

ABSTRACT
Marine mussels secret protein‐based adhesives, which enable them to anchor to various surfaces in a saline, intertidal zone. Mussel foot proteins (Mfps) contain a large abundance of a unique, catecholic amino acid, Dopa, in their protein sequences. Catechol offers robust and durable adhesion to various substrate surfaces and contributes to the curing of the adhesive plaques. In this article, we review the unique features and the key functionalities of Mfps, catechol chemistry, and strategies for preparing catechol‐functionalized polymers. Specifically, we reviewed recent findings on the contributions of various features of Mfps on interfacial binding, which include coacervate formation, surface drying properties, control of the oxidation state of catechol, among other features. We also summarized recent developments in designing advanced biomimetic materials including coacervate‐forming adhesives, mechanically improved nano‐ and micro‐composite adhesive hydrogels, as well as smart and self‐healing materials. Finally, we review the applications of catechol‐functionalized materials for the use as biomedical adhesives, therapeutic applications, and antifouling coatings. © 2016 The Authors. Journal of Polymer Science Part A: Polymer Chemistry Published by Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 9–33
Keywords: adhesives, adhesive polymers, biomaterials, biomimetic design, biopolymers, Dopa, mussel foot proteins, wet adhesion
INTRODUCTION
Marine mussels have mastered the ability to anchor to foreign surfaces in seawater through the use of adhesive proteins.1 These mussel foot proteins (Mfps) are known to cure rapidly to form adhesive plaques with high interfacial binding strength, durability, and toughness. 3,4‐Dihydroxyphenylalanine (Dopa), which is modified from tyrosine through post‐transitional hydroxylation, is one of the main constituents in Mfps.2, 3, 4 The catechol side chain of Dopa has the ability to form various types of chemical interactions and crosslinking, which imparts Mfps with the ability to solidify in situ and bind tightly to various types of surface substrates. To harvest the remarkable wet adhesive properties of these adhesive proteins, many efforts have been made to develop new adhesive materials inspired by the designs of Mfps.
Natural Mfps have been extracted and analyzed from different species of mussels in the aim of creating strong adhesive materials.5, 6 However, several thousand mussel specimens are required for extracting one gram of adhesive proteins, making the direct use of these adhesives for commercial applications highly challenging.7 This highlights the need for developing synthetic mussel‐inspired adhesive polymers with strong water‐resistant adhesive properties.
The adhesive mechanism of marine mussels and the key features of Mfps that affect adhesive properties have been extensively studied during past decades, which provide guidance for developing new synthetic biomimetic adhesives.8, 9, 10 Here, we provide an updated review on the design of adhesive materials inspired by Mfps. We first describe the unique features of adhesive plaque proteins and their key functionalities as well as strategies for preparing biomimetic adhesive polymers. We also summarize the recent developments in designing advanced mussel‐inspired materials including coacervated Dopa‐functionalized adhesives, mechanically improved nano‐ and micro‐composite adhesive hydrogels, hydrogel actuators, self‐healing hydrogels, and smart adhesives. Finally, the applications of these adhesive materials as biomedical adhesives, drug carriers for therapeutic uses, and antifouling coatings are reviewed.
CHEMISTRY OF ADHESION: MUSSEL ADHESIVE PROTEINS
Mussel adhesives proteins enable marine mussels to attach strongly to various surfaces in their turbulent, wet and saline habitats.1 These proteins are secreted in a liquid form, which then solidify to form a byssal thread and an adhesive plaque complex (Fig. 1). The byssal threads are engineered to withstand elevated mechanical loads applied by waves and currents. A byssal thread connects a mussel to the adhesive plaque that is anchored to a foreign surface.2, 11 The average force needed to dislodge a California mussel, Mytilus californianus, is estimated to be 250–300 N/mussel, with an average detachment force of 5–6 N/thread.12, 13 This remarkable surface anchoring capacity provides insights in designing synthetic polymers for interfacial applications. In this section, we review the compositions and chemistries of various plaque proteins with a specific focus on those found at the plaque‐surface interface that contribute to strong interfacial binding.
Figure 1.
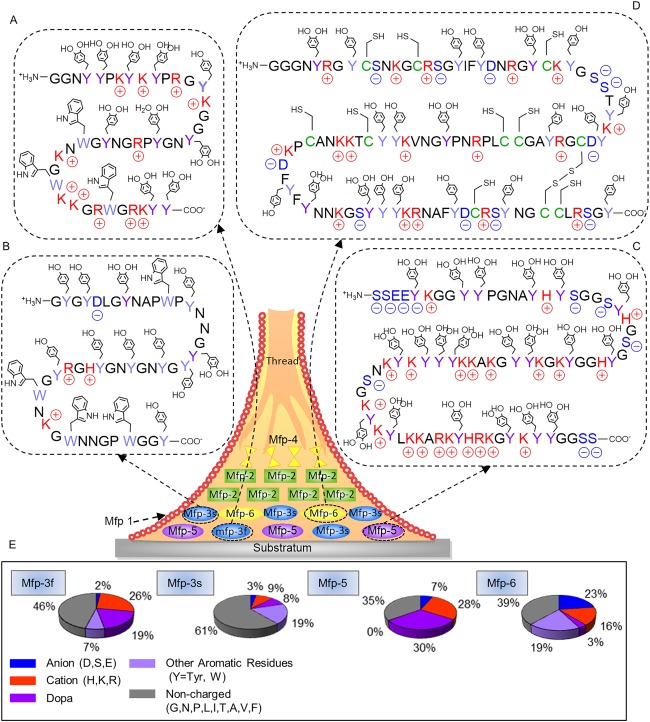
Schematic representation of a byssal thread and adhesive plaque with the approximate distribution of known Mfps. Primary sequences of Mfp‐3f (A), Mfp‐3s (B), Mfp‐5 (C), and Mfp‐6 (D). Acidic, basic, Dopa, and aromatic residues are shaded blue, red, dark purple, and light purple, respectively. Post‐translational modification of tyrosine to Dopa and the formation of disulfide cysteine could occur anywhere within the peptide sequences. Pie charts illustrating the distribution of key functionalities found in selected Mfps (E).
Mussel Foot Proteins
At least six Mfps (Mfp‐1 through Mfp‐6) have been identified from the adhesive plaques of several species of mussel (i.e., Mytilus edulis, Mytilus galloprovincialis, M. californianus, etc.).14 These proteins are characterized by a basic isoelectric point (pI) due to a high content of cationic amino acids. The pI is described as the pH in which the net charge of the protein is zero.15 Most importantly, these plaque proteins contain various amounts of the unique amino acid, Dopa.3, 4 The catechol side chain of Dopa offers robust and durable adhesion to various substrate surfaces and contributes to the curing of the adhesive plaques.4 Mfps extracted from different Mytilus species exhibited a high level of sequence homology and a similar distribution within the adhesive plaque (Fig. 1).12, 13 These findings suggest that each type of Mfp has a unique function and contributes differently to the interfacial properties of the adhesive plaque.
Mfp‐1 is a high molecular weight (e.g., 108 kDa in M. edulis) and basic protein with very little secondary structures.16 It is located in the cuticle of the byssus threads and the adhesive plaques and acts mainly as a protective varnish layer.17 Mfp‐2 is a smaller protein (e.g., 42–47 kDa in M. edulis) with highly repetitive motifs and is the most abundant protein found within the plaque (∼25 wt %).18 Mfp‐2 contains a relatively high content of cysteine residues (6 mol %) in the form of disulfide bonds, and it is believed that Mfp‐2 provides structural integrity to the adhesive plaques.18 Mfp‐4 consists of a histidine‐ (His‐) rich decapeptide tandemly repeated more than 36 times, which binds exceptionally well to transition metal ions such as copper.19 Mfp‐4 is strategically located between the adhesive plaque and the distal portion of the byssal thread, effectively linking plaque proteins (e.g., Mfp‐2) with those found within the byssal thread (e.g., preCOL).9, 19 PreCols are collagenous proteins with high Dopa and His contents and these proteins are mainly distributed throughout the byssal threads.20, 21 Specifically, Mpf‐4 is believed to interact with the His‐rich domain of preCOLs through metal ion coupling.9, 19, 22
Mfp‐3, 5, and 6 are predominantly found at the plaque–substrate interface, contributing to strong, wet adhesion. Mfp‐3 is the smallest adhesive protein among plaque proteins (e.g., molecular weight of 5–7 kDa in M. edulis and M. californianus).22, 23, 24 It is the most polymorphic adhesive protein with no known repeating sequences.24 It is reported to have over 30–35 different variants, which can be further subdivided into two separate groups known as Mfp‐3 fast and slow (Mfp‐3f and Mfp‐3s, respectively).24, 25 Based on the sequences reported for M. californianus (Table 1), both Mfp‐3f and Mfp‐3s are rich in glycine (25–29 mol %) and asparagine (10–18 mol %).24 Additionally, Mfp‐3f exhibits elevated contents of post‐translationally modified Dopa (> 20 mol %) and 4‐hydroxyarginine (1 mol %), and positively charged residues (26 mol %) [Fig. 1(A,E)], which renders Mfp‐3f highly hydrophilic.
Table 1.
Amino Acid Composition of Mussel Foot Proteins Isolated From the Plaque Represented in the Number of Residue Per 100 Residues
| Amino acids | Mfp‐3f | Mfp‐3s | Mfp‐5 | Mfp‐6 |
|---|---|---|---|---|
| Pro (P) | 6.0 | 8.0 | 3.6 | 4.9 |
| Gly (G) | 25.0 | 29.0 | 19.6 | 13.7 |
| Ala (A) | 2.0 | 1.0 | 2.7 | 2.9 |
| Cys (C) | 0 | 0 | 0 | 2.9 |
| Asp/Asn (D/N) | 10.0 | 18.0 | 3.6 | 13.4 |
| Glu (E) | 1.0 | 1.0 | 0.6 | 2.3 |
| Ser (S) | 1.0 | 2.0 | 1.2 | 4.3 |
| pSer a | 0 | 0 | 4.8 | 2.8 |
| Dopa | 19.0 | 8.0 | 30.4 | 3.2 |
| Tyr (Y) | 1.0 | 19.0 | 0.2 | 19.2 |
| Trp (W) | 6.0 | – | 0 | 0 |
| His (H) | 1.0 | 3.0 | 4.8 | 0 |
| Lys (K) | 15.0 | 4.0 | 19.8 | 9.8 |
| hArg b | 1.0 | 0 | 0 | 0 |
| Arg (R) | 9.0 | 2.0 | 3.1 | 6.5 |
| Total | 100 | 99 | 100 | 100 |
| Reference | 19 | 19 | 14 | 14 |
Phosphoserine.
Hydroxyarginine.
Acidic, basic and aromatic residues are shaded blue, red and purple, respectively
In contrast, Mfp‐3s exhibits a lower conversion of tyrosine to Dopa residues (5–10 mol %) and contains a lower charge density (9 and 3 mol % positively and negatively charged residues, respectively) when compared to Mfp‐3f, resulting in a polar but hydrophobic protein [Fig. 1(B)]. However, its Dopa content can approach 28 mol % in some variants, which may be crucial for adhesion to metal and mineral surfaces.24 This level of Dopa content is only exceeded by another plaque protein, Mfp‐5.26 In Mfp‐3s, Dopa is protected from oxidation even in footprints left behind from the removed plaques. This is surprising given the susceptibility of the Dopa to autoxidation at the pH of seawater (pH 7.5–8.4).27 We reviewed the possible reduction–oxidation (redox) chemistry of plaque proteins in the subsequent section entitled “Effect of Oxidation State of Catechol.”
Mfp‐5 has a molecular mass of 8.9 kDa and is the least polymorphic plaque proteins, consisting of one protein sequence with two closely related variants.14, 26 Mfp‐5 contains the highest amount of the adhesive Dopa (30 mol %) residues amongst all the plague proteins [Fig. 1(C)].14 Mfp‐5 is also characterized by its hydrophilicity and a basic pI due to an elevated content of cationic amino acids (27.7 mol %). Additionally, Mfp‐5 also contains variable amounts of post‐translationally modified phosphoserine (∼4.8 mol %), known for its ability to bind to calcareous mineral materials (e.g., statherin and osteopontin).28, 29 The presence of elevated Dopa content and phosphoserine suggest that Mfp‐5 plays an important role in interfacial binding.
Unlike aforementioned interfacial plaque proteins, Mfp‐6 contains a much lower amount of Dopa (3 mol %) but a higher levels of tyrosine (20 mol %) [Fig. 1(D)].14 Although the total phenolic residues content in Mfp‐6 is similar to those found in Mfp‐3 and 5, the tyrosine residues in Mfp‐6 are not efficiently converted to Dopa. Additionally, Mfp‐6 has the highest contents of charged residues (23 and 16 mol % anionic and cationic amino acids, respectively). Another unique feature of Mfp‐6 is the presence of cysteine (11 mol %) with a small portion of these residues present in the form of disulfide bonds (2 mol %). The high level of thiol gives Mfp‐6 the ability to effectively control the redox chemistry of Dopa residues present in other plaque proteins,14, 30 which is further reviewed in a later section.
Catechol Chemistry
One of the unique features of Mfps is the abundance of the catecholic amino acid, Dopa, in their protein sequences. The presence of catechol is believed to fulfill the dual role of interfacial binding and the solidification of the adhesive proteins.31 Catechol is capable of diverse chemistries, which enables it to bind to both organic and inorganic surfaces through the formation of reversible non‐covalent or irreversible covalent interactions (Fig. 2). These chemical reactions are also critical to designing in situ curable materials. In this section, we summarize various catechol side chain chemical interactions.
Figure 2.
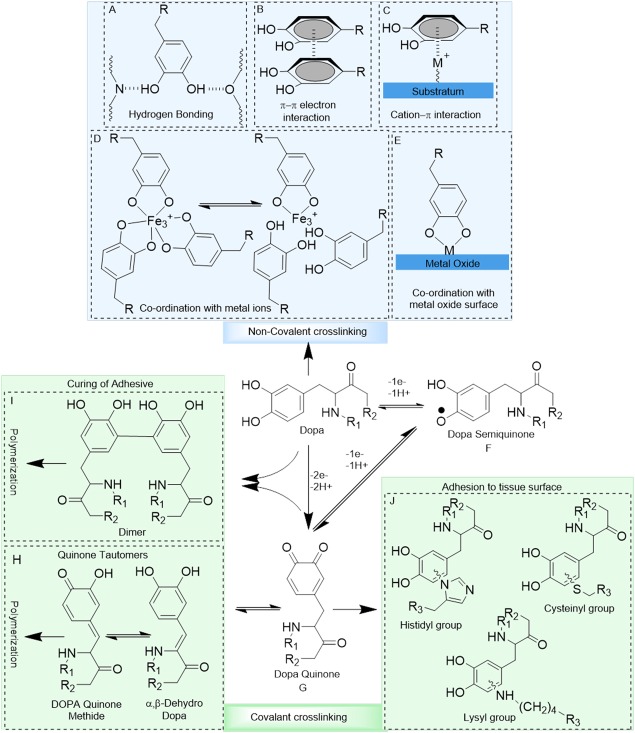
Possible interactions and reaction products of the catechol side chain of Dopa. Catechol forms hydrogen bonds through its —OH groups (A), π–π electron interaction with another benzene ring (B) and, cation–π interaction with positively charged ions (C). Catechol chelates metal ions to form self‐healing crosslinking (D) and, forms co‐ordination bonds with metal oxide surfaces (E). Dopa oxidizes to its semiquinone and quinone forms which are highly reactive (F, G). Quinone tautomerizes to form quinonemethide and α,β‐dehydrodopa, leading to the subsequent polymerization of the catechol group (H). Quinone also forms dimers with another catechol moiety, resulting in dimer formation (I). Quinone reacts with nucleophiles (i.e., –NH2, –SH) found on tissue surface, resulting interfacial covalent crosslinking (J).
Non‐Covalent Dopa Interactions
The dihydroxy functionality of catechol enables it to form strong hydrogen bonds [H‐bonds, Fig. 2(A)], which promotes its absorption to mucosal tissues32, 33 and hydroxyapatite surfaces.34, 35 The benzene ring of catechol is capable of interacting with other aromatic rings through π–π electron interaction [Fig. 2(B)],1, 11 which improves the cohesive properties of catechol‐containing polymers and enables them to attach to surfaces rich in aromatic compounds (e.g., polystyrene)36 and gold substrates.37 The aromatic ring also forms cation–π interaction with positively charged ions, which is one of the strongest non‐covalent interactions in water [Fig. 2(C)].38, 39, 40 Cation–π interaction enhances absorption of catechol to charged surfaces41 and contributes to the cohesive properties of materials rich in both aromatic and cationic functional groups.42 Since catechols are easily oxidized to its poorly adhesive quinone form in an oxygen rich and basic environment, cation–π interaction complements the underwater adhesive properties of catechol.42, 43, 44, 45
Catechol chelates metal ions to form strong, reversible complexes with various metal ions, including Cu2+, Zn2+, Mn2+, Fe3+, V3+, Ti3+, and Ti4+ [Fig. 2(D)].46, 47, 48, 49 The log stability constants of these complexes are significantly higher when compared to those of polymeric acid‐ or amino acid‐based ligands (e.g., log stability constant of 62 for Ti4+ in a tris‐catecholate complex).50, 51 Catechol forms complexes with different stoichiometry (i.e., mono‐, bis‐, and tris‐catecholate–metal ion complexes), depending on the valency of the metal ion, catechol to metal ion molar ratios, and pH.46, 52, 53 Catecholate–metal ion complexation increases the wear resistance of byssal thread cuticles during the large, cyclic strains experienced by the byssus in the turbulent intertidal zone.54, 55 This complexation chemistry has been exploited in creating self‐healing hydrogels,56, 57, 58 pH‐responsive drug carrier,59 soft actuators,60, 61, 62 and mechanically reinforced polymeric fibers.63, 64
In addition to forming strong complexes with metal ions, catechol forms strong, reversible interfacial bonds with metal oxide surfaces [Fig. 2(E)].65, 66, 67 The pull‐off force required to separate a single Dopa molecule from titanium (Ti) averaged around 800 pN, which approaches 40% the bond strength of a covalent bond (2000 pN for a carbon–silicon bond).68 This is the strongest reversible bond reported in the literature involving a biological molecule. According to density functional theory analysis, the catechol groups readily compete with water molecules and adsorb directly onto the metal oxide surfaces with binding energy of 20–30 kcal/mol.69, 70 Catechol's ability to attach to various metal substrates (e.g., Au2O3, Al2O3, SiO2 TiO2, NiTi, and stainless steel), makes it an ideal anchoring group for surface modification.71, 72, 73 However, the binding strength of the catechol to metal substrates is highly dependent on its oxidation state, and its binding strength is drastically reduced when the catechol is oxidized.66, 68, 74
Oxidation‐Mediate Covalent Crosslinking
When catechol is oxidized, it becomes highly reactive and can participate in intermolecular covalent crosslinking, resulting in the curing of Dopa‐containing adhesives. Catechol can be oxidized to form semiquinone [Fig. 2(F)] and quinone [Fig. 2(G)] by one‐electron and two‐electron oxidation, respectively, through autoxidation in the presence of molecular oxygen or the addition of either chemical (e.g., periodate) or enzymatic (e.g., tyrosinase, peroxidase) oxidants.1, 75 Quinone can tautomerize to form quinone methide and then to α,β‐dehydro‐Dopa,76 resulting in the subsequent dimer formation and polymerization of the Dopa residues [Fig. 2(H)].77 Additionally, crosslinking of the quinone with other catechol groups leads to dimers formation and subsequent polymerization of the catechol groups [Fig. 2(I)].1, 75 Finally, quinone can also react with various nucleophilic functional groups (i.e., –NH2, –SH, imidazole) found on biological substrates, forming interfacial covalent bond [Fig. 2(J)].68, 78, 79
Oxidation of catechol amine with a free primary amine group (e.g., dopamine) results in intra‐molecular cyclization and polymerization resembling melanin formation.77 The polymeric form of dopamine or polydopamine has the capability of attaching to various types of surfaces ranging from noble metals to inert polymers and ceramics80 with different geometry and sizes.81, 82, 83 The amine group found in the polydopamine film has been found to contribute to the cohesive force formed between polydopamine films.84 Additionally, polydopamine coating remains reactive and can be further modified with functional groups (i.e., —NH2, —SH, etc.) or metal ions.80, 82, 85 Antifouling polymers such as PEG and alginate can graft on polydopamine‐coated surfaces for designing antifouling surfaces that prevent non‐specific attachment of cells and bacteria.80, 86
The oxidative crosslinking of catechol is dependent on multiple factors, which include the type of oxidant, the concentration of oxidant, and pH. For example, enzyme‐induced crosslinking results in the polymerization of phenyl groups [Fig. 2(I)] and, the rate and degree of polymerization increase with increasing enzyme concentration.77 On the other hand, periodate‐mediated crosslinking involves the polymerization of α,β‐dehydro form of the catechol [Fig. 2(H)], with a maximum rate of crosslinking occurring at periodate to catechol molar ratios between 1 and 0.5.77, 87 Additionally, the rate of crosslinking increases with increasing pH, due to an elevated conversion of catechol to quinone at a more basic pH.87 On the other hand, oxidation intermediate of catechol such as quinone methide is more stable at a mild acidic condition (pH 5.7–6.7), and the rate of crosslinking is retarded at an acidic pH.87, 88 Similarly, protonation of nucleophilic functional groups (e.g., pK a of ε‐lysine ∼10) at an acidic pH limits their ability for covalent crosslinking, resulting in reduced interfacial bonding between catechol‐containing adhesive and soft tissue surface at an acidic pH.87 This reduced crosslinking capability at an acid pH may restrict the application of the catechol in contact with mildly acidic tissues (e.g., cancer cells (pH ≤ 7),89 skin (pH 4–6),90 subcutaneous tissues (pH 6.7–7.1),91 and dysoxic tissues due to extensive hemorrhage or ischemia (pH < 7).92
During catechol oxidation, reactive oxygen species (ROS) such as super oxide anion ( ¯) and hydrogen peroxide (H2O2) are generated.93, 94 Catechol‐containing hydrogel released 102−103 µM of H2O2 during a period of 48 h in culture, which reduced cell viability.93 The cytotoxic effect of ROS was prevented in the presence of catalase, indicating that the release of ROS is a source of cytotoxicity of catechol in culture.93, 95 The biological effects of ROS are highly concentration dependent and can range from beneficiary (i.e., promote wound healing, antimicrobial effects) to detrimental (i.e., chronic inflammation, tumor initiation) responses.96, 97, 98, 99 Therefore, precise regulation of ROS generated from catechol‐containing biomaterial is necessary depending on the intended application.
Contribution of Other Factors to Adhesion
Dopa is the main constituent of Mfps that contributes to strong wet adhesion.100, 101, 102, 103 However, recent findings revealed other factors such as the redox chemistry, coacervate formation, surface drying capability, and other properties of Mfps may also play a role. This section reviews the contribution of these factors on interfacial binding.
Effect of Oxidation State of Catechol
The adhesive property of catechol is highly dependent on its oxidation state.66, 74, 104 At an acidic pH where catechol exists predominantly in its reduced form, catechol exhibits elevated adhesive strength to inorganic substrates.102, 105 When the pH approaches and exceeds the first dissociation constant of the catechol –OH group (pK a1 = 9.3),106 the catechol side chain autoxidizes to its quinone form with reduced adhesive strength.66, 68, 74 The force required to separate a single molecule of Dopa from Ti surface reduces by nearly 80% with the oxidation of the catechol side chain.68 Similarly, addition of chemical oxidants (i.e., periodate) irreversibly oxidizes catechol, resulting in reduced adhesive properties.66, 107
To counteract the mildly basic condition of seawater, the presence of Mfp‐6 near the plaque‐surface interface was proposed to provide an antioxidant mechanism to preserve the reduced form of catechol for enhanced adhesion (Fig. 3). Mfp‐6 is rich in cysteine residues with an unusually low content of Dopa for an interfacial plaque protein.14, 105 The reducing capacity of Mfp‐6 was recently reported to be 17 electrons per protein, where nearly half of them (at least 8 electrons) were contributed by factors other than the cysteine residues and potentially by the presence of the Dopa moieties.108 The redox‐active Dopa in Mfp‐6 likely serves as a sacrificial antioxidant, and when these catechols oxidize [Fig. 3(A)] Dopa residues in Mfp‐3 or Mfp‐5 are reduced to its adhesive catechol state [Fig. 3(C)].108 Similarly, oxidation of thiol functional groups in the cysteine residues results in the formation of dithiol bonds [Fig. 3(B)], which serves to preserve the reduced form of Dopa in Mfp‐3 and Mfp‐5 [Fig. 3(D)].102
Figure 3.
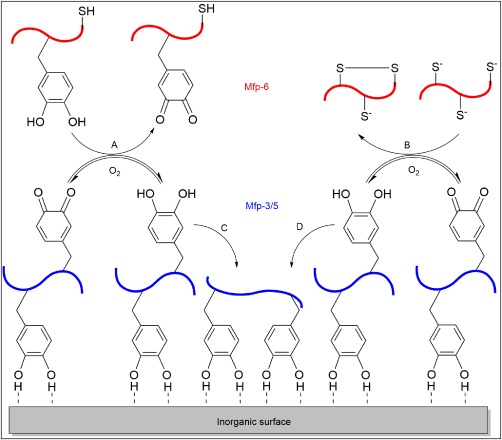
Schematic illustration of the adhesion of Mfp‐3 and 5 in the presence of Mfp‐6. Oxidation of catechol (A) and the formation of dithiol bonds (B) in Mfp‐6 counteract the autoxidation of catechol in Mfp‐3/5 to quinone to enhance the adsorption of Mfp‐3/5 to the substrate surface (C, D). Reprinted by permission from Macmillan Publishers Ltd: Nature Chemical Biology Ref. 102, copyright 2011.
Another strategy employed by marine mussels to minimize Dopa autoxidation is the presence of Mfp‐3s at the plaque‐surface interface. Mfp‐3s has a low post‐transitional modification of tyrosine to Dopa residues and a lower charge density than Mfp‐3f, making Mfp‐3s to be a polar but hydrophobic protein.4 Mfp‐3s has demonstrated a higher redox potential and significantly lower loss of H‐bonding interactions between its Dopa residues and mica substrate when compared to the hydrophilic Mfp‐3f at a slightly basic pH.4 The hydrophobic environment created by Mfp‐3s shields Dopa from the basic seawater, which retards Dopa oxidation and enhances the adhesion of plaque proteins on inorganic surfaces.4
Coacervate Formation
Coacervation is the fluid–fluid phase separation of ionic polymers or proteins from the aqueous solution.109 The coacervates are driven by the coulombic attraction and the neutralization of oppositely charged side chains found in polyelectrolytes, and are further stabilized by hydrophobic forces.110, 111 Coacervation contributes to underwater adhesion as it increases polymer concentration in the coacervates, enhances wetting properties through decreasing interfacial energy, and eases the adhesive delivery by reducing viscosity.101
Two different coacervation systems have been reported thus far; self‐coacervation and complex coacervation (Fig. 4). Self‐coacervation is the formation of coacervates by a single species of polymer, which has been demonstrated by the highly charged yet hydrophobic Mfp‐3s [Fig. 4(A)].4 Plaque proteins are delivered at an acidic pH, at which point the net positively charged Mfp‐3s is highly soluble. When the pH increases to the pI of the protein (pI ∼7.5), the ionic residues are neutralized and the decreased electrostatic repulsion between the proteins resulted in self‐coacervation.112 Similarly, increasing the ionic strength of the solution resulted in a similar effect. Additionally, the coacervates are further stabilized by the hydrophobic residues of Mfp‐3s, as more than 60% of its amino acid residues are more hydrophobic than glycine.4
Figure 4.
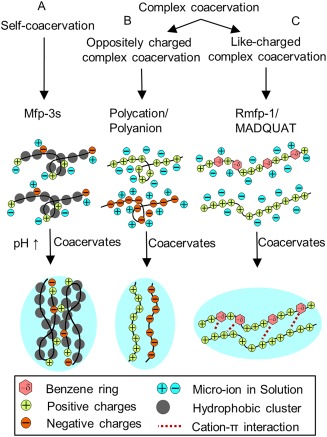
Schematic representation of self‐coacervation formed by a polyampholytic protein (i.e., Mfp‐3s) with increasing pH (A) and complex coacervation formed between oppositely charged polyelectrolytes (B) Reprint from Ref. 112, Copyright 2014, with permission from Elsevier, and like‐charged polyelectrolytes facilitated by the strong short range cation–π interactions between catechol and cationic functional groups (C).
Complex coacervation involves the formation of coacervates when a polyanion and a polycation neutralize one another due to the strong electrostatic interaction between the two oppositely charged polyelectrolytes [Fig. 4(B)]. Recently, Kim et al.113 reported the coacervation of two positively charged polyelectrolytes [Fig. 4(C)]. Like‐charged complex coacervation occurred through the formation of a strong short‐range cation–π interactions between benzene ring of Dopa in a recombinant mussel foot protein 1 (rMfp‐1) and cation in poly(2‐(trimethylamino)ethyl methacrylate, which overcame the long‐range electrostatic repulsion.
Surface Drying Properties
The presence of a layer of absorbed water in between the adhesive and the surface interferes with interfacial molecular contact and results in a strong hydration repulsive force which hinders adhesion.114 Recent findings suggest that the hydrophobic Mfp‐3s can overcome the repulsive hydration force by spontaneously “dry” the wetted surface before adhesion.115 The hydrophobic side chains such as tryptophan residues in Mfp‐3s have been found to contribute to the removal of the surface hydration layer.115
Additionally, the exceptional surface wetting properties of Dopa116 and the ability for Mfp‐3s to undergo self‐coacervation115 have also been suggested to contribute to the drying of the surface. Using homologues of Mfp‐3s, Wei et al.116 demonstrated that only Dopa‐containing coacervates have the ability to displace interfacial water molecules from the surface due to its high wetting properties which significantly improve wet‐adhesion properties of the peptide by promoting the formation of molecular hydrogen bonds. Upon removing the water molecules from the metal oxide surface, catechol–metal co‐ordination and hydrogen interactions results in the formation of inner‐sphere surface complex followed by the formation of electrostatic interactions and creating outer‐sphere complexes (Fig. 5).116
Figure 5.
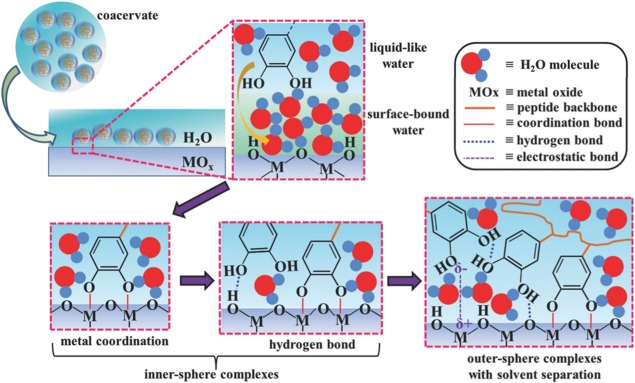
Proposed adhesion mechanism of catechol groups in a coacervate binding to underwater metal oxide surface through the displacement of interfacial water molecules, and the formation of interfacial co‐ordination and hydrogen bonds (inner‐sphere complexes), and electrostatic interactions (outer‐sphere complexes). Reproduced with permission from Ref. 116, Copyright 2016 Wiley.
Bridging Adhesion
The ability for an adhesive to bridge two adherent substrates has also been found to contribute to adhesion. Using surface forces apparatus (SFA), Mfp‐3 (16 mol% Dopa) exhibited adhesion energy of 30 mJ/m2, effectively gluing two mica surfaces together. On the other hand, minimal adhesion was detected for Mfp‐1 despite having similar Dopa content (12 mol %).117 Mfp‐3 exhibited the ability to bridge two mica surfaces, whereas Mfp‐1 was predominantly bound to only one of the two surfaces. Peptide chain length is another factor that significantly affects the bridging adhesion (Fig. 6). Shorter peptides have fewer bridging opportunities as they may attach solely to one surface.44 Doubling the peptide chain length have been reported to double the magnitude of bridging adhesion to two mica surfaces.
Figure 6.
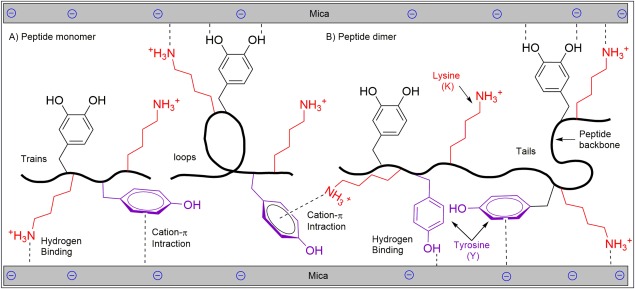
Schematic representation of peptide monomer (A) and dimer (B) adhering to mica substrates. The peptide dimer with a longer chain length is capable of bridging two mica surfaces and demonstrated enhanced adhesion.
Strong bridging adhesion can be obtained even for shorter peptides, if one of the surfaces does not solely rely on Dopa‐mediated adhesion (i.e., hydrogen and co‐ordination bindings).44 Additionally, Mfps demonstrate enhanced adhesion energy when bridging two asymmetric surfaces (e.g., mica and methyl (CH3)‐terminated self‐assembled monolayer) as opposed to between two symmetric surfaces (e.g., two mica surfaces) as the adhesive proteins can partition their domains of chemically effective residues between two dissimilar surfaces for strong interfacial binding to theses surfaces.118 Incorporation of lysine residues promoted adhesion to mineral surfaces, due to its ability to displace surface cations which assisted the catechol groups to form strong hydrogen bonding.119 Similarly, electrostatic interaction is one of the factors that promote bridging adhesion of Mfp‐5 on two mica surfaces due to its long‐range interaction in comparison with Dopa‐mediated surface absorption, which requires the geometry of attachment site for the formation of co‐ordination and hydrogen bonds.44
Catechol‐Modified Side Chain
Similar to marine mussels, Dopa has also been found in adhesive proteins secreted by sandcastle worms to cement sand fragments into tubed‐shaped dwellings.120 In these adhesive proteins, para proton (–H) in the catechol side chain is substituted with a chloro‐functional group which is considered as a natural adaptation to increase interfacial binding strength [Fig. 7(A)].120 Similarly, catechol side chain modified with an electron withdrawing nitro‐groups significantly increased the reactivity and interfacial binding strength of the catechols [Fig. 7(B)].121 Nitro‐catechol bound to an inorganic surface exhibited increased resistance to elevated temperature and oxidation when compared to an unsubstituted catechol.122, 123, 124 These modifications lowered the dissociation constants (pK a) of the catechol hydroxyl groups, which promoted the formation of catechol–metal ion complexes at a reduced pH and with a higher stoichiometry.123, 125 Additionally, nitro‐catechol exhibited increased rate of covalent crosslinking, bound to biological substrates over a wider range of pH, and increased the rate of adhesive degradation when compared to unmodified catechol.121, 126 Furthermore, nitro‐ and chloro‐functionalized catechols were reported to exhibit unique properties such as light‐induced degradation127 and antimicrobial characteristics,128 respectively. Similarly, pyridine modified quinone [Fig. 7(C)] has also formed strong metal ion complexes125 as well as enhanced interfacial binding strength to inorganic substrates.123, 124
Figure 7.
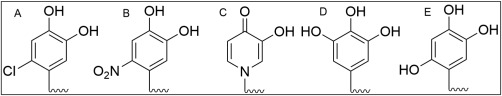
Chemical structure modified catechol side chain with the proton (‐H) at the para position replaced with a chloro‐ (A) and nitro‐ (B) functional group, the benzene ring was substituted with a pyridine group (C), and the proton substituted with a hydroxyl group at the meta (D) and para (E) positions of the benzene ring.
The adhesion of –OH modified catechol groups [Fig. 7(D,E)] has also been reported125 and these trioxyphenyl moieties have demonstrated the capability of forming strong complexes with metal ions129 and boronic acid.130 Tannic acid is a natural polyphenol consisting of trihydroxy phenol moieties. Tannic acid is capable of forming a colorless coating for subsequent surface modifications to create antibacterial and antioxidant surfaces,131 and has been utilized in creating nanoparticles for entrapping and stabilizing anticancer drug.132
STRATEGIES FOR PREPARING CATECHOL‐FUNCTIONALIZED ADHESIVE POLYMERS
Functionalizing synthetic, inert polymers with Dopa and various catecholic derivatives has been utilized to develop functional adhesive polymers with strong wet adhesive properties and the ability to cure rapidly. Dopa‐functionalized polymers have been prepared through five general strategies including direct functionalization of polymers with catechol, polymerization of catechol‐modified monomers, the use of catechol‐functionalized initiator to polymerize synthetic monomers, creating Dopa‐containing peptides through solid‐phase peptide synthesis approach, and recombinant genetic engineering techniques. These strategies are described in this section.
Protection of Catechol Groups
The first step in preparing catechol‐functionalized polymers involves the preservation of the catechol side chains against oxidation and undesirable chemical reactions during the process of synthesis, which may diminish their reactivity. A desirable protecting group should remain stable during the synthetic chemical reaction while the deprotection technique should be chosen based on the composition of the final polymer so that its functionality is not reduced during the process. For multi‐step chemical synthesis approaches, protecting groups such as acetyl,66 acetonide,133, 134 methyl ether,135 cyclic ethyl orthoformate (Ceof),136 carboxybenzyl,137 and t‐butyldimethylsilyl (TBDMS)138 have been utilized. Additionally, catechol forms pH responsive complex with boronic acid, which can act as a temporary protecting group in a basic aqueous solution.138, 139 Similarly, triethylsilane has also been used for protection of hydroxyl (—OH) and methyl ether side chains of a natural phenolic compound, eugenol,140 which has antimicrobial, antifungal, antioxidant, and anti‐inflammatory properties141, 142, 143 and has widely been studied for dental applications.144, 145
Direct Functionalization of Polymers with Catechol
Catechol such as Dopa and dopamine can be directly coupled to polymers with functional groups such as —NH2, —COOH, and —OH, through the formation of amide,77 urethane,146 and ester147 linkages (Fig. 8). This strategy can be adopted to functionalized polymers, such as poly(ethylene glycol) (PEG), with different polymer architectures (i.e., linear and branched), where catechol is attached as terminal functional groups. PEG is a biocompatible, hydrophilic, and inert polymer which has been used for various biomedical applications such as antifouling coatings,71, 146 bioadhesives, and sealants.148, 149
Figure 8.
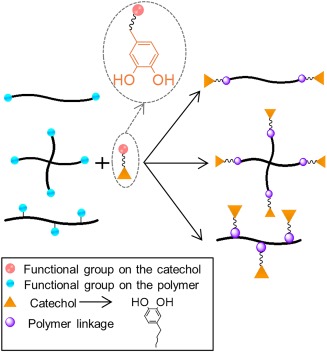
Strategies for preparing catechol‐modified polymers of different architecture with functional groups such as —NH2, —OH, and —COOH, through formation of an amide, ester, and urethane linkages.
Additionally, branched and linear PEG can be linked with polycaprolactone (PCL)150, 151 and polypropylene oxide (PPO)146, 152 polymers to form block copolymers which can be further grafted with catechol along its polymer chains. These block copolymers have demonstrated improved material properties through the self‐assembly of the hydrophobic blocks,150, 151 as well as thermal responsivity146 and desirable swelling properties.152 Similarly, block copolymers composed of PEG and polymers such as poly(methyl mehacrylate)153 and poly(methacrylate)107 as well as polyurethane of PEG and oligo alkyl chains154 have also been reported. Furthermore, acid‐ or amine‐functionalized catechol can be covalently attached to biopolymers such as dextran,155 chitosan,156 hyaluronic acid,157 gelatin,158 and alginate159 to form bioadhesives which are well suited for tissue engineering and drug‐delivery applications.
Eugenol also can be directly coupled with wide variety of polymers through its active terminal alkene group using thiol‐en chemistry.140 The protected eugenol can be end‐functionalized with thiol to create a catechol protected monomer, which can be used to link to a polymer with a desired architecture.160, 161 For example, random copolymers of poly(styrene‐co‐(4‐ethynyl styrene)), which contain an active alkyne group have been functionalized with eugenol through thiol‐yne reaction to create catechol‐functionalzed polystyrene.162
Polymerization of Catechol‐Modified Monomer
Catechol‐modified monomers, such as dopamine methacrylamide (DMA) [Fig. 9(A)],138, 139, 158 can be polymerized through heat‐activated or photo‐initiated free‐radical polymerization to form acrylate‐based polymers [Fig. 9(a)]. Various monomers such as oligomeric ethylene glycol,163 monoacryloxyethyl phosphate,164 and methoxyethyl acrylate139 have been copolymerized with DMA to form dopamine grafted polymers with a broad range of physical characteristics, molecular weights, and catechol contents. In a similar manner, polystyrene‐based copolymers can also be created through copolymerization of catechol‐containing vinyl group (e.g., 3,4‐dihyroxystyrene [Fig. 9(B)], 4‐vinylcatechol acetonide, and 3‐vinylcatechol acetonide) and styrene.135, 165, 166, 167, 168 A three dimensional polymer network can be formed from the copolymerization with a bi‐functional crosslinker (e.g., N,N′‐methylenebisacrylamide) to form a network with catechol covalently tethered to its backbone [Fig. 9(c)].93, 169
Figure 9.
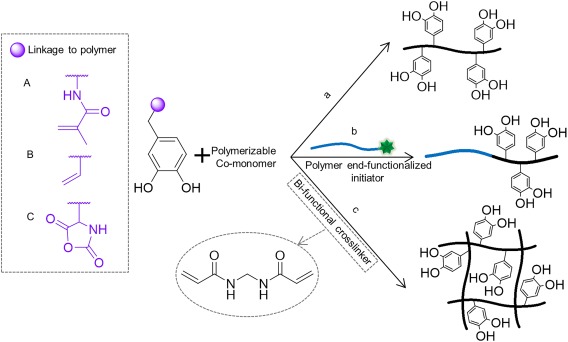
Catechol modified with polymerizable methacrylate (A), vinyl (B), and N‐carboxyanhydride (C) functional groups. Polymerization of catechol‐modified monomers to form linear homopolymer or random copolymer (a), block copolymer in the presence of polymer end‐functionalized initiator (b), and a three dimensional polymer network in the presence of bi‐functional crosslinker (c).
In the presence of molecular oxygen, the catechol side chain partially inhibits and retards free‐radical polymerization.138 As such to minimize the inhibitive effect of oxygen the use of catechol protection groups135 or the elimination of molecular oxygen139 are required to form polymers with high catechol content and molecular weight (MW). Similarly, separating catechol side chain from the polymerizable acrylate group using the ability of block copolymers to self‐assemble into hydrophilic and hydrophobic domains have also been reported to be an effective approach.66
Water soluble copolypeptides have been created through ring‐opening polymerization of N‐carboxyanhydride [NCA, Fig. 9(C)] of lysine and Dopa, which have been reported to have high bonding strength and high MW's as well as narrow MW distributions.137, 170 Additionally, in the presence of polymer end‐functionalized with an initiator (i.e., —NH2), block copolymers with poly(DOPA‐lysine) and oligomeric poly(DOPA) copolypeptide have been created [Fig. 9(b)].66
Most recently, catechol‐modified with an epoxide monomer (i.e., acetonide glycidyl ether) have been copolymerized with ethylene glycol to create linear PEG‐based block copolymers.171 Similarly, copolymerization with glycidol resulted in the formation of a hyperbranched copolymer. These copolymers have the ability to form hydrogel networks in the presence of Fe+3 metal ions under basic condition and demonstrated the ability for surface modification.
Catechol‐Functionalized Initiator for Polymer Synthesis
Catechol‐modified initiators have been utilized to create polymer end‐functionalized with the adhesive moiety (Fig. 10). Dopamine functionalized with reversible addition‐fragmentation chain transfer (RAFT) agent [Fig. 10(A)] has been used as an initiator to prepare polymers such as polystyrene, poly(N‐isopropylacrylamide), and poly(tert‐butyl acrylate) end‐modified with a catechol moiety.172, 173 Similarly, catechol functionalized with alkyl bromine [Fig. 10(B)] has been used as an initiator to synthesize poly(mathacrylate)‐ and poly(acrylate)‐based polymers through atom transfer radical polymerization (ATRP).174, 175, 176, 177 Ring‐opening metathesis polymerization (ROMP) has also been used to prepare catechol‐modified poly(pentadecafluorooctyl‐5‐norbornene‐2‐carboxylate) from dopamine functionalized with ROMP agent [Fig. 10(C)].178 Beside dopamine‐functionalized initiator, the use of poly(DOPA‐lysine) oligopeptide functionalized‐initiator has also been reported.179
Figure 10.
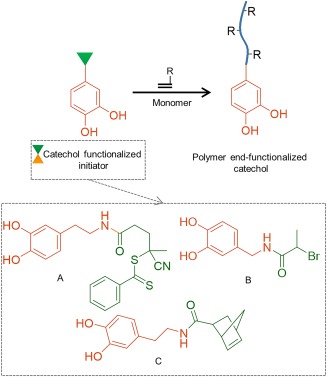
Schematic representation of catechol‐functionalized initiator to prepare polymer end‐functionalized with catechol. Chemical structures of catechol‐functionalized initiator using RAFT (A) ATRP (B), and ROMP (C) polymerization. R represents the side chain of monomers used during polymerization.
Solid‐Phase Peptide Synthesis
Dopa‐containing peptides have been prepared through solid‐phase peptide synthesis method using Dopa residues where its primary amine and the catechol side chain are protected.136, 180, 181 tert‐butyloxycarbonyl (Boc) and 9‐fluorenylmethyloxycarbonyl (Fmoc) are two of the most common protecting groups for the α‐amino group.136, 180 However, Fmoc is preferable due to a cleaner synthesis and peptide products as well as the ability to avoid the use of strong acid.182 The catechol side chain is typically protected using acid‐liable groups such as TBDMS,183 tert‐butyldiphenylsilyl,180 Ceof,136 and acetonide,133 which have been demonstrated to be compatible with solid‐phase peptide synthesis protocols.
Peptoids have a peptide‐like backbone with side chain substitution on the amide nitrogen instead of the α‐carbon, which increases their resistance against protease degradation.184 Solid‐phase peptide synthesis strategy has been used to synthesize peptoids end‐modified with Dopa‐containing peptide, which demonstrated long‐term antifouling properties when coated on a titanium surface.185
Recombinant Genetic Engineering
Dopa‐containing peptides have been prepared through recombinant DNA technology to replicate peptide sequences found in various Mfps.186, 187, 188 Peptides are produced with tyrosine residues in bacteria (e.g., Escherichia coli), which are further converted to Dopa using tyrosinase to generate functional adhesive peptides.188 Although, this technology cannot produce full‐length native foot proteins due to their length and highly repetitive sequences, recombinant peptides have been investigated as wet bioadhesives,189, 190 coatings,190 and self‐healing hydrogels.191
One of the unique features in using the recombinant technique is the ability to combine sequences from different Mfps to generate hybrid peptides. For example, sequences from both Mfp‐5 and Mfp‐1 have been used to create ABA block copolypeptide, where “A” and “B” blocks each contain separate sequences from these two proteins.189 Similarly, hybrid copolypeptides have been created to contain sequences from both Mfp‐1 and ‐3.192 Sequences from Mfp‐1 and ‐3 have also been combined with protein sequences from E. coli fiber‐forming amyloid to form a chimeric peptide that is capable of self‐assembling into higher‐ordered, adhesive nanofibers.193 Peptides containing Mfp sequences have also been created with a RGD peptide sequence found in fibronectin, to promote cell attachment.194 However, limitations of using recombinant DNA techniques to generate bioadhesives include low yields, requiring post‐transitional modification to generate catechol moiety, low efficiency in the modification of tyrosine to Dopa, and lacking other post‐translationally modified amino acids (e.g., hydroxyproline).186, 189, 195
RECENT POLYMER SYSTEMS INCORPORATING NOVEL BIOMIMETIC DESIGNS
Coacervated Dopa‐Functionalized Polymer Adhesives
Inspired by Mfp‐3s's ability to form self‐coacervate in promoting adhesion and increasing Dopa's oxidation potential,4 a series of ampholytic copolymers composed of varying amounts of amphiphilic and ionic functional groups found in Mfp‐3s (i.e., anionic, cationic, nonionic hydrophilic, and nonionic hydrophobic co‐monomers) have been synthesized to study the effect of these functional groups on microphase behavior and wet adhesion properties.110 These Mfp‐3s‐mimetic polymers demonstrated the ability to self‐coacervate and uniformly spread over a surface. Additionally, binding energy was reported to be doubled (30 mJ/m2) by increasing pH to 7 after delivering the adhesive solution at pH 4 without the use of oxidative crosslinking. These adhesive values are nearly an order of magnitude higher than those found for natural Mfp‐3s. To combine the self‐coacervation capability of Mfp‐3s and the strong adhesive properties of Mfp‐5, a low MW catecholic zwitterionic surfactant has also been synthesized, which demonstrated strong wet‐adhesion and the ability to self‐coacervate.111
Complex coacervation formed between polymers and peptides has also been explored for developing new coacervates for various biomedical applications.113, 190, 196 Coacervates have been formed between cationic recombinant peptides and anionic hyaluronic acid (HA), which demonstrated favorable adhesive spreading properties over the targeted surface and wet‐adhesion properties.190, 197 Recently, pH responsive adhesive coacervates have been developed by combining Zn2+ ion with Dopa‐modified poly(acrylic acid) (PAA‐Dopa).196 The PAA‐Dopa solution was injected at a low pH into the zinc solution which formed dense coacervates as a result of electrostatic interaction between zinc chelated mono‐catechol groups and negative charged carboxyl groups of PAA. The coacervate then gelled when the solution pH was increased to 9 due to oxidation of catechol groups which enables the formation of bis‐catecholate‐Zn+2 complexes. This coacervate system exhibited excellent adhesive and self‐healing properties after gelation, and has the potential for localized drug‐delivery application.
Nano‐ and Micro‐composite Adhesive Hydrogel
Hydrogels are highly hydrated three dimensional polymer networks and can be used in various biomedical applications, including drug‐delivery vehicles,198 actuators,199 and tissue adhesives.151 However, hydrogels are fragile, which limits their uses in most load‐bearing applications. The incorporation of inorganic nanoparticles into an organic polymer network has been shown to increase the material properties of the hydrogel network.200, 201, 202 Laponite, a nano‐silicate, was incorporated into a chemically crosslinked polyacrylamide (PAAm) nano‐composite hydrogels containing DMA.169 This nano‐composite hydrogel can be repeatedly compressed to a strain of 0.8 while demonstrating compressive stress of over 1 MPa.
Laponite was incorporated into a branched PEG‐terminated with dopamine to create an injectable bioadhesive with improved mechanical and adhesive properties (Fig. 11).203, 204 Additionally, Laponite provided binding sites for cellular attachment and promoted tissue ingrowth when the adhesive was implanted subcutaneously.203 Additionally, other types of nanoparticles (i.e., iron‐ and silver‐based) have been used to create mechanically enhanced hydrogels, which provided additional functionality into the nano‐composite system (e.g., ability to respond to applied magnetic field and antimicrobial properties, respectively).205, 206
Figure 11.
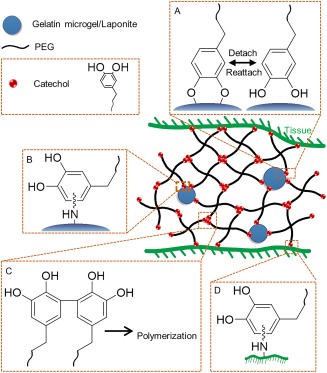
Schematic illustration of composite adhesive consisted of PEG‐catechol containing either a nano‐silicate, Laponite, or gelatin microgels. Catechol forms reversible non‐covalent crosslinking with Laponite (A) and irreversible covalent crosslinking with functional groups (e.g.,–NH2, –SH) found on the gelatin microgels, (B) contributing to the increased bulk mechanical properties of the adhesive. Catechol also polymerizes (C) and reacts with nucleophilic groups found on tissue surface (D), resulting in rapid curing of the adhesive and interfacial binding, respectively.
The mechanical properties of catechol‐containing nano‐composite hydrogels are highly dependent on the strength of interaction between the catechol moiety and the encapsulated nanoparticles.122 The oxidation state of catechol greatly influences its interfacial binding properties to inorganic surfaces.66, 74, 104 As such, catechol's adhesive properties decrease with increasing pH as it is progressively autoxidized to form the more poorly adhesive quinone.74 Shear moduli of dopamine‐modified nano‐composite hydrogel was demonstrated to be lower at pH 9 when compared to values obtained at a neutral to acidic pH.122 Similarly, when dopamine was replaced with nitrodopamine that binds more strongly with inorganic substrates, shear moduli of the nano‐composite hydrogel increased.122 Nitrodopamine was also less prone to autoxidation and exhibited stronger binding to the nanoparticles even at a basic pH.
In addition to inorganic nanoparticles, incorporation of gelatin‐based microparticles into dopamine‐functionalized PEG‐based adhesive has also demonstrated improved mechanical properties (Fig. 11).207 Gelatin is the denatured form of collagen often used for biomedical applications.208 Gelatin contains nucleophilic side chains (i.e., —NH2 and —SH from lysine and cysteine, respectively) that enabled gelatin microparticles to be chemically incorporated into the PEG network through covalent bond formation with oxidized catechol (i.e., quinone) found on dopamine [Fig. 11(B)].207 Additionally, gelatin microparticles retained the triple helix structure of collagen.209, 210 This provided reversible, physical interaction in the PEG network and improved the viscous dissipation properties to the micro‐composite hydrogel. Most importantly, gelatin provided binding sites for cellular attachment and a pocket for cellular infiltration in vivo, which are critical for improving healing responses.207
Hydrogel Actuator
Hydrogel actuators change their shapes and physical properties in response to environmental stimuli (i.e., temperature, pH, etc.).211 The ability for catechol to transition reversibly between metal ion complexes [Fig. 2(D)] of different stoichiometry (i.e., mono‐ vs. tris‐catecholate complexes) in response to pH was exploited to create a pH‐responsive actuator.62, 212 DMA‐containing hydrogel was locally ionoprinted with ferric ions (Fe+3), which increased local crosslinking density at the ionoprinting site and resulted in the sharp bending of the hydrogel.62 Hydrogels can transform into different three dimensional geometries depending on the ionoprinting pattern. Additionally, the metal ions are reversibly bound to the catechol and can be removed using ethylenediaminetetraacetic acid, so that metal ions can be reintroduced into a new pattern.62 The rate and extent to which hydrogel can actuate is considerably tunable by different factors including catechol and metal ion content, pH, hydrogel thickness, and metal ion type (Fe+3, Al+3, Cu+2, Zn+2, and Ti+4).60, 61 Specifically, using different metal ions enabled different sections of the hydrogels to actuate at different rates, achieving sequential folding.
Self‐Healing Hydrogels
The ability for catechol to form various strong reversible bonds has been utilized to design self‐healing hydrogels (Fig. 12). Metal‐catechol co‐ordination bond has been employed to design pH‐responsive self‐healing hydrogels by mixing catechol‐containing polymers and peptides with various metal ions such as Fe3+, Zn2+, V+3, Al3+, Ga3+, and In3+ under basic condition [Fig. 12(A)].56, 57, 213, 214 These self‐healing networks exhibited elastic moduli values approaching those of covalently crosslinked hydrogels at high strain rate.56 Catechol–boronate complex has also been utilized to create similar pH‐responsive self‐healing hydrogels that exhibited high stability in an alkaline pH [Fig. 12(B)].215
Figure 12.
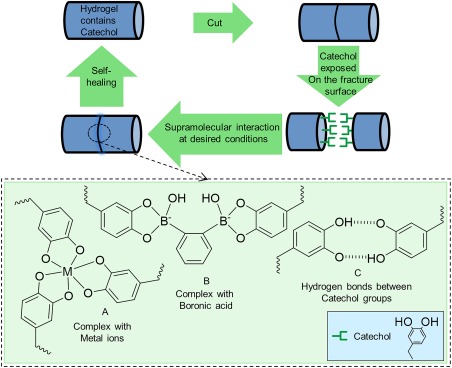
Schematic illustration of self‐healing hydrogels containing catechol through the formation of catechol–metal ion complexation (A), catechol–boronate complexation (B), and H‐bonds formed between catechol groups (C).
A mussel inspired polymer with the ability to heal under acidic conditions has also been reported.216 Polyacrylate and polymethacrylate surface‐functionalized with triethylsilyl‐protecting catechols demonstrated self‐healing properties under slight compression and acidic condition (pH 3). The catechol protecting groups were removed under acidic conditions resulted in the formation of hydrogen bonds between interfacial catechol groups and self‐healing of the polymer [Fig. 12(C)]. Recently, an injectable, thermosensitive catechol functionalized ABA tri‐block copolymer composed of poly(N‐isopropylacrylamide) as thermosensitive “A” block and PEG as water‐soluble “B” block has been prepared.217 The hydrogel self‐healed from deformation through the formation of hydrogen bonds which facilitated the self‐healing process. Compare with self‐healing hydrogels based on catechol‐metal ions co‐ordination which may be toxic when used in vivo, this metal‐free hydrogel is capable of tolerating high strain and also fast recovery of its mechanical properties after repeated deformation; thus, exhibited great potentials for various biomedical applications such as drug delivery.
Smart Adhesives
Smart adhesives can transition reversibly between its adhesive and non‐adhesive states in response to externally applied stimuli. These tunable adhesive properties have been met with significant interests in various fields of materials science and engineering, including manufacturing, robotic locomotion, and wound dressings.218, 219, 220, 221 However, existing smart adhesives are limited by the need for extreme conditions to promote transition (e.g., elevated temperature219), adhesion to a limited substrate types,222 or reduced interfacial binding strength to wetted surfaces.220 Smart adhesives that combine the moisture‐resistant adhesive properties of Dopa‐based chemistry have recently been reported. Catechol has been coupled to light‐degradable polymer consisting of di‐o‐nitrobenzaldehyde and the adhesive degraded in response to ultraviolet light irradiation.223 Similarly, nitrodopamine‐modified PEG is photosensitive and the cured adhesive degrades when exposed to light.127 Tyrosine‐functionalized adhesives can be activated to their adhesive catechol form with the addition of tyrosinase, which transforms the non‐adhesive tyrosine residues into the adhesive Dopa residues for adhesion.224, 225 Although these catechol‐containing smart adhesives demonstrated moisture‐resistant adhesive properties, they are not reversible (i.e., one‐time activation or one‐time deactivation).
Most recently, our lab exploited the reversible catechol–boronate complexation chemistry [Fig. 12(B)] to create a reversible smart adhesive that transitioned reversibly between its adhesive and non‐adhesive states.226 This adhesive contained network‐bound dopamine and phenyl boronic acid that formed strong interfacial bonds with wetted glass surface at pH 3 (Fig. 13). When the pH was increased to 9, formation of the catechol–boronate complex reduced the measured work of adhesion by more than an order of magnitude. The boronic acid also served as a protecting group to prevent the irreversible oxidation and crosslinking of catechol, so that the adhesive can reversibly transition between its adhesive and non‐adhesive states.
Figure 13.
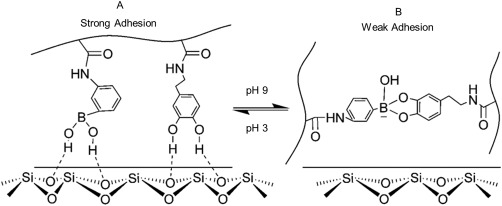
Schematic representation of the smart adhesive containing catechol and phenylboronic acid functional groups. At an acidic pH, both the catechol and borate functional groups contributed to strong interfacial binding with the wetted borosilicate substrate (A). In a basic pH, formation of catechol–boronate complexation reduced the interfacial binding strength of the adhesive (B). Changing the pH, effectively converts the smart adhesive between its adhesive and non‐adhesive states. Reprinted with permission from Ref. 226, Copyright 2016 American chemical society.
BIOMEDICAL APPLICATIONS OF BIOMIMETIC POLYMER ADHESIVES
The unique and robust interfacial chemistry of catechol provide scientists a tool to design various bioadhesive materials for a wide range of biomedical applications. Table 2 summarizes various strategies for preparing catechol‐functionalized polymers for various biomedical applications reviewed here.
Table 2.
A Brief Summary of the Preparation Strategies, Composition, and Intended Applications of Catechol‐Functionalized Adhesives Polymers
| Preparation Strategies | Composition | Intended Applications | References |
|---|---|---|---|
| Direct functionalization | Catechol‐functionalized: | ||
| • Linear and branched PEG | Biomedical adhesive and sealant | 146, 150, 151, 152 | |
| • Block copolymer of PEG and PCL or PPO | |||
| • PEG based triblock copolymer | Antifouling/cell‐resistant coating | 160 | |
| Self‐healing hydrogel | 217 | ||
| • Light‐degradable polymer | Smart adhesive | 223 | |
| Dopa‐functionalized: | |||
| • PEG‐based polymer | Self‐healing hydrogela | 56, 57, 213 | |
| • PEG hydrogel prepared with Laponite nanoparticles | Nano‐composite hydrogel | 203 | |
| Polymerization of Catecholic monomers | Copolymerization of: | Biomedical adhesives/wound healing | 93, 163, 169 |
| • DMA With various monomers | Antifouling/bacteria‐resistant | 138 | |
| Smart adhesive | 226 | ||
| Actuatorb | 60, 61, 62 | ||
| • Eugenol with other monomers | Coacervates | 110 | |
| Polymerization of: | |||
| • eugenol acrylates and eugenol methacrylates | Self‐healing (in slightly acidic condition) | 216 | |
| Ring‐opening polymerization of: | |||
| • NCA of lysine and Dopa with PEG | Antifouling coating | 227 | |
| Catechol‐functionalized initiator | Antifouling coating grown from dopamine‐functionalized: | ||
| • ATRP agent | Antifouling/cell‐resistant, bacteria‐resistant coating | 174, 175, 176, 177 | |
| • ROMP agent | Water and oil resistant coating | 178 | |
| Solid phase peptide synthesis | • Dopa‐containing peptide and antifouling polymer | Antifouling/cell‐resistant coating | 71 |
| • Peptoid modified with Dopa‐containing peptide | Antifouling/cell‐resistant coating | 185 | |
| • Short peptide analogue Mfp‐3s | Coacervate adhesive | 116 | |
| Recombinant genetic engineering | • RMfp‐3,RMfp‐5, | 187, 188, 189 | |
| • hybrid copolypeptide of Mfp‐1 and Mfp‐5 | Biomedical adhesive | ||
| • RMfp‐1 hydrogel | Self‐healing hydrogela | 191 | |
| • Peptides containing Mfp sequences with a RGD peptide sequence found in fibronectin | Coacervate adhesivec | 190, 194 | |
| Biomedical adhesive |
In the presence of metal ions.
Ionoprinting with metal ions.
In the presence of HA.
Biomedical Adhesives
Tissue adhesives can simplify surgical procedures and minimize trauma typically associated with the use of suture and staples.228, 229 However, commercially available adhesives are limited by slow degradation rate, toxicity concerns, and poor adhesive strength.230, 231, 232 PEG‐based adhesives are one of the popular synthetic adhesives that have been used for various surgical applications such as dural,233 pulmonary,234 and cardiovascular235 surgeries. Dopa‐ and catechol‐modified PEG‐based adhesives are one of the earliest synthetic bioadhesives that have been developed, which exhibited potential in various applications including sealing of fetal membrane,236 sutureless wound closure,147 and cell engineering.147 These adhesives demonstrated superior adhesive properties when compared to commercially available fibrin‐ and PEG‐based glue. However, due to the hydrophilicity of PEG, PEG‐based adhesives swell excessively, which reduces mechanical properties and may lead to complications (e.g., local nerve compression).152, 237
Incorporating thermosensitive tetronic polymers as the backbone of polymeric bioadhesives is one of the promising approaches to control the swelling behavior of the adhesive hydrogel.152, 238 Tetronic is a branched, multi‐block copolymer consisting of a central hydrophobic PPO block and peripheral hydrophilic PEG blocks and demonstrated deswelling behavior when heated. Tetronic end‐functionalized with catechol exhibited enhanced mechanical toughness and systematically controllable deswelling of the adhesive (0–25%), due to the self‐assembling capability of the PPO block.152 Enhanced bulk mechanical properties of tetronic‐based adhesive also resulted in increased lap shear adhesive strength to decellularized porcine dermis when compared to PEG‐based adhesives.
A PEG‐citrate‐based polymer functionalized with dopamine has been prepared by polycondensation reaction and demonstrated the potential for sutureless wound closure.147 The adhesive demonstrated adhesive strength that was 2.5–8.0 folds higher than that of fibrin glue, while exhibiting a tissue‐like elastomeric behavior and improved load bearing and stress transferring properties. The adhesive also exhibited controllable degradation rate (1–25 days) and excellent biocompatibility. However, the use of PEG as the diol significantly elevated equilibrium water content of the adhesive. To modulate the swelling behavior, the adhesive was formulated with hydrophobic 1,8‐octanediol, which resulted in stronger adhesive with improved swelling properties.239
Naturally occurring biopolymers such as silk fibroin, HA, and chitosan functionalized with catechol have also been investigated.240, 241, 242, 243, 244 Silk fibroin is a hydrophobic biopolymer with repeated amino acid sequence of glycine, alanine, and serine residues,245 and has recently attracted attentions as a biomaterial platform.246, 247 A silk fibroin polymer purified from silkworm fibers modified with catechol and PEG side chains has demonstrated improved adhesive strength in comparison with catechol‐free silk as well as a lower degree of swelling compared to PEG‐based adhesive.240 This silk‐based adhesive also supported the attachment and proliferation of human mesenchymal cells in culture.
HA is a natural non‐sulfated glucosaminoglycan which has widely been used for various biomedical applications, including tissue regeneration and wound healing applications.248, 249 HA grafted with dopamine demonstrated excellent biocompatibility with enhanced wet adhesive properties.244 The HA‐based adhesive successfully encapsulated two types of cells (i.e., human adipose‐derived stem cells and hepatocytes) and demonstrated the potential for minimally invasive cell transplantation. Encapsulated cell demonstrated increased viability and functionality when compared to those encapsulated in conventional hydrogels crosslinked through photopolymerization. Similar to PEG‐based biomaterials, HA swells excessively in an aqueous environment which results in poor mechanical properties. To improve the mechanical properties of HA‐based hydrogels, dopamine‐modified HA were formulated with Pluronic, which is an ABA triblock polymer consisting of hydrophilic PEG “A” blocks flanking a hydrophobic PPO “B” block.250 This composite adhesive demonstrated rapid and reversible sol–gel transition and high stability both in vitro and in vivo.
Chitosan is a natural polysaccharide which has been used in various biomedical applications such as wound closure251 and hemostatic252 applications. Hydrogels consisting of chitosan and catechol‐containing moieties (i.e., Dopa, hydrocaffeic acid (HCA), and dopamine) were broadly investigated as a mucoadhesive biomaterial.241, 242, 243 Chitosan‐based bioadhesives demonstrated strong adhesive properties to mucosal tissues with minimal cytotoxicity. Chemically crosslinking the catechol–chitosan‐based adhesives with genipin demonstrated further enhancement in mucoadhesive properties and stability.253
Therapeutic Applications
The unique ability for catechol to anchor to wide ranges of surfaces in different length scale has been utilized to create biomaterials for various anticancer and antimicrobial applications. Gold nanoparticles exhibits unique optical properties and can convert light energy into heat.254 Using polydopamine as a surface anchoring group, gold nanorods were functionalized with epidermal growth factors receptor antibodies (anti‐EGFR) to target and selectively bind to cells overexpressing EGFR, such as the cancer cells in the breast and colon.83 Subsequent light irradiation resulted in considerable heat production locally, resulting in photo‐induced cancer cell death. Similarly, polydopamine‐coated gold nanorods were coated with silver nanoparticles and antibacterial antibodies to specifically bind to both Staphylococcus epidermidis and E. coli.255 Illumination of bacteria‐bound gold nanorods resulted in plasmonic heating and triggered release of silver ions for dual antimicrobial effect.
Polydopamine coated on a sacrificial nanoparticle template has been used to create a capsule with controllable size and wall thickness after removing the template.256, 257 The capsules can be subsequently loaded with anticancer drugs for their sustained release. However, this capsule lack the ability for targeted and triggered release of the loaded drugs for enhanced therapeutic efficacy. To create a pH‐responsive capsule, doxorubicin (Dox), an anticancer drug, was immobilized onto polydopamine capsule using an acid‐labile hydrazine bond.258 Degradation of the hydrazine bond in the acidic environment (pH 5–6) of the endosomal and lysosomal compartments within a cancer cell triggers the rapid release of Dox.
Similar pH‐responsive capsule was created using a tannic acid–Fe3+ ion complexation.129 This so called metal‐phenolic network (MPN) capsules are stable at a neutral pH due to the formation of complexes with higher stoichiometry (3:1 tannic acid: Fe3+ ion). At an acidic pH, formation of a complex with a reduced stoichiometry (1:1 tannic acid: Fe3+ ion) resulted in the rapid disassembly of the capsules and release of the encapsulated drugs. MPN with the ability to target cancer cells was also created using the combination of catechol‐modified HA and PEG (Fig. 14).259, 260 Incorporation of PEG minimized nonspecific adsorption of proteins and cells, while HA enhanced the binding and targeting abilities of the capsules to cancer cells that overexpressed CD44 receptor. The HA to PEG contents can be optimized to create capsules with high targeting capability to CD44 positive cancer cells, while minimize binding to CD44 negative cells [Fig. 14(B)].260
Figure 14.
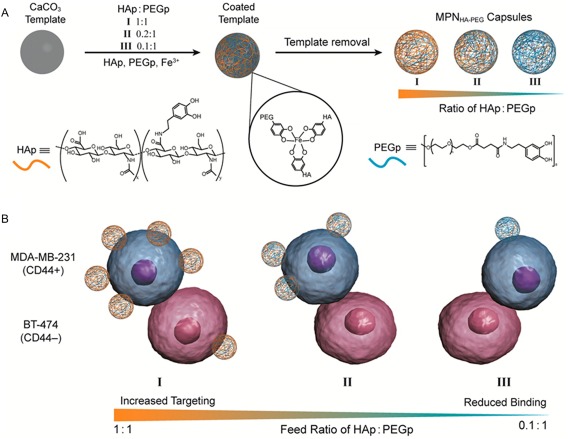
Schematic representation of metal‐phenolic network capsules (MPNHA‐PEG) assembly consists of hyaluronic acid (HA) and poly(ethylene glycol) (PEG) with three different HA/PEG ratios (A). HA enhances targeting and binding to CD44+ (cancer cell line, blue) while PEG minimizes nonspecific binding to CD44− (pink) cells (B). Reprinted with permission from Ref. 260, Copyright 2016 American chemical society.
Paclitaxel (PTX) is a common anticancer drug which has broadly been used for treating various cancers such as lung cancer, but its application is limited by its poor solubility in an aqueous solution.261 Strong complexes formed between tannic acid and Fe+3 ions have also been used to stabilize the interface of nanoparticles composed of PTX.132 The stabilized nano‐drug particles are uniform in size (100–120 nm) with high drug loading capability and long‐term colloidal stability (longer than half a year).132
Antifouling Coating
Controlling interfacial adsorption of biomolecules, proteins, and cells greatly affects the success of implanted biomaterials.262 Immobilizing antifouling polymers such as PEG on device surfaces is a common strategy to reduce nonspecific adsorption of cells and proteins.73, 185 Catechol's ability to attach to both organic and inorganic surfaces through a simple dip‐coating approach provides a robust and versatile surface anchoring technique for tethering antifouling polymers onto various types of surface.
Two polymer grafting strategies have been reported, which include “graft to” and “graft from” approaches (Fig. 15). The “graft to” approach consists of direct coating of prefabricated antifouling polymer end‐modified with a catechol moiety or a short peptide containing Dopa. PEG end‐functionalized with Dopa‐containing decapeptide sequence from Mfp‐1 [Fig. 15(A)],71 oligopeptide containing 1‐3 Dopa residues [Fig. 15(B)],73, 163, 263 and a copolypeptide containing Dopa and lysine residues227 have rendered surfaces resistant to proteins, cells, and bacteria attachment. Zwitterionic polymers have also demonstrated excellent antifouling capability due to their ionic hydration.264 Catechol‐functionalized zwitterionic polymers such as poly(carboxybetaine),176 poly(carboxybetaine methacrylate) [Fig. 15(D)],265 and poly(sulfobetaine methacrylate) (pSBMA)266 have been reported in the literature.
Figure 15.
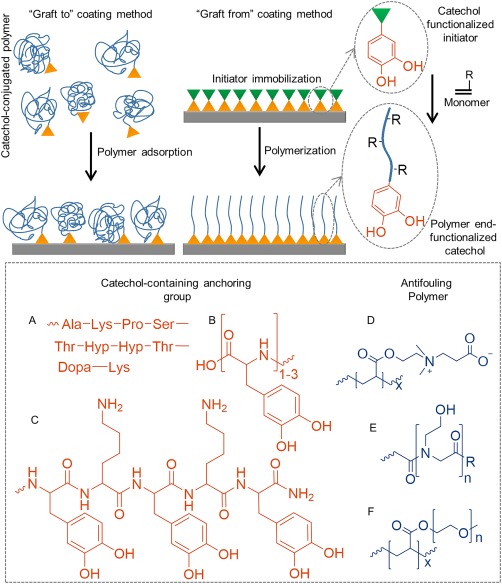
Schematic representation of “graft to” and “graft from” approach used to prepare antifouling surfaces. Chemical structures of catechol‐containing anchoring group for immobilizing coatings using the “graft to” coating method (A–C), and three examples of antifouling polymers (D–F).
Peptoid‐based antifouling coating has been created with poly(N‐methoxyethylglycine) [Fig. 15(E)], which demonstrated long‐term biofouling‐resistant properties.185, 267 Similarly, peptoids containing zwitterionic side chain268 and antimicrobial sequences269 have also been used to prepare surfaces with excellent antifouling and active bactericidal properties, respectively.
In addition to polymer brushes, ABA triblock copolymers consisted of catechol‐containing “A” blocks and antifouling “B” block (e.g., PEG, poly(N,N′‐dimethylacrylamide)) formed polymeric loop when coated onto a surface.160, 270 These coatings exhibited enhanced antifouling properties against protein and cell absorption when compared to polymer brushes with similar grafting density, due to enhanced steric hindrance associated with the neutrally charged polymer loops. Additionally, the coating also exhibited low friction coefficient, which makes it an ideal candidate as a coating for ocular lenses.160
The development of various catechol‐functionalized initiators (Fig. 10) enabled scientists to prepare coatings through surface‐initiated polymerization (SIP) process or the so‐call “graft‐from” approach (Fig. 15). SIP involves anchoring the catechol–initiator conjugates on to the surface followed by initiating monomer polymerization to create a more homogenous and denser polymer brushes when compared to the “graft to” approach.172 Surface‐initiated ATRP of various antifouling polymer brushes including oligo(ethylene glycol) methyl ether methacrylate [Fig. 15(F)],174 zwitterionic pSBMA,177, 179 and thermo‐responsive N‐isopropylacrylamide271 have been used to graft polymers onto various substrates ranging from metal to inert polymers. These coating were highly stable antifouling coatings,174, 177, 179 with excellent protein‐,272 bacterial‐,177, 179 and cell‐resistant175, 179 properties. Additionally, SIP has been used to chemically tether polymers onto colloidal and planar substrates as well as grafting polymers onto nanoparticles.271 When combined with photolithography, polymers have been grafted in various micro‐scale patterns such as lines and squares.175, 271
SUMMARY AND FUTURE OUTLOOK
The incredible moisture‐resistant adhesive property of Mfps has inspired scientists to engineer a wide array of advanced functional materials. The ability for Dopa to form strong reversible and irreversible interactions has been used to create a unique and versatile platform for developing adhesive polymers with enhanced material properties (i.e., composite hydrogels with enhanced mechanical properties, smart materials and adhesives, and self‐healing materials). Additionally, catechol‐functionalized polymers have the potential in functioning as biomedical adhesives, drug carrier for cancer therapy, and antifouling coatings. Although we have focused on the application of this unique biomimetic technology for biomedical applications, catechol‐based chemistries have also been explored in other fields (e.g., drinking water purification,272 controlled release of fertilizer,273 nano‐composite for tire rubbers,274 as well as adhesive for battery275 and plastics135).
Over the last decade or so, scientists have predominantly focused on incorporating the adhesive catechol moiety into the design of polymers for interfacial applications. However, mussel plaque proteins rely on other amino acid residues (i.e., charged, hydrophobic, and antioxidant thiol residues, etc.) and intermolecular chemical interactions between multiple Mfps to create adhesive plaques that bind tightly to the substrate surface. Additionally, byssus threads utilize strong, reversible His‐metal ion (i.e., zinc, copper) interactions to minimize permanent structural damage.55 These non‐catechol chemistries are largely neglected in the existing synthetic mimics of these adhesive proteins. The utilization of these chemistries has only been reported in the last couple of years (i.e., self‐coacervation,4 complex coacervation,103 His‐based self‐healing hydrogel276). The incorporation of these designs may yield future adhesive polymers with new and improved properties.
When designing mussel‐mimetic adhesives, there is a need to modify the design criteria based on the desired applications. For example, catechol utilizes different interfacial chemistries to adhere to inorganic and organic substrates. The reduced catechol is responsible for strong interfacial binding to inorganic surfaces. As such, incorporating features that minimize catechol oxidation (i.e., cysteine in Mfp‐6,108 hydrophobic residues in Mfp‐3s4) can potentially enhance adhesion to inorganic surfaces in an oxygen‐rich and mildly basic aqueous environment. On the other hand, Dopa needs to be oxidized to its quinone form in order to participate in intermolecular covalent crosslinking, which is critical for designing in situ curable materials and adhesion to biological substrates.68, 78, 79 Additionally, reactive oxygen species (i.e., H2O2) are released as a byproduct during the oxidation of catechol.93, 95 Given the biological responses to H2O2 is highly concentration and biological system dependent, the release of H2O2 from catechol containing adhesive needs to be carefully monitored. As such, controlling the redox reaction of catechol will be critical to the success of these biomimetic adhesive polymers.
In addition to the various chemical interactions reviewed here, the byssal thread and plaque also employ gradation in their materials properties, as well as structural and geometrical designs to minimize structural damage associated with contact deformation between two dissimilar materials (i.e., biological tissue and mineralized surface).12, 277, 278 To‐date, these features has yet to be incorporated into the development of synthetic adhesives. Specifically, an adhesive capable of bonding two dissimilar surfaces with a large discrepancy in their materials properties will be highly desirable in many fields (i.e., attaching tendon to bone).
ACKNOWLEDGMENTS
Authors acknowledge the Office of Naval Research—Young Investigator Award (N00014‐16‐1‐2463) and the National Institutes of Health (R15GM104846) for funding.
Biographies
Pegah Kord Forooshani received her bachelor's degree in chemical engineering from University of Tehran, Iran, and her master's degree in biomedical engineering from University of Malaya, Malaysia. During her master studies, she worked on the synthesis and characterization of citric acid‐based polyester elastomers for tissue engineering applications. Currently, she is perusing a PhD degree in biomedical engineering at Michigan Technological University, USA, under the supervision of Professor Bruce P. Lee. She focuses on tuning the redox chemistry of catechol for promoting wound healing.

Bruce P. Lee received a PhD degree in biomedical engineering from Northwestern University. Prior to his current appointment at Michigan Technological University, he co‐founded a startup company, Nerites Corporation, which aimed at commercializing adhesives and coating inspired by mussel adhesive proteins. His current research interests lie in utilizing the interfacial chemistries of these adhesive proteins in designing bioadhesives and smart materials. He was awarded the 2016 Young Investigator Award from the Office of Naval Research.

REFERENCES AND NOTES
- 1. Waite J. H., Int. J. Adhes. Adhes. 1987, 7, 9–14. [Google Scholar]
- 2. Waite J., Chem. Ind. 1991, 2, 607–611. [Google Scholar]
- 3. Danner E. W., Kan Y., Hammer M. U., Israelachvili J. N., Waite J. H., Biochemistry 2012, 51, 6511–6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wei W., Yu J., Broomell C., Israelachvili J. N., Waite J. H., J. Am. Chem. Soc. 2012, 135, 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Waite J. H., Andersen S. O., Biochim. Biophys. Acta. 1978, 541, 107–114. [Google Scholar]
- 6. Ninan L., Monahan J., Stroshine R. L., Wilker J. J., Shi R., Biomaterials 2003, 24, 4091–4099. [DOI] [PubMed] [Google Scholar]
- 7. Burzio L. O., Burzio V. A., Silva T., Burzio L. A., Pardo J., Curr. Opin. Biotechnol. 1997, 8, 309–312. [DOI] [PubMed] [Google Scholar]
- 8. Lee B. P., Messersmith P. B., Israelachvili J. N., Waite J. H., Annu. Rev. Mater. Res. 2011, 41, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hagenau A., Suhre M. H., Scheibel T. R., Prog. Polym. Sci. 2014, 39, 1564–1583. [Google Scholar]
- 10. Faure E., Falentin‐Daudré C., Jérôme C., Lyskawa J., Fournier D., Woisel P., Detrembleur C., Prog. Polym. Sci. 2013, 38, 236–270. [Google Scholar]
- 11. Waite J. H., Ann. NY Acad. Sci. 1999, 875, 301–309. [DOI] [PubMed] [Google Scholar]
- 12. Bell E., Gosline J., J. Exp. Biol. 1996, 199, 1005–1017. [DOI] [PubMed] [Google Scholar]
- 13. Witman J. D., Suchanek T. H., Mar. Ecol. Prog. Ser. 1984, 16, 259–268. [Google Scholar]
- 14. Zhao H., Waite J. H., J. Biol. Chem. 2006, 281, 26150–26158. [DOI] [PubMed] [Google Scholar]
- 15. Yao Y., Khoo K., Chung M., Li S., J. Chromatogr. A 1994, 680, 431–435. [Google Scholar]
- 16. Filpula D. R., Lee S. M., Link R. P., Strausberg S. L., Strausberg R. L., Biotechnol. Prog. 1990, 6, 171–177. [DOI] [PubMed] [Google Scholar]
- 17. Benedict C. V., Waite J. H., J. Morphol. 1986, 189, 261–270. [DOI] [PubMed] [Google Scholar]
- 18. Rzepecki L. M., Hansen K. M., Waite J. H., Biol. Bull. 1992, 183, 123–137. [DOI] [PubMed] [Google Scholar]
- 19. Zhao H., Waite J. H., Biochemistry 2006, 45, 14223–14231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qin X., Waite J. H., J. Exp. Biol. 1995, 198, 633–644. [DOI] [PubMed] [Google Scholar]
- 21. Qin X. X., Waite J. H., Proc. Natl. Acad. Sci. USA. 1998, 95, 10517–10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Warner S., Waite J., Marine Biol. 1999, 134, 729–734. [Google Scholar]
- 23. Papov V. V., Diamond T. V., Biemann K., Waite J. H., J. Biol. Chem. 1995, 270, 20183–20192. [DOI] [PubMed] [Google Scholar]
- 24. Zhao H., Robertson N. B., Jewhurst S. A., Waite J. H., J. Biol. Chem. 2006, 281, 11090–11096. [DOI] [PubMed] [Google Scholar]
- 25. Floriolli R. Y., von Langen J., Waite J. H., Marine Biotechnol. 2000, 2, 352–363. [DOI] [PubMed] [Google Scholar]
- 26. Waite J. H., Qin X., Biochemistry 2001, 40, 2887–2893. [DOI] [PubMed] [Google Scholar]
- 27. Proudfoot G., Ritchie I., Aust. J. Chem. 1983, 36, 885–894. [Google Scholar]
- 28. Silverman H. G., Roberto F. F., Marine Biotechnol. 2007, 9, 661–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Long J. R., Dindot J. L., Zebroski H., Kiihne S., Clark R. H., Campbell A. A., Stayton P. S., Drobny G. P., Proc. Natl. Acad. Sci. USA. 1998, 95, 12083–12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu J., In Adhesive Interactions of Mussel Foot Proteins; Springer; 2014, pp 31–41. [Google Scholar]
- 31. Waite J. H., Tanzer M. L., Science 1981, 212, 1038–1040. [DOI] [PubMed] [Google Scholar]
- 32. Schnurrer J., Lehr C. M., Int. J. Pharm. 1996, 141, 251–256. [Google Scholar]
- 33. Catron N. D., Lee H., Messersmith P. B., Biointerphases 2006, 1, 134–141. [DOI] [PubMed] [Google Scholar]
- 34. Chirdon W. M., O'Brien W. J., Robertson R. E., J. Biomed. Mater. Res. Part B: Appl. Biomater. 2003, 66, 532–538. [DOI] [PubMed] [Google Scholar]
- 35. Coombs T. L., Keller P. J., Aquatic Toxicol.. 1981, 1, 291–300. [Google Scholar]
- 36. Baty A. M., Leavitt P. K., Siedlecki C. A., Tyler B. J., Suci P. A., Marchant R. E., Geesey G. G., Langmuir 1997, 13, 5702–5710. [Google Scholar]
- 37. Baker B. R., Laiwalla A. N., Yoon J. Y., Canavate J., Garrell R. L., Polym. Mater. Sci. Eng. 2001, 85, 115–116. [Google Scholar]
- 38.J. P. Gallivan, Dougherty D. A., J. Am. Chem. Soc. 2000, 122, 870–874. [Google Scholar]
- 39. Gallivan J. P., Dougherty D. A., J. Am. Chem. Soc. 2000, 122, 870–874. [Google Scholar]
- 40. Lu Q., Oh D. X., Lee Y., Jho Y., Hwang D. S., Zeng H., Angew. Chem. Int. Ed. 2013, 52, 3944–3948. [DOI] [PubMed] [Google Scholar]
- 41. Pillai K. V., Renneckar S., Biomacromolecules 2009, 10, 798–804. [DOI] [PubMed] [Google Scholar]
- 42. Das S., Rodriguez N. R. M., Wei W., Waite J. H., Israelachvili J. N., Adv. Funct. Mater. 2015, 25, 5840–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim S., Faghihnejad A., Lee Y., Jho Y., Zeng H., Hwang D. S., J. Mater. Chem. B 2015, 3, 738–743. [DOI] [PubMed] [Google Scholar]
- 44. Wei W., Yu J., Gebbie M. A., Tan Y., Martinez Rodriguez N. R., Israelachvili J. N., Waite J. H., Langmuir 2015, 31, 1105–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hwang D. S., Zeng H., Lu Q., Israelachvili J., Waite J. H., Soft Matter 2012, 8, 5640–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sever M. J., Wilker J. J., Dalton Trans. 2006, 14, 813–822. [DOI] [PubMed] [Google Scholar]
- 47. Tyson C. A., Martell A. E., J. Am. Chem. Soc. 1968, 90, 3379–3386. [DOI] [PubMed] [Google Scholar]
- 48. Borgias B. A., Cooper S. R., Koh Y. B., Raymond K. N., Inorg. Chem. 1984, 23, 1009–1016. [Google Scholar]
- 49. Sommer L., Z. Anorg. Allg. Chem. 1963, 321, 191–197. [Google Scholar]
- 50. Morlay C., Cromer M., Mouginot Y., Vittori O., Talanta 1998, 45, 1177–1188. [DOI] [PubMed] [Google Scholar]
- 51. Borggaard O. K., Acta Chem. Scand. 1972, 26, 393–395. [DOI] [PubMed] [Google Scholar]
- 52. Taylor S. W., G. W. Luther, III , Waite J. H., Inorg. Chem. 1994, 33, 5819–5824. [Google Scholar]
- 53. Taylor S. W., Chase D. B., Emptage M. H., Nelson M. J., Waite J. H., Inorg. Chem. 1996, 35, 7572–7577. [Google Scholar]
- 54. Harrington M. J., Masic A., Holten‐Andersen N., Waite J. H., Fratzl P., Science. 2010, 328, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harrington M. J., Gupta H. S., Fratzl P., Waite J. H., J. Struct. Biol. 2009, 167, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Holten‐Andersen N., Harrington M. J., Birkedal H., Lee B. P., Messersmith P. B., Lee K. Y. C., Waite J. H., Proc. Natl. Acad. Sci. USA. 2011, 108, 2651–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Holten‐Andersen N., Jaishankar A., Harrington M. J., Fullenkamp D. E., DiMarco G., He L., McKinley G. H., Messersmith P. B., Lee K. Y. C., J. Mater. Chem. B 2014, 2, 2467–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krogsgaard M., Behrens M. A., Pedersen J. S., Birkedal H., Biomacromolecules 2013, 14, 297–301. [DOI] [PubMed] [Google Scholar]
- 59. Kim B. J., Cheong H., Hwang B. H., Cha H. J., Angew. Chem. 2015, 127, 7426–7430. [Google Scholar]
- 60. Lee B. P., Narkar A., Wilharm R., Sens. Actuator. B: Chem. 2016, 227, 248–254. [Google Scholar]
- 61. Lee B. P., Lin M. H., Narkar A., Konst S., Wilharm R., Sen. Actuator. B: Chem. 2015, 206, 456–462. [Google Scholar]
- 62. Lee B. P., Konst S., Adv. Mater. 2014, 26, 3415–3419. [DOI] [PubMed] [Google Scholar]
- 63. Kim B. J., Kim S., Oh D. X., Masic A., Cha H. J., Hwang D. S., J. Mater. Chem. B. 2015, 3, 112–118. [DOI] [PubMed] [Google Scholar]
- 64. Krogsgaard M., Andersen A., Birkedal H., Chem. Commun. 2014, 50, 13278–13281. [DOI] [PubMed] [Google Scholar]
- 65. Kummert R., Stumm W., J. Colloid Interface Sci. 1980, 75, 373–385. [Google Scholar]
- 66. Lee B. P., Chao C. Y., Nunalee F. N., Motan E., Shull K. R., Messersmith P. B., Macromolecules 2006, 39, 1740–1748. [Google Scholar]
- 67. Soriaga M. P., Hubbard A. T., J. Am. Chem. Soc. 1982, 104, 3937–3945. [Google Scholar]
- 68. Lee H., Scherer N. F., Messersmith P. B., Proc. Natl. Acad. Sci. USA. 2006, 103, 12999–13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mian S. A., Gao X., Nagase S., Jang J., Theor. Chem. Acc. 2011, 130, 333–339. [Google Scholar]
- 70. Mian S. A., Saha L. C., Jang J., Wang L., Gao X., Nagase S., J. Phys. Chem. C 2010, 114, 20793–20800. [Google Scholar]
- 71. Dalsin J. L., Hu B. H., Lee B. P., Messersmith P. B., J. Am. Chem. Soc. 2003, 125, 4253–4258. [DOI] [PubMed] [Google Scholar]
- 72. Dalsin J. L., Lin L., Messersmith P. B., Polym. Mater. Sci. Eng. Preprints 2004, 90, 247–248. [Google Scholar]
- 73. Dalsin J. L., Lin L., Tosatti S., Vörös J., Textor M., Messersmith P. B., Langmuir. 2005, 21, 640–646. [DOI] [PubMed] [Google Scholar]
- 74. Yu J., Wei W., Menyo M. S., Masic A., Waite J. H., Israelachvili J. N., Biomacromolecules 2013, 14, 1072–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McDowell L. M., Burzio L. A., Waite J. H., Schaefer J., J. Biol. Chem. 1999, 274, 20293–20295. [DOI] [PubMed] [Google Scholar]
- 76. Rzepecki L. M., Nagafuchi T., Waite J. H., Arch. Biochem. Biophys. 1991, 285, 17–26. [DOI] [PubMed] [Google Scholar]
- 77. Lee B. P., Dalsin J. L., Messersmith P. B., Biomacromolecules 2002, 3, 1038–1047. [DOI] [PubMed] [Google Scholar]
- 78. Waite J. H., Comp. Biochem. Physiol. Part B: Biochem. Mol. Biol. 1990, 97, 19–29. [DOI] [PubMed] [Google Scholar]
- 79. Sugumaran M., Dali H., Semensi V., Arch. Insect Biochem. Physiol. 1989, 11, 127–137. [DOI] [PubMed] [Google Scholar]
- 80. Lee H., Dellatore S. M., Miller W. M., Messersmith P. B., Science 2007, 318, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cheng C., Li S., Nie S., Zhao W., Yang H., Sun S., Zhao C., Biomacromolecules 2012, 13, 4236–4246. [DOI] [PubMed] [Google Scholar]
- 82. Ren Y., Rivera J. G., He L., Kulkarni H., Lee D.‐K., Messersmith P. B., BMC Biotechnol. 2011, 11, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Black K. C., Yi J., Rivera J. G., Zelasko‐Leon D. C., Messersmith P. B., Nanomedicine 2013, 8, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lim C., Huang J., Kim S., Lee H., Zeng H., Hwang D. S., Angew. Chem. Int. Ed. 2016, 55, 3342–3346. [DOI] [PubMed] [Google Scholar]
- 85. Kang S. M., Hwang N. S., Yeom J., Park S. Y., Messersmith P. B., Choi I. S., Langer R., Anderson D. G., Lee H., Adv. Funct. Mater. 2012, 22, 2949–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kim S., Moon J.‐M., Choi J. S., Cho W. K., Kang S. M., Adv. Funct. Mater., 2016, 26, 4099–4105. [Google Scholar]
- 87. Cencer M. M., Liu Y., Winter A., Murley M., Meng H., Lee B. P., Biomacromolecules 2014, 15, 2861–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Li J., Christensen B. M., J. Electroanal. Chem. 1994, 375, 219–231. [Google Scholar]
- 89. Tannock I. F., Rotin D., Cancer Res. 1989, 49, 4373–4384. [PubMed] [Google Scholar]
- 90. Ohman H., Vahlquist A., Acta Derm. Venereol. 1994, 74, 375–379. [DOI] [PubMed] [Google Scholar]
- 91. Soller B. R., Micheels R. H., Coen J., Parikh B., Chu L., Hsi C., J. Clin. Monitor. Comput. 1996, 12, 387–395. [DOI] [PubMed] [Google Scholar]
- 92. Soller B. R., Khan T., Favreau J., Hsi C., Puyana J. C., Heard S. O., J. Surg. Res. 2003, 114, 195–201. [DOI] [PubMed] [Google Scholar]
- 93. Meng H., Li Y., Faust M., Konst S., Lee B. P., Acta Biomater. 2015, 17, 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mochizuki M., Yamazaki S., Kano K., Ikeda T., Biochim. Biophys. Acta (BBA) Gen. Subjects 2002, 1569, 35–44. [DOI] [PubMed] [Google Scholar]
- 95. Chelikani P., Fita I., Loewen P. C., CMLS, Cell. Mol. Life Sci. 2004, 61, 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Brian N., Ahswin H., Smart N., Bayon Y., Wohlert S., Hunt J. A., Eur. Cells Mater. 2012, 24, 249–265. [DOI] [PubMed] [Google Scholar]
- 97. Cooper A. M., Segal B. H., Frank A. A., Holland S. M., Orme I. M., Infect. Immun. 2000, 68, 1231–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Loo A. E. K., Wong Y. T., Ho R., Wasser M., Du T., Ng W. T., Halliwell B., PLoS One 2012, 7, e49215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chigurupati S., Mughal M. R., Okun E., Das S., Kumar A., McCaffery M., Seal S., Mattson M. P., Biomaterials 2013, 34, 2194–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Martinez Rodriguez N. R., Das S., Kaufman Y., Israelachvili J. N., Waite J. H., Biofouling 2015, 31, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Stewart R., Ransom T., Hlady V.. J. Polym. Sci. Part B: Polym. Phys. 2011, 49, 757–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yu J., Wei W., Danner E., Ashley R. K., Israelachvili J. N., Waite J. H., Nat. Chem. Biol. 2011, 7, 588–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ortony J. H., Hwang D. S., Franck J. M., Waite J. H., Han S., Biomacromolecules 2013, 14, 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yu M., Hwang J., Deming T. J., J. Am. Chem. Soc. 1999, 121, 5825–5826. [Google Scholar]
- 105. Yu J., Wei W., Danner E., Israelachvili J. N., Waite J. H., Adv. Mater. 2011, 23, 2362–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Springsteen G., Wang B., Tetrahedron 2002, 58, 5291–5300. [Google Scholar]
- 107. Guvendiren M., Brass D. A., Messersmith P. B., Shull K. R., J. Adhes. 2009, 85, 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nicklisch S. C., Spahn J. E., Zhou H., Gruian C. M., Waite J. H., Biochemistry 2016, 55, 2022–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. De Kruif C. G., Weinbreck F., de Vries R., Curr Opin Colloid Interface Sci. 2004, 9, 340–349. [Google Scholar]
- 110. Seo S., Das S., Zalicki P. J., Mirshafian R., Eisenbach C. D., Israelachvili J. N., Waite J. H., Ahn B. K., J. Am. Chem. Soc. 2015, 137, 9214–9217. [DOI] [PubMed] [Google Scholar]
- 111. Ahn B. K., Das S., Linstadt R., Kaufman Y., Martinez‐Rodriguez N. R., Mirshafian R., Kesselman E., Talmon Y., Lipshutz B. H., Israelachvili J. N., Waite J. H., Nat. Commun. 2015, 6, 8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wei W., Tan Y., Rodriguez N. R. M., Yu J., Israelachvili J. N., Waite J. H., Acta Biomater. 2014, 10, 1663–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kim S., Huang J., Lee Y., Dutta S., Yoo H. Y., Jung Y. M., Jho Y., Zeng H., Hwang D. S., Proc. Natl. Acad. Sci. USA. 2016, 113, E847–E853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Moulds R., Baldwin T., Int. J. Adhes. Adhes. 1983, 3, 203–207. [Google Scholar]
- 115. Akdogan Y., Wei W., Huang K. Y., Kageyama Y., Danner E. W., Miller D. R., Martinez Rodriguez N. R., Waite J. H., Han S., Angew. Chem. 2014, 126, 11435–11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wei W., Petrone L., Tan Y., Cai H., Israelachvili J. N., Miserez A., Waite J. H., Adv. Funct. Mater. 2016, 26, 3496–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lin Q., Gourdon D., Sun C., Holten‐Andersen N., Anderson T. H., Waite J. H., Israelachvili J. N., Proc. Natl. Acad. Sci. USA. 2007, 104, 3782–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yu J., Kan Y., Rapp M., Danner E., Wei W., Das S., Miller D. R., Chen Y., Waite J. H., Israelachvili J. N., Proc. Natl. Acad. Sci. USA. 2013, 110, 15680–15685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Maier G. P., Rapp M. V., Waite J. H., Israelachvili J. N., Butler A., Science 2015, 349, 628–632. [DOI] [PubMed] [Google Scholar]
- 120. Sun C. J., Srivastava A., Reifert J. R., Waite J. H., J. Adhes. 2009, 85, 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Cencer M., Murley M., Liu Y., Lee B. P., Biomacromolecules 2015, 16, 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ding X., Vegesna G. K., Meng H., Winter A., Lee B. P., Macromol. Chem. Phys. 2015, 216, 1109–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Amstad E., Gehring A. U., Fischer H., Nagaiyanallur V. V., Hahner G., Textor M., Reimhult E., J. Phys. Chem. C 2011, 115, 683–691. [Google Scholar]
- 124. Amstad E., Gillich T., Bilecka I., Textor M., Reimhult E., Nano Lett. 2009, 9, 4042–4048. [DOI] [PubMed] [Google Scholar]
- 125. Menyo M. S., Hawker C. J., Waite J. H., Soft Matter 2013, 9, 10314–10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Anderson J., Lin M.‐H., Privette C., Flowers M., Murley M., Lee B. P., Ong K. G., ScienceJet 2015, 4, 1–14. [PMC free article] [PubMed] [Google Scholar]
- 127. Shafiq Z., Cui J., Pastor‐Pérez L., San Miguel V., Gropeanu R. A., Serrano C., del Campo A., Angew. Chem. Int. Ed. 2012, 51, 4332–4335. [DOI] [PubMed] [Google Scholar]
- 128. García‐Fernández L., Cui J., Serrano C., Shafiq Z., Gropeanu R. A., Miguel V. S., Ramos J. I., Wang M., Auernhammer G. K., Ritz S., Golriz A. A., Berger R., Wagner M., del Campo A., Adv. Mater. 2013, 25, 529–533. [DOI] [PubMed] [Google Scholar]
- 129. Ejima H., Richardson J. J., Liang K., Best J. P., van Koeverden M. P., Such G. K., Cui J., Caruso F., Science 2013, 341, 154–157. [DOI] [PubMed] [Google Scholar]
- 130. Guo J., Sun H., Alt K., Tardy B. L., Richardson J. J., Suma T., Ejima H., Cui J., Hagemeyer C. E., Caruso F., Adv. Healthcare Mater. 2015, 4, 1796–1801. [DOI] [PubMed] [Google Scholar]
- 131. Barrett D. G., Sileika T. S., Messersmith P. B., Chem. Commun. 2014, 50, 7265–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Shen G., Xing R., Zhang N., Chen C., Ma G., Yan X., ACS Nano, 2016, 10, 5720–5729. [DOI] [PubMed] [Google Scholar]
- 133. Liu Z., Hu B. H., Messersmith P. B., Tetrahedron Lett. 2008, 49, 5519–5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Black K. C. L., Liu Z., Messersmith P. B., Chem. Mater. 2011, 23, 1130–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Westwood G., Horton T. N., Wilker J. J., Maclomolecules 2007, 40, 3960–3964. [Google Scholar]
- 136. Hu B. H., Messersmith P. B., Tetrahedron Lett. 2000, 41, 5795–5798. [Google Scholar]
- 137. Yu M., Deming T. J., Macromolecules 1998, 31, 4739–4745. [DOI] [PubMed] [Google Scholar]
- 138. Lee B. P., Huang K., Nunalee F. N., Shull K. R., Messersmith P. B., J. Biomater. Sci. Polym. Ed. 2004, 15, 449–464. [DOI] [PubMed] [Google Scholar]
- 139. Lee H., Lee B. P., Messersmith P. B., Nature 2007, 448, 338–341. [DOI] [PubMed] [Google Scholar]
- 140. Heo J., Kang T., Jang S. G., Hwang D. S., Spruell J. M., Killops K. L., Waite J. H., Hawker C. J., J. Am. Chem. Soc. 2012, 134, 20139–20145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Nam H., Kim M. M., Food Chem. Toxicol. 2013, 55, 106–112. [DOI] [PubMed] [Google Scholar]
- 142. Hamed S. F., Sadek Z., Edris A., J. Oleo Sci. 2012, 61, 641–648. [DOI] [PubMed] [Google Scholar]
- 143. Bachiega T. F., de Sousa J. P. B., Bastos J. K., Sforcin J. M., J. Pharm. Pharmacol. 2012, 64, 610–616. [DOI] [PubMed] [Google Scholar]
- 144. Almaroof A., Niazi S., Rojo L., Mannocci F., Deb S., Dent. Mater. 2016, 32, 929–939. [DOI] [PubMed] [Google Scholar]
- 145. Almaroof A., Rojo L., Mannocci F., Deb S., Dent. Mater. 2016, 32, 149–160. [DOI] [PubMed] [Google Scholar]
- 146. Huang K., Lee B. P., Ingram D. R., Messersmith P. B., Biomacromolecules 2002, 3, 397–406. [DOI] [PubMed] [Google Scholar]
- 147. Mehdizadeh M., Weng H., Gyawali D., Tang L., Yang J., Biomaterials 2012, 33, 7972–7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Brubaker C. E., Kissler H., Wang L. J., Kaufman D. B., Messersmith P. B., Biomaterials 2010, 31, 420–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Perrini M., Barrett D., Ochsenbein‐Koelble N., Zimmermann R., Messersmith P., Ehrbar M., J. Mech. Behav. Biomed. Mater., 2016, 58, 57–64. [DOI] [PubMed] [Google Scholar]
- 150. Murphy J. L., Vollenweider L., Xu F., Lee B. P., Biomacromolecules 2010, 11, 2976–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. B. Michael, V. Laura, L. M. John, X. Fangmin, L. Arinne, D. L. William, P. L. Bruce , Biomed. Mater. 2011, 6, 015014. 21266745 [Google Scholar]
- 152. Barrett D. G., Bushnell G. G., Messersmith P. B., Adv. Healthcare Mater. 2013, 2, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Guvendiren M., Messersmith P. B., Shull K. R., Biomacromolecules 2008, 9, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sun P., Wang J., Yao X., Peng Y., Tu X., Du P., Zheng Z., Wang X., ACS Appl. Mater. Interfaces 2014, 6, 12495–12504. [DOI] [PubMed] [Google Scholar]
- 155. Park J. Y., Yeom J., Kim J. S., Lee M., Lee H., Nam Y. S., Macromol. Biosci. 2013, 13, 1511–1519. [DOI] [PubMed] [Google Scholar]
- 156. Kim K., Ryu J. H., Lee D. Y., Lee H., Biomater. Sci. 2013, 1, 783–790. [DOI] [PubMed] [Google Scholar]
- 157. Neto A. I., Cibrão A. C., Correia C. R., Carvalho R. R., Luz G. M., Ferrer G. G., Botelho G., Picart C., Alves N. M., Mano J. F., Small 2014, 10, 2459–2469. [DOI] [PubMed] [Google Scholar]
- 158. Lee B. P. (Knc Ner Acquisition Sub, Inc.). Patent U.S. 8,030,413, October 4, 2011.
- 159. Lee C., Shin J., Lee J. S., Byun E., Ryu J. H., Um S. H., Kim D. I., Lee H., Cho S. W., Biomacromolecules 2013, 14, 2004–2013. [DOI] [PubMed] [Google Scholar]
- 160. Kang T., Banquy X., Heo J. H., Lim C. N., Lynd N. A., Lundberg P., Oh D. X., Lee H. K., Hong Y. K., Hwang D. S., Waite J. H., Israelachvili J. N., Hawker C. J., Acs Nano 2016, 10, 930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mattson K. M., Latimer A. A., McGrath A. J., Lynd N. A., Lundberg P., Hudson Z. M., Hawker C. J., J. Polym. Sci. Part A: Polym. Chem. 2015, 53, 2685–2692. [Google Scholar]
- 162. Sprafke J. K., Spruell J. M., Mattson K. M., Montarnal D., McGrath A. J., Pötzsch R., Miyajima D., Hu J., Latimer A. A., Voit B. I., J. Polym. Sci. Part A: Polym. Chem. 2015, 53, 319–326. [Google Scholar]
- 163. Pechey A., Elwood C. N., Wignall G. R., Dalsin J. L., Lee B. P., Vanjecek M., Welch I., Ko R., Razvi H., Cadieux P. A., J. Urol. 2009, 182, 1628–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Shao H., Bachus K. N., Stewart R. J., Macromol. Biosci. 2009, 9, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Matos‐Perez C. R., White J. D., Wilker J. J., J. Am. Chem. Soc. 2012, 134, 9498–9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Jenkins C. L., Meredith H. J., Wilker J. J., ACS Appl. Mater. Interfaces 2013, 5, 5091–5096. [DOI] [PubMed] [Google Scholar]
- 167. Matos‐Pérez C. R., Wilker J. J.. Macromolecules 2012, 45, 6634–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Leibig D., Müller A. H., Frey H., Macromolecules 2016, 49, 4792–4801. [Google Scholar]
- 169. Skelton S., Bostwick M., O'Connor K., Konst S., Casey S., Lee B. P., Soft Matter 2013, 9, 3825–3833. [Google Scholar]
- 170. Deming T. J., Nature 1997, 390, 386–389. [DOI] [PubMed] [Google Scholar]
- 171. Niederer K., Schüll C., Leibig D., Johann T., Frey H., Macromolecules 2016, 49, 1655–1665. [Google Scholar]
- 172. Liu J., Yang W., Zareie H. M., Gooding J. J., Davis T. P., Macromolecules 2009, 42, 2931–2939. [Google Scholar]
- 173. Zobrist C., Sobocinski J., Lyskawa J., Fournier D., Miri V., Traisnel M., Jimenez M., Woisel P., Macromolecules 2011, 44, 5883–5892. [Google Scholar]
- 174. Fan X., Lin L., Messersmith P. B., Biomacromolecules 2006, 7, 2443–2448. [DOI] [PubMed] [Google Scholar]
- 175. Fan X., Lin L., Dalsin J. L., Messersmith P. B., J. Am. Chem. Soc. 2005, 127, 15843–15847. [DOI] [PubMed] [Google Scholar]
- 176. Gao C., Li G., Xue H., Yang W., Zhang F., Jiang S., Biomaterials 2010, 31, 1486–1492. [DOI] [PubMed] [Google Scholar]
- 177. Li G., Cheng G., Xue H., Chen S., Zhang F., Jiang S., Biomaterials 2008, 29, 4592–4597. [DOI] [PubMed] [Google Scholar]
- 178. Ye Q., Wang X., Li S., Zhou F., Macromolecules 2010, 43, 5554–5560. [Google Scholar]
- 179. Kuang J., Messersmith P. B., Langmuir 2012, 28, 7258–7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Sever M. J., Wilker J. J., Tetrahedron 2001, 57, 6139–6146. [Google Scholar]
- 181. Behnam M. A., Sundermann T. R., Klein C. D., Org. Lett. 2016, 18, 2016–2019. [DOI] [PubMed] [Google Scholar]
- 182. Bodanszky M., Principles of Peptide Synthesis; Springer Science & Business Media: New York, 2012. [Google Scholar]
- 183. Akemi Ooka A., Garrell R. L., Biopolymers 2000, 57, 92–102. [DOI] [PubMed] [Google Scholar]
- 184. Miller S. M., Simon R. J., Ng S., Zuckermann R. N., Kerr J. M., Moos W. H., Drug Dev. Res. 1995, 35, 20–32. [Google Scholar]
- 185. Statz A. R., Meagher R. J., Barron A. E., Messersmith P. B., J. Am. Chem. Soc. 2005, 127, 7972–7973. [DOI] [PubMed] [Google Scholar]
- 186. Zeng H., Hwang D. S., Israelachvili J. N., Waite J. H., Proc. Natl. Acad. Sci. USA. 2010, 107, 12850–12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Hwang D. S., Gim Y., Cha H. J., Biotechnol. Prog. 2005, 21, 965–970. [DOI] [PubMed] [Google Scholar]
- 188. Hwang D. S., Yoo H. J., Jun J. H., Moon W. K., Cha H. J., Appl. Environ Microbiol. 2004, 70, 3352–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Hwang D. S., Gim Y., Yoo H. J., Cha H. J., Biomaterials 2007, 28, 3560–3568. [DOI] [PubMed] [Google Scholar]
- 190. Hwang D. S., Zeng H., Srivastava A., Krogstad D. V., Tirrell M., Israelachvili J. N., Waite J. H., Soft Matter 2010, 6, 3232–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Kim B. J., Oh D. X., Kim S., Seo J. H., Hwang D. S., Masic A., Han D. K., Cha H. J., Biomacromolecules 2014, 15, 1579–1585. [DOI] [PubMed] [Google Scholar]
- 192. Lim S., Choi Y. S., Kang D. G., Song Y. H., Cha H. J., Biomaterials 2010, 31, 3715–3722. [DOI] [PubMed] [Google Scholar]
- 193. Zhong C., Gurry T., Cheng A. A., Downey J., Deng Z., Stultz C. M., Lu T. K., Nat Nano 2014, 9, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Hwang D. S., Sim S. B., Cha H. J., Biomaterials 2007, 28, 4039–4046. [DOI] [PubMed] [Google Scholar]
- 195. Wilker J. J., Nat. Chem. Biol. 2011, 7, 579–580. [DOI] [PubMed] [Google Scholar]
- 196. Wang W., Xu Y., Li A., Li T., Liu M., von Klitzing R., Ober C. K., Kayitmazer A. B., Li L., Guo X., RSC Adv. 2015, 5, 66871–66878. [Google Scholar]
- 197. Miller D. R., Das S., Huang K. Y., Han S., Israelachvili J. N., Waite J. H., ACS Biomater. Sci. Eng. 2015, 1, 1121–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Kim M. R., Park T. G., J. Controlled Release 2002, 80, 69–77. [DOI] [PubMed] [Google Scholar]
- 199. Jain M. K., Grimes C. A., Sens. Actuator. A: Phys. 2002, 100, 63–69. [DOI] [PubMed] [Google Scholar]
- 200. Wang T., Liu D., Lian C., Zheng S., Liu X., Tong Z., Soft Matter 2012, 8, 774–783. [Google Scholar]
- 201. Gaharwar A. K., Dammu S. A., Canter J. M., Wu C. J., Schmidt G., Biomacromolecules 2011, 12, 1641–1650. [DOI] [PubMed] [Google Scholar]
- 202. Xiang X., Long F., Narkar A., Kinnunen R. E., Shahbazian‐Yassar R., Lee B. P., Heiden P. A., J. Appl. Polym. Sci., 2016, 133, 1–12. [Google Scholar]
- 203. Liu Y., Meng H., Konst S., Sarmiento R., Rajachar R., Lee B. P., ACS Appl. Mater. Interfaces 2014, 6, 16982–16992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Liu Y., Zhan H., Skelton S., Lee B. P., MRS Proc. 2013, 1569, 33–38. [Google Scholar]
- 205. Jaiswal M. K., Xavier J. R., Carrow J. K., Desai P., Alge D., Gaharwar A. K., ACS Nano 2015, 10, 246–256. [DOI] [PubMed] [Google Scholar]
- 206. GhavamiNejad A., Park C. H., Kim C. S., Biomacromolecules 2016, 17, 1213–1223. [DOI] [PubMed] [Google Scholar]
- 207. Li Y., Meng H., Liu Y., Narkar A., Lee B. P., ACS Appl. Mater. Interfaces 2016, 8, 11980–11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Li Y., Meng H., Liu Y., Lee B. P., Sci. World J. 2015, 2015, 685690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Djabourov M., Papon P., Polymer 1983, 24, 537–542. [Google Scholar]
- 210. de Carvalho W., Djabourov M., Rheol. Acta 1997, 36, 591–609. [Google Scholar]
- 211. Ionov L., Adv. Funct. Mater. 2013, 23, 4555–4570. [Google Scholar]
- 212. Lee B. P., Liu Y., Konst S., MRS Proc. 2014, 1710, mrss14‐1710‐xx1708‐1701. [Google Scholar]
- 213. Krogsgaard M., Hansen M. R., Birkedal H., J. Mater. Chem. B 2014, 2, 8292–8297. [DOI] [PubMed] [Google Scholar]
- 214. Grindy S. C., Learsch R., Mozhdehi D., Cheng J., Barrett D. G., Guan Z., Messersmith P. B., Holten‐Andersen N., Nat. Mater. 2015, 14, 1210–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. He L., Fullenkamp D. E., Rivera J. G., Messersmith P. B., Chem. Commun. 2011, 47, 7497–7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Ahn B. K., Lee D. W., Israelachvili J. N., Waite J. H., Nat. Mater. 2014, 13, 867–872. [DOI] [PubMed] [Google Scholar]
- 217. Li L., Yan B., Yang J., Chen L., Zeng H., Adv. Mater. 2015, 27, 1294–1299. [DOI] [PubMed] [Google Scholar]
- 218. Heinzmann C., Coulibaly S., Roulin A., Fiore G. L., Weder C., ACS Appl. Mater. Interfaces 2014, 6, 4713–4719. [DOI] [PubMed] [Google Scholar]
- 219. Northen M. T., Greiner C., Arzt E., Turner K. L.. Adv. Mater. 2008, 20, 3905–3909. [Google Scholar]
- 220. Luo X., Lauber K. E., Mather P. T., Polymer 2010, 51, 1169–1175. [Google Scholar]
- 221. Banea M., da Silva L., Campilho R., Ann. Dunarea de Jos Univ. Galati. Fascicle XII: Weld. Equip. Technol. 2013, 24, 11–14. [Google Scholar]
- 222. Sudre G., Olanier L., Tran Y., Hourdet D., Creton C., Soft Matter 2012, 8, 8184–8193. [Google Scholar]
- 223. Wang Y. Z., Li L., Du F. S., Li Z. C., Polymer 2015, 68, 270–278. [Google Scholar]
- 224. Wilke P., Helfricht N., Mark A., Papastavrou G., Faivre D., Börner H. G., J. Am. Chem. Soc. 2014, 136, 12667–12674. [DOI] [PubMed] [Google Scholar]
- 225. Wilke P., Börner H. G., ACS Macro Lett. 2012, 1, 871–875. [DOI] [PubMed] [Google Scholar]
- 226. Narkar A. R., Barker B., Clisch M., Jiang J., Lee B. P., Chem. Mater. 2016, 28, 5432–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Dalsin J. L., Sherman D. L., Lee B. P., Messersmith P. B., Polym. Mater. Sci. Eng. Preprints 2006, 94, 854–855. [Google Scholar]
- 228. Mehdizadeh M., Yang J., Macromol. Biosci. 2013, 13, 271–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Peng H. T., Shek P. N., Expert Rev. Med. Devices 2010, 7, 639–659. [DOI] [PubMed] [Google Scholar]
- 230. Fortelny R., Petter‐Puchner A., Walder N., Mittermayr R., Öhlinger W., Heinze A., Redl H., Surg. Endosc. 2007, 21, 1781–1785. [DOI] [PubMed] [Google Scholar]
- 231. Seyednejad H., Imani M., Jamieson T., Seifalian A., Br. J. Surg. 2008, 95, 1197–1225. [DOI] [PubMed] [Google Scholar]
- 232. P. Klimo, Jr. , Khalil A., Slotkin J. R., Smith E. R., Scott R. M., Goumnerova L. C., Neurosurgery 2007, 60, 305–309. [DOI] [PubMed] [Google Scholar]
- 233. Than K. D., Baird C. J., Olivi A., Neurosurgery 2008, 63, ONS182–ONS187. [DOI] [PubMed] [Google Scholar]
- 234. Allen M. S., Wood D. E., Hawkinson R. W., Harpole D. H., McKenna R. J., Walsh G. L., Vallieres E., Miller D. L., Nichols F. C., Smythe W. R., Ann. Thorac. Surg. 2004, 77, 1792–1801. [DOI] [PubMed] [Google Scholar]
- 235. Napoleone C. P., Valori A., Crupi G., Ocello S., Santoro F., Vouhé P., Weerasena N., Gargiulo G., Interact. Cardiovasc. Thorac. Surg. 2009, 9, 978–982. [DOI] [PubMed] [Google Scholar]
- 236. Bilic G., Brubaker C., Messersmith P. B., Mallik A. S., Quinn T. M., Haller C., Done E., Gucciardo L., Zeisberger S. M., Zimmermann R., Am. J. Obstet. Gynecol. 2010, 202, 85. e81–85. e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Haller C., Buerzle W., Kivelio A., Perrini M., Brubaker C., Gubeli R., Mallik A., Weber W., Messersmith P., Mazza E., Acta Biomater. 2012, 8, 4365–4370. [DOI] [PubMed] [Google Scholar]
- 238. Alvarez‐Lorenzo C., Gonzalez‐Lopez J., Fernandez‐Tarrio M., Sandez‐Macho I., Concheiro A., Eur. J. Pharm. Biopharm. 2007, 66, 244–252. [DOI] [PubMed] [Google Scholar]
- 239. Ji Y., Ji T., Liang K., Zhu L., J. Mater. Sci. Mater. Med. 2016, 27, 1–9. [DOI] [PubMed] [Google Scholar]
- 240. Burke K. A., Roberts D. C., Kaplan D. L., Biomacromolecules 2015, 17, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Ryu J. H., Lee Y., Kong W. H., Kim T. G., Park T. G., Lee H., Biomacromolecules 2011, 12, 2653–2659. [DOI] [PubMed] [Google Scholar]
- 242. Kim K., Kim K., Ryu J. H., Lee H., Biomaterials 2015, 52, 161–170. [DOI] [PubMed] [Google Scholar]
- 243. Xu J., Soliman G. M., Barralet J., Cerruti M., Langmuir 2012, 28, 14010–14017. [DOI] [PubMed] [Google Scholar]
- 244. Shin J., Lee J. S., Lee C., Park H. J., Yang K., Jin Y., Ryu J. H., Hong K. S., Moon S. H., Chung H. M., Adv. Funct. Mater. 2015, 25, 3814–3824. [Google Scholar]
- 245. Samal S. K., Kaplan D. L., Chiellini E., Macromol. Mater. Eng. 2013, 298, 1201–1208. [Google Scholar]
- 246. Yucel T., Lovett M. L., Kaplan D. L., J. Controlled Release 2014, 190, 381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Kundu B., Rajkhowa R., Kundu S. C., Wang X., Adv. Drug Deliv. Rev. 2013, 65, 457–470. [DOI] [PubMed] [Google Scholar]
- 248. Ghosh K., Shu X. Z., Mou R., Lombardi J., Prestwich G. D., Rafailovich M. H., Clark R. A., Biomacromolecules 2005, 6, 2857–2865. [DOI] [PubMed] [Google Scholar]
- 249. Jia X., Yeo Y., Clifton R. J., Jiao T., Kohane D. S., Kobler J. B., Zeitels S. M., Langer R., Biomacromolecules 2006, 7, 3336–3344. [DOI] [PubMed] [Google Scholar]
- 250. Lee Y., Chung H. J., Yeo S., Ahn C. H., Lee H., Messersmith P. B., Park T. G., Soft Matter 2010, 6, 977–983. [Google Scholar]
- 251. Azad A. K., Sermsintham N., Chandrkrachang S., Stevens W. F., J. Biomed. Mater. Res. Part B Appl. Biomater. 2004, 69, 216–222. [DOI] [PubMed] [Google Scholar]
- 252. Rao S. B., Sharma C. P., J. Biomed. Mater. Res. 1997, 34, 21–28. [DOI] [PubMed] [Google Scholar]
- 253. Xu J., Strandman S., Zhu J. X., Barralet J., Cerruti M., Biomaterials 2015, 37, 395–404. [DOI] [PubMed] [Google Scholar]
- 254. Harris N., Ford M. J., Cortie M. B., J. Phys. Chem. B 2006, 110, 10701–10707. [DOI] [PubMed] [Google Scholar]
- 255. Black K. C. L., Sileika T. S., Yi J., Zhang R., Rivera J. G., Messersmith P. B., Small 2014, 10, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Ochs C. J., Hong T., Such G. K., Cui J., Postma A., Caruso F., Chem. Mater. 2011, 23, 3141–3143. [Google Scholar]
- 257. Cui J., Wang Y., Postma A., Hao J., Hosta‐Rigau L., Caruso F., Adv. Funct. Mater. 2010, 20, 1625–1631. [Google Scholar]
- 258. Cui J., Yan Y., Such G. K., Liang K., Ochs C. J., Postma A., Caruso F., Biomacromolecules 2012, 13, 2225–2228. [DOI] [PubMed] [Google Scholar]
- 259. Ju Y., Cui J., Müllner M., Suma T., Hu M., Caruso F., Biomacromolecules, 2015, 16, 807–814. [DOI] [PubMed] [Google Scholar]
- 260. Ju Y., Cui J., Sun H., Müllner M., Dai Y., Guo J., Bertleff‐Zieschang N., Suma T., Richardson J. J., Caruso F., Biomacromolecules, 2012, 17, 2268–2276. [DOI] [PubMed] [Google Scholar]
- 261. Zhang Y., Huo M., Zhou J., Yu D., Wu Y., Carbohydr. Polym. 2009, 77, 231–238. [Google Scholar]
- 262. Damodaran V. B., Murthy N. S., Biomater. Res. 2016, 20, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Ko R., Cadieux P. A., Dalsin J. L., Lee B. P., Elwood C. N., Razvi H., J. Endourol. 2008, 22, 1153–1160. [DOI] [PubMed] [Google Scholar]
- 264. Jiang S., Cao Z., Adv. Mater. 2010, 22, 920–932. [DOI] [PubMed] [Google Scholar]
- 265. Brault N. D., Gao C., Xue H., Piliarik M., Homola J., Jiang S., Yu Q., Biosens. Bioelectron. 2010, 25, 2276–2282. [DOI] [PubMed] [Google Scholar]
- 266. Yang W., Sundaram H. S., Ella J.‐R., He N., Jiang S., Acta Biomater., 2010, 40, 92–99. [DOI] [PubMed] [Google Scholar]
- 267. Statz A. R., Barron A. E., Messersmith P. B., Soft Matter 2008, 4, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Lau K. H. A., Sileika T. S., Park S. H., Sousa A. M. L., Burch P., Szleifer I., Messersmith P. B., Adv. Mater. Interfaces 2015, 2, 1400225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Statz A. R., Park J. P., Chongsiriwatana N. P., Barron A. E., Messersmith P. B., Biofouling 2008, 24, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Li L., Yan B., Zhang L., Tian Y., Zeng H., Chem. Commun. 2015, 51, 15780–15783. [DOI] [PubMed] [Google Scholar]
- 271. Wang X., Ye Q., Gao T., Liu J., Zhou F., Langmuir 2012, 28, 2574–2581. [DOI] [PubMed] [Google Scholar]
- 272. Lee M., Rho J., Lee D. E., Hong S., Choi S. J., Messersmith P. B., Lee H., ChemPlusChem 2012, 77, 987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Jia X., Ma Z. Y., Zhang G. X., Hu J. M., Liu Z. Y., Wang H. Y., Zhou F., J. Agric. Food Chem. 2013, 61, 2919–2924. [DOI] [PubMed] [Google Scholar]
- 274. Pan X. D., Qina Z., Yana Y. Y., Sadhukhana P., Polymer 2010, 51, 3453–3461. [Google Scholar]
- 275. Ryou M. H., Kim J., Lee I., Kim S., Jeong Y. K., Hong S., Ryu J. H., Kim T. S., Park J. K., Lee H.. Choi J. W., Adv. Mater. 2013, 25, 1570–1570. [DOI] [PubMed] [Google Scholar]
- 276. Fullenkamp D. E., He L., Barrett D. G., Burghardt W. R., Messersmith P. B., Macromolecules 2013, 46, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Tamarin A., Lewis P., Askey J., J. Morphol. 1976, 149, 199–221. [DOI] [PubMed] [Google Scholar]
- 278. Desmond K. W., Zacchia N. A., Waite J. H., Valentine M. T., Soft Matter 2015, 11, 6832–6839. [DOI] [PubMed] [Google Scholar]