

Abstract
Objectives
We recently observed a decrease in deoxyribonucleotide (dNTP) pools in HIV‐infected individuals on antiretroviral therapy (ART). Alterations in dNTPs result in mutations in mitochondrial DNA (mtDNA) in cell culture and animal models. Therefore, we investigated whether ART is associated with mitochondrial genome sequence variation in peripheral blood mononuclear cells (PBMCs) of HIV‐infected treatment‐experienced individuals.
Methods
In this substudy of a case−control study, 71 participants were included: 22 ‘cases’, who were HIV‐infected treatment‐experienced patients with mitochondrial toxicity, 25 HIV‐infected treatment‐experienced patients without mitochondrial toxicity, and 24 HIV‐uninfected controls. Total DNA was extracted from PBMCs and purified polymerase chain reaction (PCR) products were subjected to third‐generation sequencing using the PacBio Single Molecule Real‐Time (SMRT) sequencing technology. The sequences were aligned against the revised Cambridge reference sequence for human mitochondrial DNA (NC_012920.1) for detection of variants.
Results
We identified a total of 123 novel variants, 39 of them in the coding region. HIV‐infected treatment‐experienced patients with and without toxicity had significantly higher average numbers of mitochondrial variants per participant than HIV‐uninfected controls. We observed a higher burden of mtDNA large‐scale deletions in HIV‐infected treatment‐experienced patients with toxicity compared with HIV‐uninfected controls (P = 0.02). The frequency of mtDNA molecules containing a common deletion (mt.δ4977) was higher in HIV‐infected treatment‐experienced patients with toxicity compared with HIV‐uninfected controls (P = 0.06). There was no statistically significant difference in mtDNA variants between HIV‐infected treatment‐experienced patients with and without toxicity.
Conclusions
The frequency of mtDNA variants (mutations and large‐scale deletions) was higher in HIV‐infected treatment‐experienced patients with or without ART‐induced toxicity than in uninfected controls.
Keywords: Antiretroviral therapy, HIV, mutagensis, mitochondrial haplogroups, mitochondrial toxicity, peripheral blood mononuclear cells
Introduction
The prevalence of antiretroviral therapy (ART)‐related toxicities is reported to be as high as 47% and 27% for clinical and laboratory manifestations, respectively 1. During the era of monotherapy with zidovudine (ZDV), a nucleoside reverse transcriptase inhibitor (NRTI), some patients developed skeletal muscle myopathies 2. Histological examination of their muscle biopsies revealed mitochondrial pathologies 3. With widespread use of NRTIs, other clinical manifestations such as lactic acidosis, lipodystrophy, peripheral neuropathies, cardiomyopathies, and pancytopaenia were observed 4, 5, 6. Inhibition of polymerase gamma (Pol‐γ), the enzyme responsible for replication of mtDNA, leading to depletion of mitochondrial DNA (mtDNA) content and subsequent mitochondrial dysfunction, was implicated as the underlying mechanism for these toxicities 5, 7. In a recent review, Apostolova et al. concluded from emerging reports that mitochondrial dysfunction cannot be explained solely by Pol‐γ inhibition 8, 9, 10, 11. Moreover, protease inhibitors (PIs) and nonnucleoside reverse transcriptase inhibitors (NNRTIs) do not inhibit Pol‐γ and yet they also cause mitochondrial dysfunction 12, 13.
In a case−control study, we found that HIV‐uninfected controls had a statistically significantly lower absolute mtDNA copy number per 100 ng of genomic DNA compared with HIV‐infected treatment‐experienced patients either with or without ART‐induced toxicity 14. Thus, we did not observe mtDNA depletion in HIV‐infected treatment‐experienced patients with toxicity as expected. To our surprise, HIV‐infected treatment‐experienced patients with toxicity had significantly higher mRNA expression of Pol‐γ in comparison with HIV‐infected treatment‐experienced patients without toxicity (P < 0.05) and HIV‐uninfected controls (P < 0.01). This contradicts the Pol‐γ inhibition theory. Interestingly, we observed a decrease in ribonucleotide (rNTP) and deoxyribonucleotide (dNTP) pool sizes in HIV‐infected treatment‐experienced patients diagnosed with ART‐induced mitochondrial toxicity compared with age‐, gender‐ and race/ethnicity‐matched HIV‐uninfected controls. Consistent with Apostolova et al. 8, we surmise that ART‐induced mitochondrial toxicity may not be explained by inhibition of Pol‐γ alone. Interestingly, alteration in dNTP pools has been associated with mtDNA mutagenesis in cell culture and animal models 15, 16. Several mitochondrial disorders, with manifestations that mirror clinical manifestations of ART‐induced mitochondrial toxicity, have been associated with mutations in the mitochondrial genome. Examples of these disorders are mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) 17, Alpers' syndrome 18, Alzheimer's and Parkinson's diseases 19, and progressive external ophthalmoplegia (PEO) 20. Moreover, the normal aging process is associated with increasing accumulation of mtDNA mutations 21, 22. There are emerging data on the association between ART and accelerated aging in HIV‐infected treatment‐experienced individuals 6, 23. However, there is a paucity of studies on the effect of ART on the evolution of the mitochondrial genome sequence 24, 25. We investigated mtDNA sequence alterations in peripheral blood mononuclear cells (PBMCs) of HIV‐infected treatment‐experienced individuals.
Methods
Study design and procedures
This was a subanalysis of a case−control study; the details of the study design have been presented previously 14. In brief, a ‘case’ was defined as an HIV‐infected treatment‐experienced individual on a stable NRTI‐based ART regimen diagnosed with ART‐associated mitochondrial toxicity by a clinician based on the presence of one or more clinical or laboratory toxicities associated with mitochondrial toxicity 4. After a case was identified, two controls were recruited: 14 an HIV‐infected treatment‐experienced individual on stable NRTI‐based ART without any of the observed clinical or laboratory toxicities and 1 a healthy HIV‐uninfected control. The controls were matched to the cases by age, gender and race. Exclusion criteria were any acute illness within 2 weeks of study enrolment, the presence of an active opportunistic infection, a history of a chronic disease such as muscular dystrophy, lupus, diabetes, renal failure or hepatic failure prior to the diagnosis of HIV infection or initiation of the NRTI‐based regimen, and active alcohol or substance abuse.
At study enrolment, participants completed a brief survey asking questions about demographic characteristics and significant past medical history. Medical records of HIV‐infected participants were reviewed and disease characteristics and laboratory data were extracted. Each participant gave about 20 ml of venous blood at the time of enrolment. We isolated PBMCs from whole blood within 2 h of collection using a Ficoll gradient (Ficoll‐Hypaque, (Pharmacia Biotech, Uppsala, Sweden)) according to the manufacturer's instructions. Aliquots of PBMCs were stored at −80°C prior to extraction of DNA for the experiments.
The HIV‐infected individuals were recruited from the Adult AIDS Care Programs at Yale−New Haven Hospital and the HIV‐uninfected volunteers were recruited through posting of advertisements at Yale University and Yale−New Haven Hospital, New Haven, CT. The study protocol was approved by the Institutional Review Board of the Yale School of Medicine. All participants gave their written informed consent before participation in the study.
Mitochondrial DNA isolation and amplification
Total DNA was extracted from PBMCs using TRIzol® Reagent (Invitrogen, CA, USA) according to the manufacturer's instructions. Eight pairs of overlapping barcoded primers were used in a polymerase chain reaction (PCR) reaction to amplify the entire mitochondrial genome (16.6 kb) as described previously 26. The sizes of the PCR products ranged from 1.6 to 2.6 kb. The sequences of the barcodes and primers were subjected to a blast search to make sure they did not amplify contaminating, nuclear‐embedded pseudogenes 27. The primer and barcode sequences are listed in Supporting Information Table S1. The PCR products were confirmed by agarose gel electrophoresis and purified with the QIAquick PCR purification kit (Qiagen, Valencia, CA, USA).
PacBio sequencing and sequence analysis
The purified PCR products were subjected to third‐generation sequencing using the PacBio Single Molecule Real‐Time (SMRT) (PacBio, Menlo Park, CA) sequencing technology 28. In brief, SMRT bell libraries were prepared for each sample by ligation of hairpin adaptors to both ends using PacBio DNA Template Prep Kit 2.0 (250 bp to 3 kbp). DNA/Polymerase Binding Kit 2.0 (PacBio) was used for setting up enzyme template complexes and libraries were loaded onto the PacBio RS sequencer using MagBeads according to the manufacturer's instructions. Sequencing was then carried out using Sequencing Kit 2.0 (PacBio). PacBio technology overcomes some of the limitations of current next‐generation sequencing platforms by providing significantly longer reads (> 1 kb), single molecule sequencing, and a single‐pass error rate of < 15%. Moreover, SMRT sequencing exceeds the consensus accuracy achieved by other sequencing methods because of the random nature of the errors. The SMRT sequencing achieves results with > 99.999% accuracy 28. The sequences were demultiplexed and aligned against the revised Cambridge reference sequence (NC_012920.1) for the human mitochondrial genome. Variant detection was performed using SMRT pipeline V2.2.0 (PacBio; https://github.com/PacificBiosciences/SMRT-Analysis). Phylotree‐based haplogroup assignments were performed by submitting the FASTA sequence to validated haplofind software (https://haplofind.unibo.it/). The variant annotation and effect prediction were performed with snpeff V4.0e (http://www.ncbi.nlm.nih.gov/pubmed/22728672).
Mitochondrial DNA large‐scale deletion assay
A region of mitochondrial encoded NADH dehydrogenase 4 gene (MT‐ND4) (MT‐ND4) harbours mtDNA deletions (82% of large‐scale deletions and 96% of multiple deletions are found in this region), whereas a region of the MT‐ND1 gene is rarely deleted 29. The ratio of MT‐ND4 to MT‐ND1 is used as a surrogate measure of deletions in mtDNA 29, 30. We estimated the proportion of mtDNA molecules containing large‐scale deletions using real‐time quantitative PCR (qPCR) by comparing the amplification of a template in MT‐ND4 to that of a template in MT‐ND1 using the same amount of DNA as described previously 29, 30. The sequences of the primers used in the qPCR reactions have been published previously 30 (Table S1). The PCR parameters were as follows: a 25‐μL reaction mixture comprising 12.5 μL of 2 × power SYBR Green Supermix (Applied Biosystems, Foster City, CA, USA), 0.3 μL of 10 μM primers and 50 ng of DNA. The PCR protocol comprised 95°C for 10 min, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. The relative amount of MT‐ND4 to MT‐ND1 was calculated using the formula R = 2−∆Ct, where ∆Ct is the difference in the cycle thresholds of amplification of MT‐ND4 and MT‐ND1 (CtMT‐ND4 − CtMT‐ND1). Without deletions in ND4, the ratio is about 1 and it is < 1 when there are deletions.
Mitochondrial common deletion (CD; mt.δ4977) quantification
The mitochondrial DNA common deletion (CD) is the most common mtDNA deletion in mitochondrial disorders 31, comprising a deletion of 4977 bases (mt.δ4977) in mtDNA spanning mt.8470 to 13447. We determined the prevalence of mtDNA CD in our study participants using a validated qPCR 32. Details of the primers used have been published previously (Table S1) 30. The primers targeting the deleted region were designed to span mt.8393–13509. A 139‐bp amplicon was amplified only in the presence of the CD, which occurs from mt.8470 to13447. The amplification of a conserved MT‐ND1 gene was used as a control, as described above. The qPCR reaction and conditions were the same as described above. The freqency of CDs per million molecules of mtDNA was calculated using the formula R = 1 × 106 × 2−∆Ct, where ∆Ct is CtCD – CtND1.
Statistical analysis
The data are presented as medians with interquartile ranges (IQRs) and as frequencies with percentages for continuous and categorical variables, respectively. The Wilcoxon rank sum or Mann−Whitney test and the χ2 test were used to compare continuous and categorical variables among the study groups, respectively. These nonparametric tests have fewer assumptions than parametric tests regarding the data distribution and are generally more robust when analysing genetic variation data. The comparison of nonsynonymous and synonymous mutations in coding regions was carried out using a Poisson regression model. A deviance test and overdispersion test 33 were used to check the goodness of fit and Poisson distribution assumption. The comparison of mutations in the whole genome was performed using zero inflated Poisson regression, as the deviance test and overdispersion test showed that the Poisson regression model did not fit the data well. Spearman's rank correlations were used to examine bivariate associations between study outcomes. The cut‐off for significance was set at P < 0.05.
Results
Characteristics and mitochondrial haplogroups of participants
Data for 71 of 75 participants enrolled in the original study were used in this subanalysis; 22 HIV‐infected treatment‐experienced individuals with toxicity, 25 HIV‐infected treatment‐experienced individuals without toxicity, and 24 HIV‐uninfected controls had sufficient amounts and quality of DNA for amplification and sequencing of the entire mitochondrial genome. The median (IQR) number of reads per sample was 328 (201–581). Table 1 illustrates the demographic and disease characteristics of the study participants. HIV‐infected treatment‐experienced individuals with and without toxicity were not significantly different with respect to HIV viral load, CD4 count and current NRTI type (Table 1). In contrast, HIV‐infected treatment‐experienced patients without toxicity had a longer time since HIV diagnosis (P = 0.002) and a longer duration of current NRTI use (P = 0.02) compared with HIV‐infected treatment‐experienced patients with toxicity.
Table 1.
Demographic and clinical characteristics of study participants
| HIV‐uninfected | HIV‐infected treatment‐experienced without toxicity (n = 25) | HIV‐infected treatment‐experienced with toxicity (n = 22) | P‐value | |
|---|---|---|---|---|
| Gender | ||||
| Female | 41.7 | 40 | 36.4 | >0.05 |
| Male | 58.3 | 60 | 63.6 | >0.05 |
| Age (years) | 51 (49–56) | 53 (51–57) | 53 (50–57) | >0.05 |
| Race | ||||
| White non‐Hispanic | 25 | 28.0 | 27.2 | >0.05 |
| White Hispanic | 12.5 | 12.0 | 4.6 | >0.05 |
| African American | 62.5 | 60.0 | 68.2 | >0.05 |
| CD4 count (cells/uL) | NA | 500 (291–792) | 593 (676–895) | 0.06a |
| Viral load (copies/mL) | NA | 20 (20–20) | 20 (20–89.75) | >0.05a |
| Duration of HIV infection (years) | NA | 16 (9–18) | 9 (6–10) | 0.002a , * |
| Duration of current NRTI exposure (years) | NA | 5 (4–6) | 3 (1.9–5.5) | 0.02a , * |
| Current NRTI | ||||
| TDF | NA | 80 | 72.7 | >0.05a |
| FTC | NA | 80 | 72.7 | >0.05a |
| 3TC | NA | 20 | 27.3 | >0.05a |
| EFV | NA | 40 | 22.7 | >0.05a |
| ABC | NA | 4 | 0 | >0.05a |
| ZDV | NA | 4 | 4.5 | >0.05a |
| Toxicity | ||||
| Single toxicity | NA | NA | 50 | – |
| Multiple toxicity | NA | NA | 50 | – |
Data are shown as percentage or median (IQR). P‐values were calculated using the paired t‐test for continuous variables and the χ2 test for categorical variables.
a P‐value for the comparison between HIV‐infected treatment‐experienced patients without and with toxicity only.
*P‐values are two‐sided and considered significant if <0.05.
The limit of detection of the viral load assay was 20 copies/mL; patients with undetectable viral load were assigned a value of 20 copies/mL.
NA, not applicable; TDF, tenofovir; FTC, emtricitabine; 3TC, lamivudine; EFV, efavirenz;. ABC, abacavir; ZDV, zidovudine; NRTI, nucleoside reverse transcriptase inhibitor.
Forty‐five, 19 and seven participants self‐identified as African Americans, white non‐Hispanics and white Hispanics, respectively. The final distribution of African haplogroups was L2, 40%; L3, 29%; L1, 29%; and L0, 3%. The majority (60%) of European sequences were haplogroup T, followed in frequency by haplogroups K (13%), H (10%), U (10%), J (3%) and X (3%).
Antiretroviral toxicity and mitochondrial haplogroups
We next investigated whether there was an association between ART‐induced mitochondrial toxicity and mitochondrial haplogroup as reported previously 34, 35. We found no significant association (P = 0.58) between mitochondrial toxicity and haplogroup using a multivariate logistic model that incorporated age, gender, duration of HIV infection, duration of ART, CD4 T‐cell count and HIV viral load. Of interest, 4917A>G, a nonsynonymous polymorphism in the MT‐ND2 region of mitochondrial DNA previously associated with neurodegenerative phenotypes 36, was associated with hyperlipidaemia in participants with an African haplogroup in our study. Fifty‐five per cent (six of 12) and 25% (two of nine) of participants with African and European haplogroups, respectively, had hyperlipidaemia. Among participants with hyperlipidaemia (n = 8), 67% (four of six) of those with African haplogroups had the 4917A>G polymorphism, but none of those with European haplogroups (zero of two) had this polymorphism.
Mitochondrial point mutations among study participants
We identified a total of 123 non‐haplogroup‐associated variants; there were 75 variants in the control region (D‐loop; mt.16024‐576) (Table 2), six in the ribosomal RNA (rRNA), three in the transfer RNA (tRNA) (data not shown), and 39 in the coding region (mt.577‐16023; 14 synonymous and 25 nonsynonymous changes; Table 3). As expected, about 61% of the variants occurred in the D‐loop, the hypervariable region. HIV‐infected treatment‐experienced patients had significantly higher average numbers of mitochondrial variants per participant compared with HIV‐uninfected controls (Fig. 1a). These variants were predominantly heteroplasmic mutations, that is, co‐existing wild‐type and mutant variants (Table 3). HIV‐infected treatment‐experienced patients with toxicity (P = 0.04) and HIV‐infected treatment‐experienced patients without toxicity (P = 0.04) had a significantly higher prevalence of nonsynonymous variants in the coding region compared with HIV‐uninfected controls (Fig. 1b and Table 4). Although not statistically significant, HIV‐infected treatment‐experienced individuals with toxicity had a higher prevalence of synonymous variants than HIV‐infected treatment‐experienced individuals without toxicity and HIV‐uninfected controls.
Table 2.
Mitochondrial variants in the control region (D‐loop; mt.16024–576)
| mtDNA locus | Nucleotide variant | Participants with variant [n (%)] | Reported* |
|---|---|---|---|
| 23 | T>C | 1 (1) | Yes |
| 32 | A>G | 6 (8) | No |
| 34 | G>T | 7 (10) | No |
| 35 | G>A | 3 (4) | Yes |
| 35 | G>T | 13 (18) | No |
| 36 | G>T | 1 (1) | No |
| 36 | G>C | 11 (15) | No |
| 37 | A>T | 1 (1) | No |
| 38 | G>T | 13 (18) | No |
| 40 | T>A | 6 (8) | No |
| 95 | A>G | 1 (1) | No |
| 95 | A>C | 2 (3) | Yes |
| 115 | T>C | 1 (1) | Yes |
| 179 | T>C | 1 (1) | Yes |
| 356 | C>G | 6 (8) | No |
| 483 | C>A | 1 (1) | No |
| 16346 | G>C | 1 (1) | Yes |
The mitochondrial sequences were aligned against the revised Cambridge reference sequence (NC_012920.1).
*Reported to the Mitomap database (http://www.mitomap.org).
Table 3.
Nucleotide changes in the mitochondrial coding region (mt.577−16 023)
| Subject ID* | Nucleotide variant | Percentage of variant allele† | Amino acid change | Gene¶ | Reported‡ |
|---|---|---|---|---|---|
| N01 | 5331C>A | 72 | Leu288Ile | ND2 | Yes |
| N03 | 10587 C>T | 75 | Leu40 | ND4L | Yes |
| N05 | 14334C>T | 36 | Val114Ile | ND6 | Yes |
| N23 | 6860A>G | 51 | Lys319 | CO1 | No |
| N25 | 15302C>A | 14 | Pro186Thr | CYB | No |
| P02 | 9555C>A | 13 | Pro117Thr | CO3 | No |
| P04 | 4890A<G | 23 | Ile141Val | ND2 | Yes |
| P06 | 7754G>A | 83 | Asp57Asn | CO2 | Yes |
| P06 | 11242C>G | 89§ | Leu161 | ND4 | Yes |
| P07 | 8233A>G | 70 | Leu216 | CO2 | Yes |
| P12 | 5331C>A | 77 | Leu288Ile | ND2 | Yes |
| P13 | 14754C>G | 21 | Pro3Arg | CYB | No |
| P15 | 4234A>G | 80 | Thr310Ala | ND1 | Yes |
| P15 | 5331C>A | 55 | Leu288Ile | ND2 | No |
| P19 | 15302C>A | 16 | Pro186Thr | CYB | Yes |
| P19 | 8204A>G | 18 | Met207Val | CO2 | No |
| P20 | 8896G>A | 72 | Ala124Thr | ATP6 | Yes |
| P20 | 9590A>G | 61 | Glu128 | CO3 | Yes |
| P24 | 4217A>G | 21 | Tyr304Cys | ND1 | No |
| P24 | 9482T>C | 53 | Phe92 | CO3 | Yes |
| C01 | 4217A>G | 18 | Tyr304Cys | ND1 | No |
| C01 | 12540A>G | 74 | Trp68 | ND5 | Yes |
| C09 | 3978C>T | 21 | Phe224 | ND1 | Yes |
| C10 | 4060C>G | 17 | Pro252Ala | ND1 | No |
| C10 | 9081C>T | 80 | Asn185 | ATP6 | Yes |
| C10 | 14122A>G | 21 | Ile596Val | ND5 | Yes |
| C10 | 14131C>G | 15 | Leu599Val | ND5 | No |
| C12 | 6918C>T | 98§ | Leu339Phe | CO1 | Yes |
| C12 | 10083A>G | 84 | Ile9Val | ND3 | Yes |
| C12 | 12936A>G | 79 | Gln200 | ND5 | No |
| C12 | 14079A>G | 42 | Lys581 | ND5 | Yes |
| C15 | 15472A>G | 46 | Leu242 | CYB | Yes |
| C19 | 3510C>A | 59 | Ile68Met | ND1 | Yes |
| C19 | 9193C>T | 39 | His223Tyr | ATP6 | Yes |
| C21 | 8382C>T | 20 | Thr6Ile | ATP8 | Yes |
| C23 | 3495C>A | 71 | Pro63 | ND1 | Yes |
| C23 | 6431 A>G | 71 | Met176 | CO1 | Yes |
| C24 | 5293G>A | 81 | Ser275Asn | ND2 | Yes |
| C25 | 4217A>G | 18 | Tyr304Cys | ND1 | No |
The mitochondrial sequences were aligned against the revised Cambridge reference sequence (NC_012920.1).
*Prefix N, negative control; P, positive control; C, case.
†The percentage of reads with variant allele was calculated as: R = number of reads with variant allele/(number of reads with variant allele + number of reads with reference allele) × 100.
‡Reported in the Mitomap database (http://www.mitomap.org).
§Regarded as a homoplastic change with GATK integrated SMRT pipeline V2.2.0.
¶ND, NADH dehydrogenase; CO, cytochrome c oxidase; ATP, ATP synthase; CYB, cytochrome b.
Figure 1.
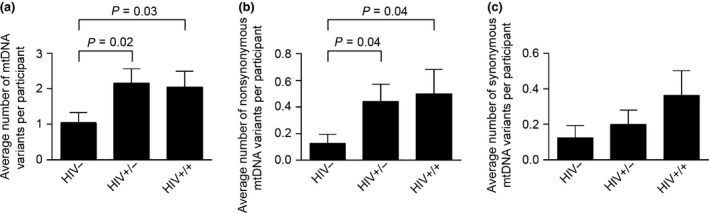
Mitochondrial point mutation frequency among study participants. Mitochondrial DNA (mtDNA) was extracted from peripheral blood mononuclear cells (PBMCs) and amplified with eight pairs of overlapping primers. The PCR products were purified and sequenced with SMRT sequencing technology as described in the ‘Methods’ section. The sequences were demultiplexed and aligned against the revised Cambridge reference sequence for the human mitochondrial genome (NC_012920.1). (a) Average number of mtDNA variants per participant. (b) Average number of nonsynonymous mtDNA variants per participant. (c) Average number of synonymous mtDNA variants per participant. Comparison of synonymous or nonsynonymous changes in the coding region was carried out using the χ2 test. Data are presented as mean ± standard error of the mean. The cut‐off for significance was set at P < 0.05. HIV−, HIV‐uninfected controls; HIV+/−, HIV‐infected treatment‐experienced patients without toxicity; HIV+/+, HIV‐infected treatment‐experienced patients with toxicity.
Table 4.
Multivariate analysis of mitochondrial DNA point mutations among study participants
| HIV+/+ vs. HIV+/− | HIV+/+ vs. HIV− | HIV+/− vs. HIV− | HIV+* vs. HIV− | |||||
|---|---|---|---|---|---|---|---|---|
| Predictor | Est (95% CI) | P‐value | Est (95% CI) | P‐value | Est (95% CI) | P‐value | Est (95% CI) | P‐value |
| Total variants† | ||||||||
| Group | −0.101 (−0.55, 0.35) | 0.66 | 0.608 (0.05, 1.16) | 0 . 03 § | 0.649 (0.08, 1.21) | 0 . 02 | 0.681 (0.17, 1.19) | 0.01 |
| Age | 0.003 (−0.03, 0.03) | 0.86 | −0.004 (−0.04, 0.04) | 0.86 | 0 (−0.04, 0.04) | 0.98 | 0.002 (−0.03, 0.03) | 0.88 |
| Gender | 0.297 (−0.22, 0.82) | 0.26 | 0.671 (−0.08, 1.42) | 0.08 | −0.091 (−0.6, 0.42) | 0.73 | 0.194 (−0.25, 0.63) | 0.39 |
| Coverage | 0.001 (0, 0) | 0.31 | 0 (0, 0) | 0.63 | 0.003 (0, 0) | 0.01 | 0.001 (0, 0) | 0.10 |
| Nonsynonymous variants in coding region‡ | ||||||||
| Group | 0.09 (−0.75, 0.93) | 0.83 | 1.316 (0.03, 2.60) | 0.04 | 1.359 (0.07, 2.65) | 0.04 | 1.364 (0.15, 2.58) | 0.03 |
| Age | −0.038 (−0.1, 0.02) | 0.19 | 0.012 (−0.06, 0.09) | 0.76 | −0.04 (−0.11, 0.03) | 0.25 | −0.025 (−0.08, 0.03) | 0.37 |
| Gender | 0.471 (−0.47, 1.41) | 0.33 | 1.271 (−0.23, 2.77) | 0.10 | −0.135 (−1.21, 0.94) | 0.81 | 0.476 (−0.40, 1.35) | 0.29 |
| Coverage | −0.002 (−0.01, 0.002) | 0.36 | −0.001 (−0.005, 0.003) | 0.61 | −0.001 (−0.005, 0.003) | 0.57 | −0.001 (−0.005, 0.003) | 0.37 |
| Synonymous variants in coding region‡ | ||||||||
| Group | 0.546 (−0.58, 1.67) | 0.34 | 1.346 (−0.21, 2.90) | 0.09 | 0.962 (−0.68, 2.61) | 0.25 | 1.18 (−0.31, 2.67) | 0.12 |
| Age | 0.001 (−0.08, 0.08) | 0.98 | 0.025 (−0.06, 0.11) | 0.57 | −0.068 (−0.15, 0.02) | 0.11 | −0.01 (−0.08, 0.06) | 0.79 |
| Gender | 1.191 (−0.32, 2.7) | 0.12 | 19.029 (−6604.4, 6642.4) | 0.10 | 0.284 (−1.44, 2.01) | 0.75 | 1.412 (−0.08, 2.90) | 0.06 |
| Coverage | −0.001 (−0.01, 0.003) | 0.64 | −0.001 (−0.01, 0.005) | 0.84 | 0.001 (−0.005, 0.007) | 0.64 | −0.001 (−0.005, 0.003) | 0.75 |
Significant values are shown in bold.
HIV−, HIV‐uninfected controls; HIV+/−, HIV‐infected treatment‐experienced patients without toxicity; HIV+/+, HIV‐infected treatment‐experienced patients with toxicity; Est, estimate.
*HIV+’ is comprised of HIV+/+ and HIV+/− patients.
†Comparison of total mutations in the mitochondrial genome and mutations in the control region was carried out using the zero‐inflated Poisson (ZIP) regression model.
‡Comparison of synonymous or nonsynonymous changes in the coding region was carried out using the normal Poisson regression model.
§The cut‐off for significance was set at P < 0.05.
In a multivariable analysis (Table 4), we found a statistically significant difference in the overall mitochondrial variant burden between HIV‐infected treatment‐experienced patients with toxicity and HIV‐uninfected controls (P = 0.03); between HIV‐infected treatment‐experienced patients without toxicity and HIV‐uninfected controls (P = 0.02); and between HIV‐infected treatment‐experienced participants (with and without toxicity) and HIV‐uninfected controls (P = 0.01). There was no statistically significant difference in the variants observed in the rRNA, the tRNA, and the control (D‐Loop) regions of the mitochondrial DNA among study participants (data not shown). There was a statistically significant difference in nonsynonymous changes in the coding region between HIV‐infected treatment‐experienced patients with toxicity and HIV‐uninfected controls (P = 0.04), and between HIV‐infected treatment‐experienced participants (with and without toxicity) and HIV‐uninfected controls (P = 0.03).
Mitochondrial DNA large‐scale deletions among study participants
Large‐scale mitochondrial DNA deletions are reported to be more common than point mutations 37; therefore, we determined deletion mutations in the mtDNA of the study participants. There were more deletions in mitochondrial ND4 in HIV‐infected treatment‐experienced individuals with toxicity compared with HIV‐uninfected controls (P = 0.02) (Fig. 2a). There was no statistically significant difference in the frequency of MT‐ND4 deletions between HIV‐infected treatment‐experienced patients without toxicity and HIV‐uninfected controls. The frequency of mtDNA molecules containing CD mutations tended to be higher in HIV‐infected treatment‐experienced individuals with toxicity compared with HIV‐uninfected controls (P = 0.06) (Fig. 2b).
Figure 2.
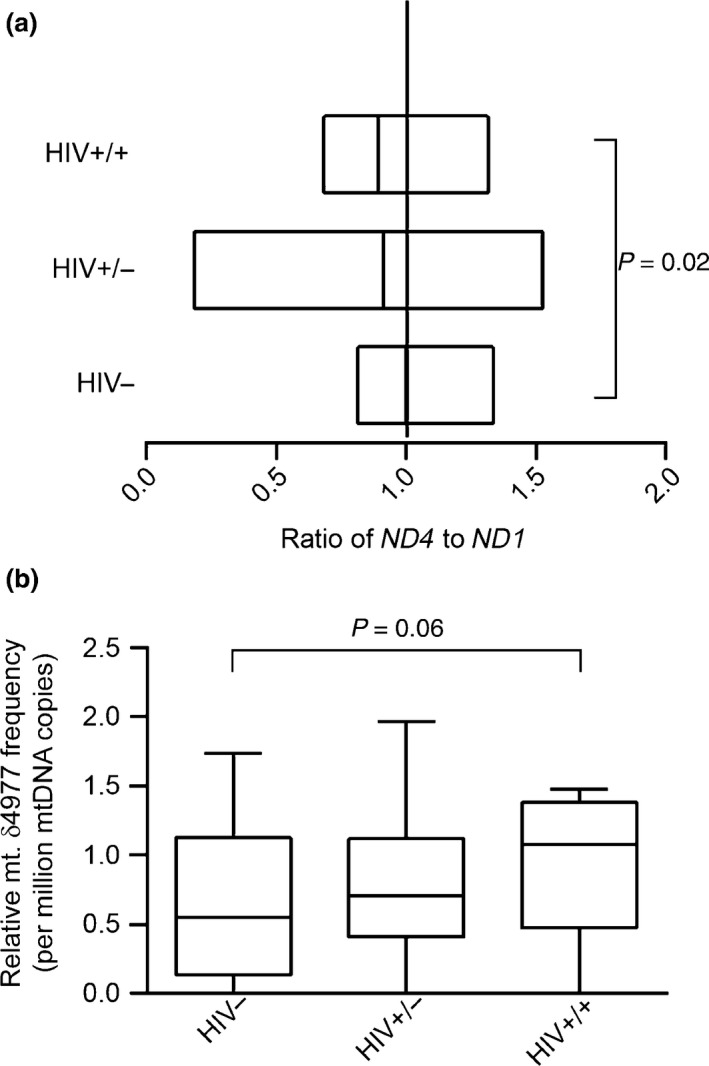
Mitochondrial DNA large‐scale deletions among study participants. Mitochondrial DNA large‐scale deletions among study participants were determined by comparing the cycle thresholds of amplification of same‐size amplicons in the MT‐ND4 and MT‐ND1 regions. (a) mtDNA large‐scale deletions. The relative amount of ND4 to ND1 was calculated using the following equation: R = 2−∆Ct, where ∆Ct is CtMT‐ND 4 − CtMT‐ND 1. Data are presented as box plots, in which boxes represent the median and the second and third quartile ratios. The vertical line represents an MT‐ND 4 to MT‐ND1 ratio of 1; this is when there are no deletions detected in the MT‐ND4 region. With deletions, the ratio is < 1. (b) The mitochondrial DNA common deletion (CD; mt.δ4977). The frequency of CDs per million molecules of mtDNA was calculated using the following formula: R = 1 × 106 × 2−∆Ct, where ∆Ct is CtCD – CtND 1. Data are represented as box‐and‐stem plots, in which boxes represent the median values and the second and third quartiles, and stems extend to the 10th and 90th percentiles. The cut‐off for significance was set at P < 0.05. HIV−, HIV‐uninfected controls; HIV+/−, HIV‐infected treatment‐experienced patients without toxicity; HIV+/+, HIV‐infected treatment‐experienced patients with toxicity.
Discussion
We investigated mtDNA sequence variation (i.e. mutations and deletions) in PBMCs of HIV‐infected treatment‐experienced individuals in a case−control study. We found that HIV‐infected treatment‐experienced individuals had a significantly higher frequency of mtDNA point mutations per participant than HIV‐uninfected individuals. However, we did not find a statistically significant difference in mtDNA variants between HIV‐infected treatment‐experienced individuals with and without toxicity. Furthermore, we observed a statistically significantly higher frequency of mtDNA large‐scale deletions in HIV‐infected treatment‐experienced individuals with mitochondrial toxicity compared with HIV‐uninfected controls. We previously reported that, in our cohort, HIV‐infected treatment‐experienced individuals had significantly lower dNTP pools (DNA precursors) than HIV‐uninfected controls 14. Associations between dNTP pool size asymmetries and a high frequency of mitochondrial DNA mutations have been reported in cell culture and animal models 15, 16. Our findings suggest that it is plausible that this association may also exist in humans. Of note, uridine supplementation was associated with improvement in lipoatrophy scores in HIV‐infected patients and a transient improvement in limb fat in patients with lipoatrophy in small clinical studies 38. ART‐induced mitochondrial dysfunction leads to depletion of intracellular pyrimidine through effects on the activity of dihydroorotate dehydrogenase (DHODH) 39. Dihydroorotate dehydrogenase converts dihydroorotate to orotate, a rate‐limiting step of the synthesis of pyrimidine. Therefore, supplementation with exogenous uridine, a precursor for pyrimidine synthesis, salvages pyrimidine synthesis and attenuates mitochondrial toxicity 40.
This is the first study, to the best of our knowledge, to report large‐scale mtDNA deletions in PBMCs of HIV‐infected treatment‐experienced individuals. We observed a higher frequency of deletions in the ND4 region, a region prone to deletions 27, in HIV‐infected treatment‐experienced patients with toxicity compared with HIV‐uninfected controls. Maagaard et al. found a statistically significantly higher frequency of large‐scale mtDNA deletions in the skeletal muscles of HIV‐infected treatment‐experienced patients compared with HIV‐infected treatment‐naïve patients and HIV‐uninfected controls 41. However, they did not find any mtDNA deletions in the PBMCs of those patients. Also, McComsey et al. did not find large‐scale mtDNA deletions in the PBMCs of HIV‐infected treatment‐experienced individuals 42. The difference between our findings and those of Maagaard et al. and McComsey et al. might have resulted from the different methods used. For detection of mtDNA deletions, we used primers designed to amplify only in the presence of deletions in a qPCR reaction, while Maagaard et al. and McComsey et al. used long‐range PCR with subsequent gel electrophoresis and Southern blot, respectively. mtDNA deletions are sporadic and have been associated with inherited mitochondrial diseases such as Kearns−Sayre syndrome (KSS), progressive external ophthalmoplegia (PEO) and Pearson's syndrome 43, 44. The high frequency of the mtDNA CD and other large‐scale deletions in our HIV‐infected treatment‐experienced cohort is consistent with the hypothesis that truncated mtDNA templates may have a replicative advantage over wild‐type sequences and thus are positively selected for in the presence of ART pressure 45. PBMCs have previously been discounted as a relevant surrogate tissue for investigating mitochondrial toxicity 41. However, if our findings are validated, the use of PBMCs for studies of mtDNA deletions and mutations will be attractive in view of the greater convenience of sampling PBMCs compared with invasive tissue biopsy.
Although the number of studies is limited, the association between mtDNA point mutations and ART has been reported in HIV‐infected treatment‐experienced individuals 24, 42. Our observation that HIV‐infected treatment‐experienced individuals had a higher frequency of mtDNA point mutations per participant compared with HIV‐uninfected controls is, therefore, consistent with those of previous studies. We identified 123 novel variants, most of which occurred in the D‐loop region, the hypervariable region. HIV‐infected treatment‐experienced patients with toxicity had a significantly higher number of mtDNA variants in the coding region of the mitochondrial DNA compared with HIV‐uninfected controls. Most of the mtDNA variants in our cohort were heteroplasmic mutations. Heteroplasmic mtDNA mutations are considered recessive and require an extremely high mutation burden to exhibit a clinical phenotype 46. Usually, the proportion of mtDNA heteroplasmic mutations in the coding region has to exceed 90% to 95% (threshold) of the mitochondrial variants for clinical manifestation 47. Thus, normal individuals might carry subthreshold levels of potentially pathogenic mtDNA mutations. Also, HIV‐infected treatment‐experienced individuals tended to have a higher frequency of synonymous mutations. These mutations could be transient and set the stage for further base changes resulting in nonsynonymous mutations. It is plausible that ART provides a conducive environment for the accumulation of pathogenic mtDNA mutations. Different classes of ART regimens might cause mtDNA depletion, proliferation of mtDNA, and/or an oxidative stress environment resulting in damage and defective repair of mtDNA 48. However, the natural variations in mtDNA sequences, as a result of a high degree of polymorphic diversity, have to be considered before assigning pathogenicity to mtDNA sequence variants. Nevertheless, it is tempting to speculate that these mutations could have pathogenic consequences for the following reasons: 1 mutations in mtDNA have been associated with inherited mitochondrial diseases that share manifestations with ART‐induced mitochondrial toxicities 46, 47; 2 there are emerging data on accelerated aging in HIV‐infected treatment‐experienced individuals 6, 23, 30; the normal aging process is associated with the accumulation of mtDNA mutations 21, 22; and 3 there is a preponderance of findings from several studies of mtDNA mutations in PBMCs and large‐scale deletions in tissues of HIV‐infected treatment‐experienced individuals. Future studies, particularly longitudinal studies, will be needed to establish the temporal association between these mtDNA variants and ART or ART‐induced toxicity. The bioenergetic and pathophysiological consequences of such mutations also have to be established.
Our study has several strengths compared with previous studies; these include sequencing and analysis of the entire mitochondrial genome. It is also the first study to observe mtDNA CD mt.δ4977 in association with ART using PBMCs. However, our study, like most cross‐sectional studies, has several limitations. First, we cannot prove causality between ART and mtDNA variants or between mtDNA variants and ART‐induced mitochondrial toxicity. Secondly, designation of mitochondrial toxicity was based on clinical manifestations and laboratory test results which were not confirmed with tissue diagnosis. Therefore, some of the HIV‐infected treatment‐experienced patients with toxicity could have been misclassified. Thirdly, we did not analyse mutations in other tissues to corroborate our findings in PBMCs. Fourthly, although there was no statistically significant difference in HIV viral load between HIV‐infected patients with and without toxicity, one cannot rule out the contribution of HIV infection as we did not include treatment‐naïve HIV‐infected individuals. Fifthly, in view of the limited sample size, our study is mainly exploratory and hypothesis‐generating. Further studies are needed to elucidate the underlying mechanisms of ART‐induced mitochondrial DNA mutations and their possible association with clinically manifest mitochondrial toxicity syndromes.
In conclusion, we found a high frequency of mtDNA variants in HIV‐infected treatment‐experienced individuals. ART appears to perturb dNTP pools, as reported previously 14; the concentrations of various dNTPs at replication sites are important determinants of the rate and the fidelity of mtDNA replication 49. Therefore, perturbation of dNTP pools during ART may be responsible for the high frequency of mtDNA mutations observed in tissue culture and animal models 15, 16, 49. ART‐induced mutations may be involved in the causal pathway of ART‐induced mitochondrial toxicity or accelerated aging in HIV‐infected treatment‐experienced individuals 23. There are currently two proposed mechanisms to explain the association between mtDNA mutations and ART: 14 ART directly or indirectly provides a permissive environment for sporadic mtDNA mutagenesis 48; and 1 ART pressure leads to clonal expansion of existing mutations 30. Further studies are needed to clarify the underlying mechanisms of mtDNA mutations in HIV‐infected treatment‐experienced individuals.
Acknowledgements
We are grateful to the patients at Nathan Smith Clinic, Yale−New Haven Hospital for their cooperation. We thank all the providers (doctors and nurses) and staff at Nathan Smith Clinic for making the study possible. This study was supported by a grant from the National Institutes of Health (KO8AI074404 to EP). We thank Dr Warren Andiman for his critical reading of the manuscript.
Conflicts of interest: All authors declare that they have no conflicts of interest.
Funding: This study was supported by a grant from the National Institutes of Health (KO8AI074404 to EP). The content of the paper is solely the responsibility of the authors and does not necessarily represent the official view of NIH.
References
- 1. Fellay J, Boubaker K, Ledergerber B et al Prevalence of adverse events associated with potent antiretroviral treatment: Swiss HIV cohort study. Lancet 2001; 358: 1322–1327. [DOI] [PubMed] [Google Scholar]
- 2. Gertner E, Thurn JR, Williams DN et al Zidovudine‐associated myopathy. Am J Med 1989; 86 (6 Pt 2): 814–818. [DOI] [PubMed] [Google Scholar]
- 3. Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, Griffin JL. Mitochondrial myopathy caused by long‐term zidovudine therapy. N Engl J Med 1990; 322: 1098–1105. [DOI] [PubMed] [Google Scholar]
- 4. Brinkman K, ter Hofstede HJ, Burger DM, Smeitink JA, Koopmans PP. Adverse effects of reverse transcriptase inhibitors: mitochondrial toxicity as common pathway. AIDS 1998; 12: 1735–1744. [DOI] [PubMed] [Google Scholar]
- 5. Lewis W, Dalakas MC. Mitochondrial toxicity of antiviral drugs. Nat Med 1995; 1: 417–422. [DOI] [PubMed] [Google Scholar]
- 6. Gardner K, Hall PA, Chinnery PF, Payne BA. HIV Treatment and Associated Mitochondrial Pathology: review of 25 Years of in Vitro, Animal, and Human Studies. Toxicol Pathol 2014; 42: 811–822. [DOI] [PubMed] [Google Scholar]
- 7. Chen CH, Vazquez‐Padua M, Cheng YC. Effect of anti‐human immunodeficiency virus nucleoside analogs on mitochondrial DNA and its implication for delayed toxicity. Mol Pharmacol 1991; 39: 625–628. [PubMed] [Google Scholar]
- 8. Apostolova N, Blas‐Garcia A, Esplugues JV. Mitochondrial interference by anti‐HIV drugs: mechanisms beyond Pol‐gamma inhibition. Trends Pharmacol Sci 2011; 32: 715–725. [DOI] [PubMed] [Google Scholar]
- 9. Hoschele D. Cell culture models for the investigation of NRTI‐induced mitochondrial toxicity. Relevance for the prediction of clinical toxicity. Toxicol In Vitro 2006; 20: 535–546. [DOI] [PubMed] [Google Scholar]
- 10. Sievers M, Walker UA, Sevastianova K et al Gene expression and immunohistochemistry in adipose tissue of HIV type 1‐infected patients with nucleoside analogue reverse‐transcriptase inhibitor‐associated lipoatrophy. J Infect Dis 2009; 200: 252–262. [DOI] [PubMed] [Google Scholar]
- 11. Galluzzi L, Pinti M, Troiano L et al Changes in mitochondrial RNA production in cells treated with nucleoside analogues. Antivir Ther 2005; 10: 191–195. [PubMed] [Google Scholar]
- 12. Deng W, Baki L, Yin J, Zhou H, Baumgarten CM. HIV protease inhibitors elicit volume‐sensitive Cl‐ current in cardiac myocytes via mitochondrial ROS. J Mol Cell Cardiol 2010; 49: 746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blas‐Garcia A, Apostolova N, Ballesteros D et al Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology 2010; 52: 115–125. [DOI] [PubMed] [Google Scholar]
- 14. Selvaraj S, Ghebremichael M, Li M et al Antiretroviral therapy‐induced mitochondrial toxicity: potential mechanisms beyond polymerase‐gamma inhibition. Clin Pharmacol Ther 2014; 96: 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song S, Pursell ZF, Copeland WC, Longley MJ, Kunkel TA, Mathews CK. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc Natl Acad Sci USA 2005; 102: 4990–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Song S, Wheeler LJ, Mathews CK. Deoxyribonucleotide pool imbalance stimulates deletions in HeLa cell mitochondrial DNA. J Biol Chem 2003; 278: 43893–43896. [DOI] [PubMed] [Google Scholar]
- 17. Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999; 283: 689–692. [DOI] [PubMed] [Google Scholar]
- 18. Naviaux RK, Nguyen KV. POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann Neurol 2004; 55: 706–712. [DOI] [PubMed] [Google Scholar]
- 19. Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron 2011; 70: 1033–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vu TH, Tanji K, Pallotti F et al Analysis of mtDNA deletions in muscle by in situ hybridization. Muscle Nerve 2000; 23: 80–85. [DOI] [PubMed] [Google Scholar]
- 21. Pallotti F, Chen X, Bonilla E, Schon EA. Evidence that specific mtDNA point mutations may not accumulate in skeletal muscle during normal human aging. Am J Hum Genet 1996; 59: 591–602. [PMC free article] [PubMed] [Google Scholar]
- 22. Simonetti S, Chen X, DiMauro S, Schon EA. Accumulation of deletions in human mitochondrial DNA during normal aging: analysis by quantitative PCR. Biochim Biophys Acta 1992; 1180: 113–122. [DOI] [PubMed] [Google Scholar]
- 23. Effros RB, Fletcher CV, Gebo K et al Aging and infectious diseases: workshop on HIV infection and aging: what is known and future research directions. Clin Infect Dis 2008; 47: 542–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin AM, Hammond E, Nolan D et al Accumulation of mitochondrial DNA mutations in human immunodeficiency virus‐infected patients treated with nucleoside‐analogue reverse‐transcriptase inhibitors. Am J Hum Genet 2003; 72: 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McComsey G, Bai RK, Maa JF, Seekins D, Wong LJ. Extensive investigations of mitochondrial DNA genome in treated HIV‐infected subjects: beyond mitochondrial DNA depletion. J Acquir Immune Defic Syndr 2005; 39: 181–188. [PubMed] [Google Scholar]
- 26. Taylor RW, Taylor GA, Durham SE, Turnbull DM. The determination of complete human mitochondrial DNA sequences in single cells: implications for the study of somatic mitochondrial DNA point mutations. Nucleic Acids Res 2001; 29: E74–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taylor RW, Taylor GA, Morris CM, Edwardson JM, Turnbull DM. Diagnosis of mitochondrial disease: assessment of mitochondrial DNA heteroplasmy in blood. Biochem Biophys Res Commun 1998; 251: 883–887. [DOI] [PubMed] [Google Scholar]
- 28. Carneiro MO, Russ C, Ross MG, Gabriel SB, Nusbaum C, DePristo MA. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genom 2012; 13: 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He L, Chinnery PF, Durham SE et al Detection and quantification of mitochondrial DNA deletions in individual cells by real‐time PCR. Nucleic Acids Res 2002; 30: e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Payne BA, Wilson IJ, Hateley CA et al Mitochondrial aging is accelerated by anti‐retroviral therapy through the clonal expansion of mtDNA mutations. Nat Genet 2011; 43: 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 2005; 39: 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meissner C, Mohamed SA, Klueter H, Hamann K, von Wurmb N, Oehmichen M. Quantification of mitochondrial DNA in human blood cells using an automated detection system. Forensic Sci Int 2000; 113: 109–112. [DOI] [PubMed] [Google Scholar]
- 33. Cameron ACT, Pravin K. Regression‐based tests for overdispersion in the Poisson Model. Journal of Econometrics 1990; 46: 347–364. [Google Scholar]
- 34. Hendrickson SL, Kingsley LA, Ruiz‐Pesini E et al Mitochondrial DNA haplogroups influence lipoatrophy after highly active antiretroviral therapy. J Acquir Immune Defic Syndr 2009; 51: 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sinxadi PZ, Dave JA, Samuels DC et al Mitochondrial genomics and antiretroviral therapy‐associated metabolic complications in HIV‐infected Black South Africans: a pilot study. AIDS Res Hum Retroviruses 2013; 29: 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Canter JA, Haas DW, Kallianpur AR et al The mitochondrial pharmacogenomics of haplogroup T: MTND2*LHON4917G and antiretroviral therapy‐associated peripheral neuropathy. Pharmacogenomics J 2008; 8: 71–77. [DOI] [PubMed] [Google Scholar]
- 37. Fayet G, Jansson M, Sternberg D et al Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord 2002; 12: 484–493. [DOI] [PubMed] [Google Scholar]
- 38. McComsey GA, Walker UA, Budhathoki CB et al Uridine supplementation in the treatment of HIV lipoatrophy: results of ACTG 5229. AIDS 2010; 24: 2507–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Setzer B, Lebrecht D, Walker UA. Pyrimidine nucleoside depletion sensitizes to the mitochondrial hepatotoxicity of the reverse transcriptase inhibitor stavudine. The American Journal of Pathology 2008; 172: 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Walker UA, Venhoff N, Koch EC, Olschewski M, Schneider J, Setzer B. Uridine abrogates mitochondrial toxicity related to nucleoside analogue reverse transcriptase inhibitors in HepG2 cells. Antivir Ther 2003; 8: 463–470. [PubMed] [Google Scholar]
- 41. Maagaard A, Holberg‐Petersen M, Kollberg G, Oldfors A, Sandvik L, Bruun JN. Mitochondrial (mt)DNA changes in tissue may not be reflected by depletion of mtDNA in peripheral blood mononuclear cells in HIV‐infected patients. Antivir Ther 2006; 11: 601–608. [PubMed] [Google Scholar]
- 42. McComsey G, Tan DJ, Lederman M, Wilson E, Wong LJ. Analysis of the mitochondrial DNA genome in the peripheral blood leukocytes of HIV‐infected patients with or without lipoatrophy. AIDS 2002; 16: 513–518. [DOI] [PubMed] [Google Scholar]
- 43. Moraes CT, DiMauro S, Zeviani M et al Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns‐Sayre syndrome. N Engl J Med 1989; 320: 1293–1299. [DOI] [PubMed] [Google Scholar]
- 44. Pearson HA, Lobel JS, Kocoshis SA et al A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr 1979; 95: 976–984. [DOI] [PubMed] [Google Scholar]
- 45. Wang H, Lemire BD, Cass CE et al Zidovudine and dideoxynucleosides deplete wild‐type mitochondrial DNA levels and increase deleted mitochondrial DNA levels in cultured Kearns‐Sayre syndrome fibroblasts. Biochim Biophys Acta 1996; 1316: 51–59. [DOI] [PubMed] [Google Scholar]
- 46. Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet 2012; 13: 878–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schapira AH. Mitochondrial diseases. Lancet 2012; 379: 1825–1834. [DOI] [PubMed] [Google Scholar]
- 48. Lewis W, Copeland WC, Day BJ. Mitochondrial dna depletion, oxidative stress, and mutation: mechanisms of dysfunction from nucleoside reverse transcriptase inhibitors. Lab Invest 2001; 81: 777–790. [DOI] [PubMed] [Google Scholar]
- 49. Mathews CK, Song S. Maintaining precursor pools for mitochondrial DNA replication. FASEB Journal 2007; 21: 2294–2303. [DOI] [PubMed] [Google Scholar]