐activated receptor gamma (PPAR‐gamma) in advanced prostate cancer. Anticancer Res 32:3479–3483. [PubMed] [Google Scholar]
- Sabichi AL, Subbarayan V, Llansa N, Lippman SM, Menter DG. 2004. Peroxisome proliferator‐activated receptor‐gamma suppresses cyclooxygenase‐2 expression in human prostate cells. Cancer Epidemiol Biomarkers Prev 13:1704–1709. [PubMed] [Google Scholar]
- Santen RJ. 1992. Clinical review 37: Endocrine treatment of prostate cancer. J Clin Endocrinol Metab 75:685–689. [DOI] [PubMed] [Google Scholar]
- Segawa Y, Yoshimura R, Hase T, Nakatani T, Wada S, Kawahito Y, Kishimoto T, Sano H. 2002. Expression of peroxisome proliferator‐activated receptor (PPAR) in human prostate cancer. Prostate 51:108–116. [DOI] [PubMed] [Google Scholar]
- Singh R, Artaza JN, Taylor WE, Braga M, Yuan X, Gonzalez‐Cadavid NF, Bhasin S. 2006. Testosterone inhibits adipogenic differentiation in 3T3‐L1 cells: Nuclear translocation of androgen receptor complex with beta‐catenin and T‐cell factor 4 may bypass canonical Wnt signaling to down‐regulate adipogenic transcription factors. Endocrinology 147:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Artaza JN, Taylor WE, Gonzalez‐Cadavid NF, Bhasin S. 2003. Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor‐mediated pathway. Endocrinology 144:5081–5088. [DOI] [PubMed] [Google Scholar]
- Spiegelman BM. 1998. PPAR‐gamma: Adipogenic regulator and thiazolidinedione receptor. Diabetes 47:507–514. [DOI] [PubMed] [Google Scholar]
- Strand DW, DeGraff DJ, Jiang M, Sameni M, Franco OE, Love HD, Hayward WJ, Lin‐Tsai O, Wang AY, Cates JM, Sloane BF, Matusik RJ, Hayward SW. 2013. Deficiency in metabolic regulators PPARgamma and PTEN cooperates to drive keratinizing squamous metaplasia in novel models of human tissue regeneration. Am J Pathol 182:449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand DW, Jiang M, Murphy TA, Yi Y, Konvinse KC, Franco OE, Wang Y, Young JD, Hayward SW. 2012. PPARgamma isoforms differentially regulate metabolic networks to mediate mouse prostatic epithelial differentiation. Cell Death Dis 3:e361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarayan V, Sabichi AL, Kim J, Llansa N, Logothetis CJ, Lippman SM, Menter DG. 2004. Differential peroxisome proliferator‐activated receptor‐gamma isoform expression and agonist effects in normal and malignant prostate cells. Cancer Epidemiol Biomarkers Prev 13:1710–1716. [PubMed] [Google Scholar]
- Sun F, Chen HG, Li W, Yang X, Wang X, Jiang R, Guo Z, Chen H, Huang J, Borowsky AD, Qiu Y. 2014. Androgen receptor splice variant AR3 promotes prostate cancer via modulating expression of autocrine/paracrine factors. J Biol Chem 289:1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. 2008. Fat and beyond: The diverse biology of PPARgamma. Annu Rev Biochem 77:289–312. [DOI] [PubMed] [Google Scholar]
- van der Steen T, Tindall DJ, Huang H. 2013. Posttranslational modification of the androgen receptor in prostate cancer. Int J Mol Sci 14:14833–14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, Janne OA, Balk SP, Mehra R, Han B, Chinnaiyan AM, Rubin MA, True L, Fiorentino M, Fiore C, Loda M, Kantoff PW, Liu XS, Brown M. 2009. Androgen receptor regulates a distinct transcription program in androgen‐independent prostate cancer. Cell 138:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CC, Wang YC, Wei S, Lin LF, Chen CS, Lee CC, Lin CC, Chen CS. 2007. Peroxisome proliferator‐activated receptor gamma‐independent suppression of androgen receptor expression by troglitazone mechanism and pharmacologic exploitation. Cancer Res 67:3229–3238. [DOI] [PubMed] [Google Scholar]
- Yoon HG, Wong J. 2006. The corepressors silencing mediator of retinoid and thyroid hormone receptor and nuclear receptor corepressor are involved in agonist‐ and antagonist‐regulated transcription by androgen receptor. Mol Endocrinol 20:1048–1060. [DOI] [PubMed] [Google Scholar]
- Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. 2005. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator‐activated receptor gamma transcriptional activity and repress 3T3‐L1 adipogenesis. J Biol Chem 280:13600–13605. [DOI] [PubMed] [Google Scholar]