

Abstract
The total synthesis of dehydromicrosclerodermin B and microsclerodermin J is described. Efficient approaches to the unusual amino acids in the target molecules were developed on the basis of a Negishi coupling (for Trp‐2‐CO2H) and Blaise reaction (for Pyrr). An incorrect assignment of the pyrrolidinone stereochemistry of both compounds was confirmed by synthesizing epimers of the proposed structures. The spectroscopic data of these epimers were in complete agreement with those for the naturally derived material.
Keywords: cross-coupling, cyclic peptides, pyrrolidinones, structure elucidation, total synthesis
Microsclerodermins A–M are a family of 13 macrocyclic peptides comprising a 23‐membered ring, which contains six amino acids, three or four of which (depending on the family member) are unique to the microsclerodermins. Microsclerodermins A–I were isolated by Faulkner and co‐workers from the marine sponges Microscleroderma and Theonella between 1994 and 2000.1 In 2012, Li and co‐workers reported the structures of microsclerodermins J and K, isolated from the marine sponge Microscleroderma herdmani,2 as well as the concomitant isolation of microsclerodermins A and B. Moreover, in a personal communication, Li also reported that microsclerodermin B was readily dehydrated during HPLC purification to dehydromicrosclerodermin B. Recently, intriguing studies on the isolation of microsclerodermins L, M, and D from the terrestrial myxobacteria Jahnella, Chondromyces, and Sorangium were reported by Müller and co‐workers, together with a coherent biosynthesis.3 Members of the family show strong antifungal activity against Candida albicans and cytotoxicity against the HCT‐116 cell line.
The microsclerodermin macrocycle contains six amino acids: a tryptophan derivative (Trp), sarcosine (Sar), a pyrrolidinone unit (Pyrr), a polyhydroxylated β‐amino acid, γ‐amino‐β‐hydroxybutanoic acid (GABOB), and glycine (Gly; Scheme 1). Throughout the family, the tryptophan, β‐amino acid, and GABOB units possess stereocenters with the same configurations. However, the pyrrolidinone fragment is reported to have different configurations depending on the family member. Microsclerodermins A and B are the only members of the family with the proposed 44S,45S configuration, and microsclerodermins J and K are the only others with the 45S configuration (for this comparison, numbering is taken from microsclerodermin B). In all nine other natural products, both C44 and C45 have been assigned as having the R configuration by both NMR and degradation experiments analogous to those performed on microsclerodermin A. Furthermore the R configuration of the pyrrolidinone stereocenter of microsclerodermin E was confirmed by total synthesis.4
Scheme 1.
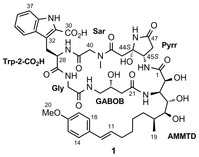
Proposed structure of microsclerodermin B.
The assignments of the C44 and C45 stereocenters of microsclerodermins B, J, and K were made by analogy to microsclerodermin A, and degradation experiments were not performed on these compounds. Another point related to the pyrrolidinone stereochemistry is that an inversion of the configuration at C45 during the biosynthesis of microsclerodermin M has been noted by Müller and co‐workers.3 With these observations in mind, we suspected that the stereochemical assignment of the pyrrolidinone unit in microsclerodermins B, J, and K might be incorrect.
The most challenging unit to prepare of any family member is the polyhydroxylated β‐amino acid residue, as this unit possesses four or five contiguous stereocenters and variation in the side chains. Several groups have reported studies towards the synthesis of these β‐amino acids.5 For example, we used tethered aminohydroxylation (TA) as the key step in a synthesis of the AMMTD fragment.6 To date the only total synthesis of any microsclerodermin was reported by Zhu and Ma for microsclerodermin E, which possesses a simpler side chain than AMMTD (AETD) and an unsaturated R‐configured pyrrolidinone core.4 The pyrrolidinone aminal moiety at C44 is extremely sensitive to basic or acidic conditions1 and as yet has not been incorporated into a total synthesis. Therefore, the original target of our research was microsclerodermin B (proposed structure 1; Scheme 1).
After preliminary studies, we planned to introduce the sensitive C44 hydroxy group at a late stage of the synthesis of 1 by using a two‐step procedure to hydrolyze the pyrrolidinone double bond: hydroxybromination of the alkene and subsequent debromination (Scheme 2). To perform this process selectively, it was decided that the terminal styrene group should be installed by cross‐metathesis after hydration of the pyrrolidinone unit to avoid competing hydroxybromination of the styrene alkene. The remaining retrosynthesis of 2 involved disconnections at the amide bonds with macrolactamization at the least hindered C25–N24 site.
Scheme 2.
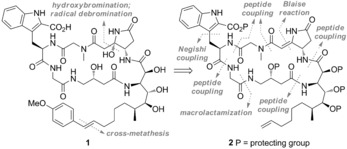
Retrosynthetic analysis of microsclerodermin B (1).
Previously we reported the construction of the AMMTD fragment by a Sharpless AD reaction to install the C4–C5 diol and a TA protocol for the C2–C3 amino alcohol.6 Substrate 3 was obtained in 16 steps from the S Roche ester6 and converted into amino alcohol 4, ready for connection with the GABOB fragment (Scheme 3). The GABOB amino acid was prepared following the route reported by the research groups of Shioiri and Ma, whereby azide 5 was synthesized in four steps from (R)‐dimethyl malate.4, 5d The methyl ester was then hydrolyzed, followed by the reduction of the azide functionality and subsequent Fmoc protection of the generated amine. Formation of the C21–N3 amide bond was achieved via acid fluoride 7 in high yield (76 %). The primary alcohol of 8 was then oxidized to the carboxylic acid, which was required for coupling of the AMMTD–GABOB dipeptide with the northern hemisphere.
Scheme 3.
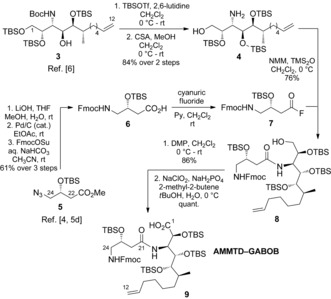
Synthesis of AMMTD–GABOB dipeptide 9. Boc=tert‐butoxycarbonyl, CSA=camphorsulfonic acid, DMP=Dess–Martin periodinane, Fmoc=9‐fluorenylmethoxycarbonyl, NMM=N‐methylmorpholine, Py=pyridine, Su=succinimide, TBS=tert‐butyldimethylsilyl, Tf=trifluoromethanesulfonyl, TMS=trimethylsilyl.
For construction of the northern hemisphere of 2, new approaches to target the tryptophan derivative and pyrrolidinone residues were developed. The key step in the synthesis of Trp‐2‐CO2R was based on a Negishi coupling between N‐Boc‐protected 3‐indole bromide 11 and organozinc reagent 12, derived from an iodoalanine derivative (Scheme 4).7 The effectiveness of our route to Trp‐2‐CO2R is twofold:4 It does not require harsh reaction conditions and allows potential variation of the indole substitution pattern. The ee value of 13 was confirmed by analysis of the corresponding Mosher amides (see the Supporting Information). Trp‐2‐CO2R 13 was incorporated into tripeptide 17 by the consecutive removal of the protecting groups and coupling with sarcosine 14 and then glycine 16, respectively.
Scheme 4.
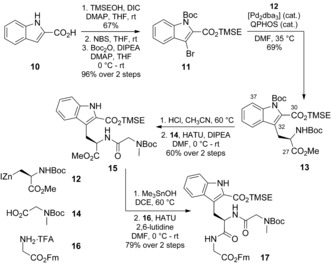
Synthesis of tripeptide 17. Bn=benzyl, dba=dibenzylideneacetone, DCE=1,2‐dichloroethane, DIC=N,N′‐diisopropylcarbodiimide, DIPEA=diisopropylethylamine, DMF=N,N‐dimethylformamide, Fm=9‐fluorenylmethyl, HATU=1‐[bis(dimethylamino)methylene]‐1H‐1,2,3‐triazolo[4,5‐b]pyridinium 3‐oxide hexafluorophosphate, NBS=N‐bromosuccinimide, QPHOS=1,2,3,4,5‐pentaphenyl‐1′‐(di‐tert‐butylphosphanyl)ferrocene, TFA=trifluoroacetic acid, TMSE=2‐(trimethylsilyl)ethyl.
For construction of the pyrrolidinone unit, a Blaise reaction was employed as the key step,8 which involved the attack of a tert‐butyl bromoacetate derived organozinc reagent onto nitrile 19 and subsequent lactamization to give 20 in 68 % yield with 95 % ee (determined by HPLC analysis on a chiral stationary phase; see the Supporting Information). This approach provided amino acid 21, ready for coupling, in 5 steps (Scheme 5), and thus compares favorably to the previously reported approach.4
Scheme 5.
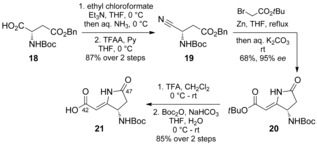
Synthesis of pyrrolidinone 21. TFAA=trifluoroacetic anhydride.
For completion of the northern hemisphere, the N‐Boc group was cleaved from 17 and the amine thus generated coupled with acid 21 (Scheme 6). Coupling of Gly‐Trp‐2‐CO2R‐Sar‐Pyrr tetrapeptide 22 with AMMTD–GABOB dipeptide 9 furnished a 1:1 mixture of linear hexapeptides 23 and 24 in 63 % yield, whereby the spontaneous loss of one of the TBS groups had occurred (23 and 24 were readily separable by column chromatography). Liberation of the C‐ and N‐termini for macrolactamization was achieved in unison with piperidine and was followed by cyclization in the presence of the phosphonium reagent PyAOP.
Scheme 6.
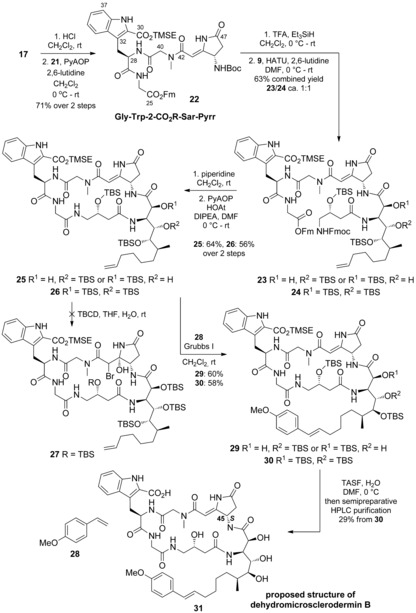
Synthesis of 31, the originally proposed structure of dehydromicrosclerodermin B. PyAOP=(7‐azabenzotriazol‐1‐yloxy)tripyrrolidinophosphonium hexafluorophosphate, HOAt=1‐hydroxy‐7‐azabenzotriazole, TBCD=2,4,4,6‐tetrabromo‐2,5‐cyclohexadienone, TASF=tris(dimethylamino)sulfonium difluorotrimethylsilicate.
According to our retrosynthetic plan, the next step was hydroxybromination of the pyrrolidinone double bond by a previously developed procedure. Unfortunately, when 26 was subjected to bromination (TBCD) in THF/H2O, the desired bromohydrin 27 was not observed. Although the starting material was consumed, only undefined and inseparable side products were obtained. After several unsuccessful attempts, the bromohydrin‐based end game had to be abandoned. However, because of the availability of advanced intermediates, our attention switched to the completion of a synthesis of dehydromicrosclerodermin B (31).
To prepare 31, we introduced the styrene moiety through cross‐metathesis of 25/26 with 4‐methoxystyrene, followed by global deprotection. For this cross‐metathesis reaction, a stoichiometric amount of the Grubbs I catalyst and a large excess of the styrene partner were required, probably owing to inhibition of the Ru catalyst as a result of metal ligation to the amide groups. Nonetheless, cross‐metathesis products 29 and 30 were obtained in good yields (60 and 58 %). The global deprotection was then performed with TASF. A sample of dehydromicrosclerodermin B was kindly provided by Li together with the original 13C NMR data (see the Supporting Information);2 this data allowed us to compare the synthesized structure 31 with the naturally derived compound.
Analysis of the NMR data for 31 provided unexpected results, as two rotameric forms were observed, in a ratio of approximately 3:1, although the NMR data for natural dehydromicrosclerodermin B do not show the presence of rotamers.9 When synthetic 31 was compared to dehydromicrosclerodermin B prepared by Li and co‐workers by 13C NMR spectroscopy,10 a large discrepancy in the data was observed for two regions of the molecule: the pyrrolidinone core and the GABOB unit (Δδ C up to 2.3 ppm; Figure 1). Furthermore, HPLC studies on synthetic 31 and the authentic sample (and a 1:1 mixture) clearly showed that these were two different compounds (see the Supporting Information).
Figure 1.
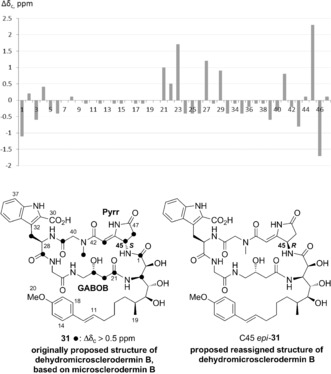
Comparison of the 13C NMR spectra of synthetic 31 and naturally derived dehydromicrosclerodermin B (note: C30, C31, and C32 were not identified in the spectrum of natural dehydromicrosclerodermin B; C31 and C32 were not identified in the spectrum of synthetic 31) and reassigned structure of dehydromicrosclerodermin B, C45 epi‐31.
Faulkner and co‐workers previously reported NOE correlations on related dehydromicrosclerodermin A.1a Interestingly, weak NOE correlations between the Pyrr and GABOB units were observed, thus suggesting that they are close in space. Consequently, we thought that stereochemical misassignment of either C45 or C23 might result in the observed discrepancy between the 13C NMR spectra. Given the lack of clarity regarding the configuration at C44–C45 in the microsclerodermins, we came to the conclusion that the C45 stereocenter may have been assigned incorrectly.1a To validate this idea, we pursued the synthesis of the C45 R epimer, C45 epi‐31.
Therefore, the 45R tetrapeptide C45 epi‐22 a was synthesized by using the protocol described above for the 45S‐configured analogue. As the route involving late‐stage selective hydroxybromination of the pyrrolidinone double bond had been discarded, the styrene moiety of the AMMTD unit was introduced earlier in the synthesis to avoid the problematic late‐stage cross‐metathesis (Scheme 7). By the use of the same reagents and conditions as for the AMMTD substrate containing the terminal alkene,6 cross‐metathesis product 33 was advanced to 34 and further to C45 epi‐31 (see the Supporting Information for details).
Scheme 7.
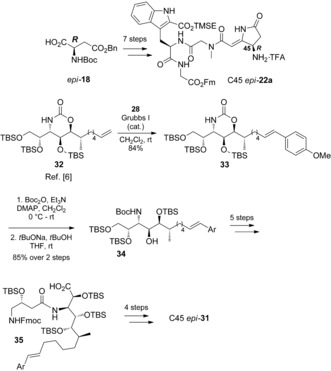
Synthesis of C45 epi‐31.
The NMR data for synthetic C45 epi‐31 was promising; the presence of rotamers was not observed. Comparison of the 13C NMR data of synthetic C45 epi‐31 and authentic dehydromicrosclerodermin B showed all signals in good agreement (Figure 2). For most of the signals, the difference does not exceed 0.1 ppm, including those for the pyrrolidinone and GABOB regions. Some signals, such as those for C28, C36, C38, C39, C40, and C41, differ by 0.2–0.3 ppm.
Figure 2.
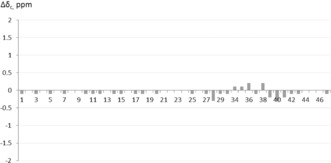
Comparison of the 13C NMR spectra of synthetic C45 epi‐31 and natural dehydromicrosclerodermin B.
Further evidence that synthetic C45 epi‐31 matched the naturally derived compound was secured by 1H NMR and HPLC analysis of a 1:1 mixture of the two samples; complete overlap of the peaks was observed (see the Supporting Information). On the basis of this analysis, it was concluded that structure 31 proposed for dehydromicrosclerodermin B, and, by extension, structure 1 proposed for microsclerodermin B are incorrect, and that both compounds have the R‐configured C45 stereocenter.
The incorrect assignment of microsclerodermin B mandates reassignment of the C44 stereocenter of structurally close microsclerodermin J (proposed structure 36; Scheme 8);11 the configuration of this compound was originally assigned by Li and co‐workers, in part by comparison with the incorrect structure of microsclerodermin B.2b We thus suspected that the correct structure of microsclerodermin J was actually C44 epi‐36.
Scheme 8.
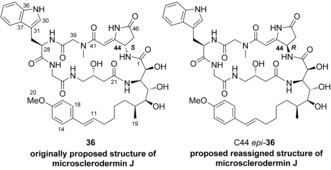
Originally proposed structure of microsclerodermin J, 36, and proposed reassigned structure of microsclerodermin J, C44 epi‐36.
The synthesis of C44 epi‐36 was similar to that used for dehydromicrosclerodermin B. Tryptophan derivative 37 was incorporated into the synthesis of Gly‐Trp‐Sar tripeptide 39 by peptide‐bond formation and protecting‐group removal (Scheme 9). The synthesis of C44 epi‐36 was completed by using the protocol described above for C45 epi‐31 (see the Supporting Information for details).
Scheme 9.
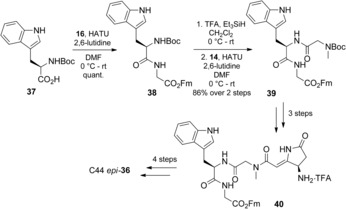
Synthesis of the reassigned structure of microsclerodermin J, C44 epi‐36.
No rotameric signals were evident in the NMR spectra of C44 epi‐36. Direct comparison of the 13C NMR data of C44 epi‐36 to that of natural microsclerodermin J was aided by a copy of the 13C NMR spectrum provided by Li. This spectrum was critical to our analysis, as there were minor inaccuracies in the data listings given in the original isolation paper (see the Supporting Information for a copy of the 13C NMR spectrum and a list of inaccuracies in the published 13C NMR data for microsclerodermin J).2a This comparison showed a complete match between the 13C NMR spectra of synthetic C44 epi‐36 and the natural material, with the differences in chemical shift not exceeding 0.1 ppm. From these data, it was confirmed that microsclerodermin J also has the R configuration at the C44 stereocenter.
To conclude, the first total synthesis of the proposed structure 31 of dehydromicrosclerodermin B was accomplished. The originally proposed C45 configuration for the parent compound 1 was reassigned from 45S to 45R, and this configuration was confirmed by synthesizing C45 epi‐31, whose data were in complete agreement with those for naturally derived dehydromicrosclerodermin B. We also reassigned the C44 configuration of an analogous member of the family—microsclerodermin J—as 44R by completing the first total synthesis of C44 epi‐36. Owing to our unsuccessful efforts to construct the sensitive pyrrolidinone aminal moiety through a hydroxybromination sequence, alternative hydration strategies will be pursued in the future for the total synthesis of microsclerodermin B.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Hill Foundation and the Society of Chemical Industry (E.Y.M.), the German Academic Exchange Service (DAAD; C.W.), and the EPSRC (R.D.C.P.) for funding. We also thank Professor Li for his kind cooperation in providing a sample and NMR spectra of dehydromicrosclerodermin B as well as NMR spectra of microsclerodermin J, H. Xu for preliminary experiments, and Dr. T. Parsons for help with HPLC analysis.
E. Y. Melikhova, R. D. C. Pullin, C. Winter, T. J. Donohoe, Angew. Chem. Int. Ed. 2016, 55, 9753.
References
- 1.
- 1a. Bewley C. A., Debitus C., Faulkner D. J., J. Am. Chem. Soc. 1994, 116, 7631; [Google Scholar]
- 1b. Schmidt E. W., Faulkner D. J., Tetrahedron 1998, 54, 3043; [Google Scholar]
- 1c. Qureshi A., Colin P. L., Faulkner D. J., Tetrahedron 2000, 56, 3679. [Google Scholar]
- 2.
- 2a. Zhang X., Jacob M. R., Rao R. R., Wang Y. H., Agarwal A. K., Newman D. J., Khan I. A., Clark A. M., Li X. C., Res. Rep. Med. Chem. 2012, 2, 7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Zhang X., Jacob M. R., Rao R. R., Wang Y. H., Agarwal A. K., Newman D. J., Khan I. A., Clark A. M., Li X. C., Res. Rep. Med. Chem. 2013, 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoffmann T., Müller S., Nadmid S., Garcia R., Müller R., J. Am. Chem. Soc. 2013, 135, 16904. [DOI] [PubMed] [Google Scholar]
- 4. Zhu J., Ma D., Angew. Chem. Int. Ed. 2003, 42, 5348; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 5506. [Google Scholar]
- 5.
- 5a. Sasaki S., Hamada Y., Shioiri T., Tetrahedron Lett. 1997, 38, 3013; [Google Scholar]
- 5b. Sasaki S., Hamada Y., Shioiri T., Tetrahedron Lett. 1999, 40, 3187; [Google Scholar]
- 5c. Sasaki S., Hamada Y., Shioiri T., Synlett 1999, 4, 453; [Google Scholar]
- 5d. Shioiri T., Sasaki S., Hamada Y., ARKIVOC, 2003, 103; [Google Scholar]
- 5e. Chandrasekhar S., Sultana S. S., Tetrahedron Lett. 2006, 47, 7255; [Google Scholar]
- 5f. Shuter E. C., Duong H., Hutton C. A., McLeod M. D., Org. Biomol. Chem. 2007, 5, 3183; [DOI] [PubMed] [Google Scholar]
- 5g. Hjelmgaard T., Faure S., Lemoine P., Viossat B., Aitken D. J., Org. Lett. 2008, 10, 841; [DOI] [PubMed] [Google Scholar]
- 5h. Tarrade-Matha A., Valle M. S., Tercinier P., Dauban P., Dodd R. H., Eur. J. Org. Chem. 2009, 5, 673; [Google Scholar]
- 5i. Davies S. G., Fletcher A. M., Foster E. M., Lee J. A., Roberts P. M., Thomson J. E., J. Org. Chem. 2013, 78, 2500; [DOI] [PubMed] [Google Scholar]
- 5j. Burnett C. M., Williams R. M., Tetrahedron Lett. 2009, 50, 5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pullin R. D. C., Rathi A. H., Melikhova E. Y., Winter C., Thompson A. L., Donohoe T. J., Org. Lett. 2013, 15, 5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tanaka M., Hikawa H., Yokoyama Y., Tetrahedron 2011, 67, 5897. [Google Scholar]
- 8. Hoang C. T., Bouillere F., Johannesen S., Zulauf A., Panel C., Pouilhes A., Gori D., Alezra V., Kouklovsky C., J. Org. Chem. 2009, 74, 4177. [DOI] [PubMed] [Google Scholar]
- 9.Similar rotameric NMR data were observed for protected cyclic peptides 25, 26, 29 and 30.
- 10.The original copies of the 1H NMR data provided by Li for natural dehydromicrosclerodermin B and natural microsclerodermin J were not informative owing to the presence of broad peaks. Consequently, comparison was best achieved by using 13C NMR data.
- 11.The original stereochemical assignment of the pyrrolidinone unit of microsclerodermins A and K is also questionable. However, owing to the presence of the C46 hydroxy group in these microsclerodermins, their synthesis was not pursued in the current study.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary