

Summary
Human mucosal melanoma (MM), an uncommon, aggressive and diverse subtype, shares characteristics with spontaneous MM in dogs. Although BRAF and N‐RAS mutations are uncommon in MM in both species, the majority of human and canine MM evaluated exhibited RAS/ERK and/or PI3K/mTOR signaling pathway activation. Canine MM cell lines, with varying ERK and AKT/mTOR activation levels reflective of naturally occurring differences in dogs, were sensitive to the MEK inhibitor GSK1120212 and dual PI3K/mTOR inhibitor NVP‐BEZ235. The two‐drug combination synergistically decreased cell survival in association with caspase 3/7 activation, as well as altered expression of cell cycle regulatory proteins and Bcl‐2 family proteins. In combination, the two drugs targeted their respective signaling pathways, potentiating reduction of pathway mediators p‐ERK, p‐AKT, p‐S6, and 4E‐BP1 in vitro, and in association with significantly inhibited solid tumor growth in MM xenografts in mice. These findings provide evidence of synergistic therapeutic efficacy when simultaneously targeting multiple mediators in melanoma with Ras/ERK and PI3K/mTOR pathway activation.
Keywords: trametinib, dactolisib, BRAF and NRAS wild type, preclinical model, dog, melanoma
Significance.
Frequent activation of one or both RAS/ERK and PI3K/mTOR in human and canine MM is analogous to human cutaneous melanomas. Pathway inhibitor mechanisms of action in canine MM mimicked known drug actions for several human cancers and denote a likely therapeutic benefit for affected dogs and humans. Therefore, naturally occurring canine melanoma, considered favorable for piloting human therapies, particularly for non‐BRAF and non‐NRAS mutated tumors, is further validated as a surrogate‐clinical trial translational model. Capabilities exist to develop combined targeted interventions in treatment‐naive and disease recurrence settings in dogs for devising improved outcomes in humans and dogs with melanoma.
Introduction
Mucosal melanoma (MM) is a rare form of melanoma comprising about 1% of human melanomas. MM may arise from any mucosal surface, primarily involving head and neck, anorectal and vulvovaginal sites. The biology of MM is distinct from cutaneous melanoma (Mclaughlin et al., 2005; Siegel et al., 2012). MM is typically invasive and malignant with a higher frequency of regional lymph node involvement than cutaneous melanoma (Chang et al., 1998). MM patient prognosis is poor due to its aggressive behavior, initially asymptomatic characteristics and relatively occult anatomic locations (Carvajal et al., 2012; Chang et al., 1998). Well‐characterized druggable targets have not been defined conclusively, and responses of MM to experimental kinase inhibitors and immunotherapy vary greatly; the small number of patients makes it difficult to accrue sufficient cohorts for clinical trials aimed at improving MM treatment effectiveness (Tacastacas et al., 2014). Lack of well‐characterized human MM cell lines and preclinical animal models further inhibits therapeutic optimization.
Numerous clinical and histopathological similarities between human and canine MM were recognized by a Comparative Melanoma Tumor Board consensus study on the human relevance of MM in pet dogs (Simpson et al., 2014). Here, we investigated naturally occurring canine MM for its preclinical potential to inform treatment regimens for both human and canine MM. Canine melanomas from all anatomic sites affect roughly 19, 000 dogs a year in the United States (Bosenberg et al., 2014). Spontaneously occurring melanomas are commonly present clinically in the skin, oral cavity, eye, and on ungual (toenail) tissue of dogs. Canine cutaneous melanocytic lesions are mostly benign, while the majority of oral and toenail/acral melanomas are malignant and frequently metastasize to regional lymph nodes or lungs, analogous to human melanomas (Smedley et al., 2011). In addition to sharing most clinical and histopathological features of human MM, both human and canine MM are, or become, resistant to chemotherapy and radiation treatment and exhibit similar propensities to also metastasize to brain and other visceral organs (Bergman, 2007; Carvajal et al., 2012; Gillard et al., 2014; Simpson et al., 2014).
While more than 50% of human cutaneous melanomas harbor BRAF mutations, typical BRAF and N‐RAS hot spot mutations are uncommon in human MM (Tacastacas et al., 2014). Similarly, orthologous N‐RAS and BRAF mutations do not appear to be significant drivers of canine MM (Fowles et al., 2015; Shelly et al., 2005). Mutation or amplification of c‐Kit has been reported in human MM (Carvajal et al., 2011; Curtin et al., 2006). Elevated c‐Kit expression has also been reported in canine MM; however, its relationship to c‐Kit mutation remains to be defined (Chu et al., 2013; Gomes et al., 2012; Murakami et al., 2011; Newman et al., 2012). Despite deficits in current understanding of the underlying genetic aberrations in most MM, the many characteristics common to MM in humans and dogs provided the rationale to test targeting signaling events associated with tumor growth in both species using a preclinical approach (Fowles et al., 2015; Simpson et al., 2014).
In the current study, we demonstrate signs of activation for multiple mediators of one or both of the RAS/extracellular signal‐regulated kinase (ERK) and phosphoinositide‐3 kinase (PI3K)/mammalian target of rapamycin (mTOR) pathways in most primary canine and human MM examined. This led to the investigation of combined signaling pathway inhibition on the growth of canine MM cells in vitro and as established subcutaneous tumors in preclinical mouse studies. Directed at these goals, GSK1120212, an allosteric inhibitor of mitogen‐activated protein kinase kinase (MEK) was selected for its ability to inhibit activation and the function of MEK. GSK1120212 significantly improved the progression‐free survival of human patients receiving chemotherapies from 1.5 to 4.8 months (Flaherty et al., 2012). GSK1120212 (Trametinib) was approved by the US Food and Drug Administration in 2014 for treating unresectable or metastatic melanoma having BRAFV600E or BRAFV600K mutations, and has been used alone or in combination with Dabrafenib, a BRAF inhibitor. As BRAF mutation is rare in human and canine MM (Tacastacas et al., 2014), we combined GSK1120212 with NVP‐BEZ235 (Dactolisib), a dual PI3K and mTOR inhibitor. NVP‐BEZ235 has been used to treat solid tumors in preclinical and clinical trials (http://www.cancer.gov/clinicaltrials/search/results?protocolsearchid=7039873). In addition to cytotoxic/cytostatic effect, NVP‐BEZ235 has been shown to sensitize tumor cells to other treatments (Kwei et al., 2012; Yasumizu et al., 2014). The combined targeted approach employed in this study led to signaling pathway modulation in association with reduced MM cell growth in vivo and in vitro. This represents a step toward improving current management of MM and establishing naturally occurring melanoma in the dog as a clinical surrogate for developing human melanoma therapeutics.
Results
Patterns of RAS/ERK and PI3K/AKT/mTOR pathway activation are similar in canine and human primary mucosal melanoma
The activation status of RAS/ERK and PI3K/AKT/mTOR was assessed in a panel of human and canine melanomas by immunohistochemistry (IHC) on tissue microarrays (TMA), in preparation for testing the feasibility of targeting downstream effectors of aberrant receptor tyrosine kinase signaling in MM therapy. Primary canine and human MM have similar patterns of activation in AKT, mTOR, and ERK (Figure 1A). Canine and human MM were immunopositive for p‐AKT, at a frequency of 41/43 (95%) dogs and 31/40 (78%) humans, respectively. p‐ERK 1/2 was immunopositive in 33/43 (77%) of canine and 21/37 (57%) of human MM (Figure 1B). Activation of mTOR and its downstream effectors was nearly universal in canine and human MM. Activation of mTOR occurred at frequencies of 43/43 (100%) dogs, 35/40 (88%) humans; p‐S6 at 43/43 (100%) dogs and 35/40 (88%) humans; and eIF4E at 43/43 (100%) dogs, and 33/40 (83%) humans (Figure 1B). Based upon the MM TMAs, both species show similar diverse combinations of RAS/ERK and AKT/mTOR signaling, although frequencies of signaling activation were greater for dogs than for humans. The manner with which surgical specimens are procured can impact preservation of labile tissue antigens. All canine melanomas were obtained according to a uniform sampling and preservation biospecimen protocol, which may have contributed to the comparatively greater frequency of immunopositive signaling detected in canine MM.
Figure 1.
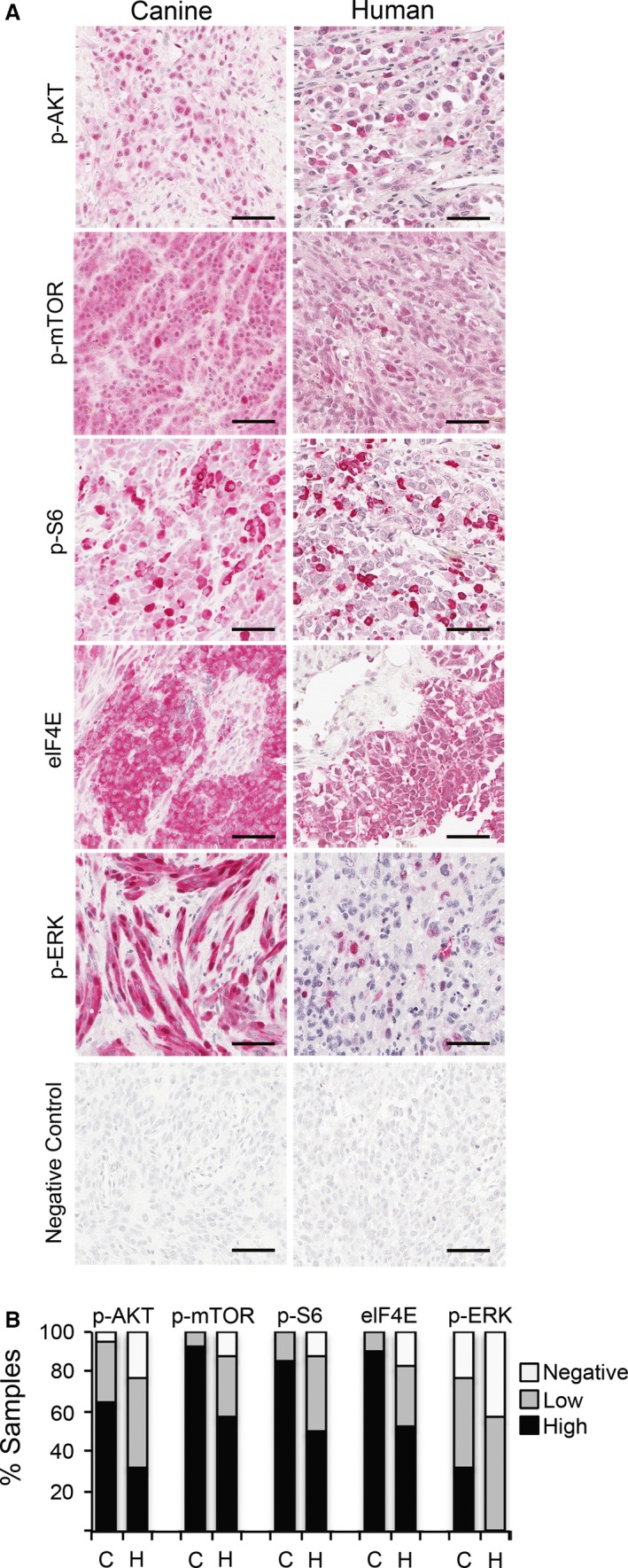
RAS/ERK and PI3K/AKT/mTOR signal transduction pathway activation in human and canine mucosal melanoma detected by immunohistochemistry on tissue arrays. (A) Representative immunopositive reactions (red chromogen, hematoxylin counterstain) for multiple pathway mediators are illustrated, as labeled. Bars = 50 μm. (B) Percentages of immunopositive melanomas for selected canine (C, n = 43) and human (H, n = 40) signal transduction mediators. Relative intensity and percentages of immunolabeled cells were considered in scores, assigned as negative, low, and high (see Methods).
To further document the activities of RAS and PI3K signaling in canine MM, phosphorylation of ERK and AKT were analyzed from 28 flash frozen primary tumors and seven MM cell lines from dogs by Western blot (WB) (Figures 2A and S1). ERK activation was evident in 19/28 (68%) of these canine patient tumors while 12/28 (43%) exhibited detectable AKT activation; prominently activated AKT and ERK occurred together in 9/28 (32%) of the primary canine melanomas examined, and 6/28 (21%) showed minimal activities in either protein. Overall, clear evidence of ERK and/or AKT activation occurred in 22/28 (79%) of the primary tumor samples. The activation states of ERK and AKT were similarly variable among canine MM cell lines (Figure 2B).
Figure 2.
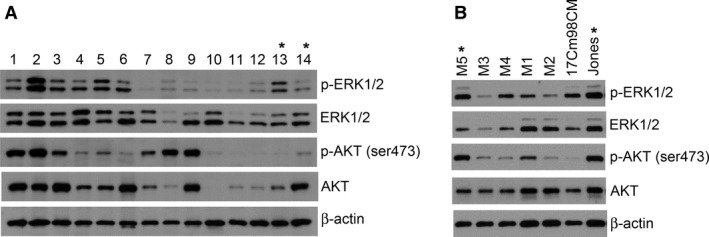
Canine melanomas exhibit a diverse range of ERK1/2 and AKT activities. To explore the potential of targeting MEK and PI3K/mTOR signaling cascades in canine melanomas, the activation status of effectors ERK1/2 and AKT, respectively, were analyzed by WB in (A) a panel of primary canine melanomas and (B) cell lines derived from canine melanomas. *Specimens or cell lines harboring N‐RAS Q61 mutation. No other RAS or BRAF hot spot mutations were found. Lane numbers indicate unique canine patient primary tumor samples.
BRAF V600 mutation, commonly associated with human cutaneous melanoma, was not detected by sequence analysis in primary canine MM, benign oral melanocytic tumors, and in melanoma cell lines (data not shown), a finding consistent with most human MM. N‐RAS Q61 mutation occurred in two primary canine melanoma samples and also in 2 MM cell lines (M5 and Jones) (data not shown). Canine MM cell lines M5 and Jones exhibited relatively greater ERK activities than the other five cell lines examined (Figure 2B). However, the presence or absence of N‐RAS mutations did not appear to be predictive of downstream ERK activities in primary tumor samples. In the two tumors with N‐RAS mutations, basal ERK activities were not greater than other tumors harboring wild‐type Ras. Two additional primary tumors that had an E62N mutation in N‐RAS also did not exhibit greater level of ERK activities.
Synergistic cytotoxicity was observed by targeting MEK and PI3K/mTOR simultaneously
Taking advantage of the diverse ERK and AKT activation status in canine melanoma cell lines, we tested the potential therapeutic value of targeting RAS/ERK and PI3K/mTOR signaling pathways in MM. The inhibitory effect of NVP‐BEZ235, a dual PI3K/mTOR inhibitor, on AKT phosphorylation was dose‐ and time‐dependent (Figure S2). Five canine melanoma cells were treated with graded dose concentrations of NVP‐BEZ235 for 4 and 24 h. Reduced AKT activity was observed at 4 h. At 24 h, the level of p‐AKT in treated cells remained low at the greater NVP‐BEZ235 drug concentrations, while AKT activation in cells exposed to less concentrated doses appeared near basal levels (Figure S2). ERK activity was assessed to gauge the efficacy of GSK1120212, a MEK inhibitor. No detectable ERK activity was observed 4 and 24 h at all doses tested (Figure S2).
Five canine melanoma cell lines with different levels of ERK and AKT activation were used to test whether or not the intrinsic ERK and AKT activity would influence potential for targeted inhibitors to impact cell growth. In addition, targeting both signaling pathways simultaneously was investigated to assess potential for increased efficacy. Cells in culture were treated with GSK1120212 and NVP‐BEZ235 individually or in combination at a 1:1 molar ratio, for 72 h. Individually, both drugs had dose‐dependent inhibitory effects on the growth of all five canine MM cell lines tested regardless of the variation in their basal levels of ERK or AKT activities (Figure 3A). NVP‐BEZ235 exhibited minimal potency at concentrations of 10 nM and below; a clear dose concentration effect was observed at doses above 10 nM. In contrast, GSK1120212 showed effects on three of five MM cell lines at doses <10 nM. Maximum growth inhibition was reached at approximately 125 nM. When the two treatments were combined, increased efficacy compared to either individual agent was observed for all five MM cell lines at approximately ≥50 nM concentration (Figure 3A). A Chou‐Talalay analysis revealed combination indices (CI) <1.0 for all five MM cell lines (Table 1), indicating synergistic inhibition by the two drugs in combination, compared to single‐agent treatment. All MM lines responded to the combination therapy in a similar manner, suggesting that the level of ERK and AKT activation in MM was not predictive of the synergistic efficacy of the two inhibitors.
Figure 3.
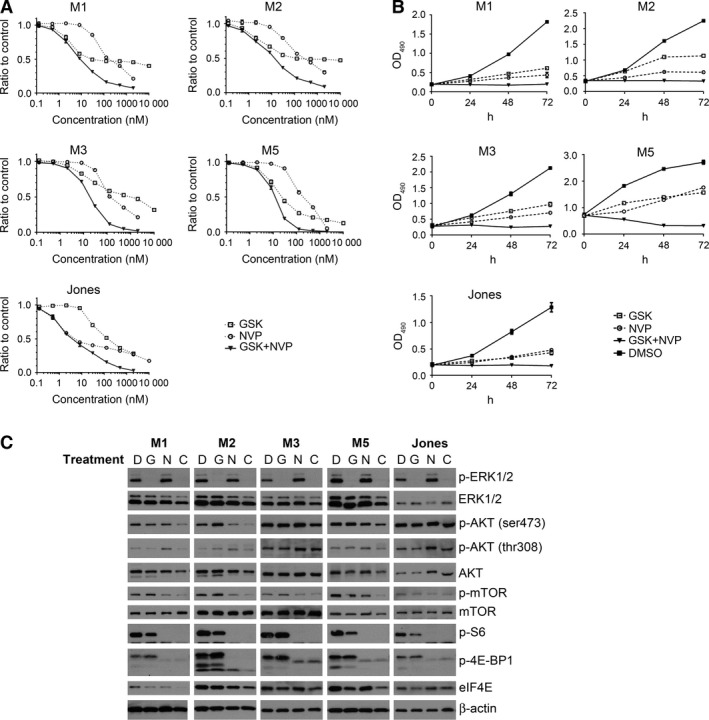
Diminished survival and pathway mediator target inhibition in five MM cell lines exposed to the combination of GSK1120212 and NVP‐BEZ235 (GSK+NVP, or C), compared to single‐agent treatment (GSK or NVP). All experiments involved the addition of drug(s) once at time 0 followed by incubation. (A) MM cell lines, M1, M2, M3, M5, and Jones, were treated with escalated doses of NVP‐BEZ235, GSK1120212, or a combination of the two drugs at 1:1 molar ratio. Cells were treated for 72 h and MTS assay was performed to determine the cell titer. Ratios of inhibitor‐treated cells to DMSO‐treated control cell titers were calculated; average of six wells for each indicated inhibitor concentration was plotted. (B) Cells were treated for 24, 48, and 72 h at a concentration near IC 50 determined in (A), or DMSO control. Relative MTS viability assay (formazan product detected at OD 490 in supernatants) was plotted to show cell titers at different time points. All treatments were significantly different from control treatment at 24 h and later (P < 0.05). (C) The inhibitory effect of the two drugs, as either single agents (G or N) or in combination (C), on the signaling effectors of RAS/ERK and PI3K/AKT/mTOR pathways was investigated. Cells were treated once at time 0 and incubated for 24, 48, and 72 h at a concentration near IC 50 of either drugs, or DMSO control (D). Data shown for 48‐hour treatment.
Table 1.
Synergistic effects of MEK and PI3K/mTOR inhibitors on canine mucosal melanoma cell lines
| Cell Line | CI at fa50 | CI at fa90 |
|---|---|---|
| UCDK9M1 | 0.14 | 0.09 |
| UCDK9M2 | 0.065 | 0.037 |
| UCDK9M3 | 0.12 | 0.089 |
| UCDK9M5 | 0.15 | 0.024 |
| Jones | 0.21 | 0.03 |
Combination indices (CI) of Chou‐Talalay computation, shown for 50% and 90% affected fraction (fa) values at 72 h post‐exposure in vitro, for five mucosal melanomas, provide quantitative definition for drug additive effect (CI = 1), synergism (CI < 1), and antagonism (CI > 1) in combination drug therapy.
Drug inhibitory kinetics of MM cell proliferation/survival was evaluated next. Cells were treated with individual inhibitors, or in combination, for 24, 48, and 72 h; a dose near the 50% inhibitory concentration (IC50) of each agent at 72 h was used. MTS assays were used to assess cell survival titer, and the activation status of targeted pathway signaling events was evaluated by WB. Individual drug treatments resulted in reduced cell growth over 72 h, while vehicle (DMSO)‐treated control cells grew steadily (Figure 3B). Combination treatment inhibited growth in all five MM cell lines, evident through static or declining cell titers over 72 h compared to the initial pretreatment cell titer and control‐treated cells (Figure 3B), indicating cell death or cell cycle arrest. Results for the three time points were similar for all five MM cell lines.
Target modulation of MAPK and AKT signaling pathway nodes was assessed at three time points over 72 h of exposure to single drugs and drug combination. GSK1120212 abolished ERK1/2 phosphorylation in each MM cell line (Figure 3C, all data shown for 48 h treatment). GSK1120212 had no consistent or apparent effect on AKT and mTOR activities, which remained relatively comparable with vehicle control‐treated cells, or slightly greater in some cases of AKT, at all three time points for all cell lines tested. In some cell lines, GSK1120212 diminished p‐S6, a downstream effector for both MAPK and AKT signaling pathways, although the effect was limited.
The inhibitory effect of NVP‐BEZ235 alone on PI3K/AKT and mTOR appeared to be less distinct than the GSK1120212 effect on ERK signaling. NVP‐BEZ235 reduced mTOR phosphorylation at 24 h, and this suppression was generally sustained to 72 h (Figure 3C, 48‐hour treatment data shown). NVP‐BEZ235 inhibition of p‐AKT was transient and variable (Figures 3 and S2). Inhibitor compound degradation in culture medium was eliminated as an explanation for the less than robust influence on p‐AKT. Consistent levels of NVP‐BEZ235 were detected by mass spectrometry in cultures at 6, 24, and 48 h following the addition of inhibitor, while maximal decline in NVP‐BEZ235 concentration at 72 h was <15% (data not shown). Despite the apparent weaker effect on more proximate signaling nodes PI3K/AKT and mTOR, NVP‐BEZ235 exhibited a more pronounced effect on PI3K/AKT/mTOR pathway downstream effectors (Figure 3C). p‐S6 was considerably moderated upon the treatment with NVP‐BEZ235. This implicates mTOR as the dominant effector on S6 activity in MM. AKT/mTOR inhibition also reduced the level of phosphorylated 4E‐BP1, suggesting an inhibitory effect on eIF4E function and perhaps on the level of translation initiation. In addition, NVP‐BEZ235 treatment (IC50) was associated with increased p‐ERK at 24 or 48 h in four of five cell lines (Figures 3C and S2). This apparent compensatory activation is consistent with cross talk among signaling pathways and may explain a potential for tumor resistance when NVP‐BEZ235 was used alone. Combination therapy retained the inhibitory effects on both pathways modulated by the individual drugs and generated further reduction in phosphorylation of signaling effectors examined (Figure 3). Importantly, no reciprocal activation was observed when both signaling pathways were inhibited simultaneously. The inhibitory effects of individual drug treatments were further potentiated, supporting the presence of a synergistic relationship between the two inhibitors in MM.
Apoptosis was induced in canine MM cells treated with GSK1120212; small shrunken cell aggregates appeared after 24 h along with activation of caspase 3/7 (Figure 4A–C). Percentages of active caspase 3/7 positive cells were significantly greater compared to DMSO‐exposed cells (Figure 4C). A population of sub‐G1 cell cycle events was also evident by flow cytometry in three of four MM cell lines tested, further indicating that GSK1120212 induced apoptosis (Figure S4). By contrast, NVP‐BEZ235 at IC50 impacted cell density, which was supported by MTS assay (Figure 3), but only minimally induced cell death (Figure 4A–C). Cell cycle analysis showed a G0/G1 population increase, indicating possible cell cycle arrest in NVP‐BEZ235‐treated cells (Figure S4). Exposure to the combination of GSK1120212 and NVP‐BEZ235 induced apoptosis, as supported by reduced cell density, shrunken morphology, and significant caspase 3/7 activation for all tested cells (Figure 4C).
Figure 4.
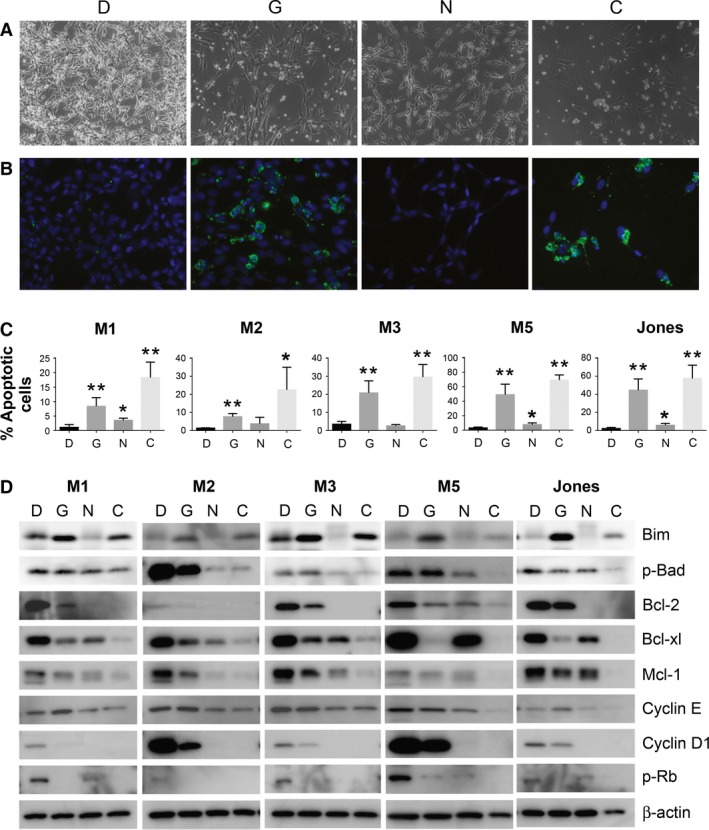
Diminished survival of MM cell lines exposed in vitro to GSK1120212 and NVP‐BEZ235 individually, or as a combination includes evidence of apoptosis, cell cycle arrest, as well as altered cyclins and Bcl‐2 family cell survival regulatory proteins. (A) Representative bright field contrast images of cultured M5 cells treated with GSK1120212 (G), NVP‐BEZ235 (N), or combination of the two agents (C), or DMSO control (D) for 48 h revealed varied cell densities. (B) Green fluorescence microscopy revealed cells positive for active caspase 3/7 in GSK1120212 (G) or combination‐treated cells (C), indicative of apoptosis. (C) Percentages of apoptotic cells assessed using fluorescent microscopy are plotted for each MM cell line exposed to (G) and (N), or the two agent combination (C). Differences significant from DMSO (D) control (*P < 0.05) or (**P < 0.005) are indicated. (D) Treated cells were analyzed by Western blot for the expression level of Bcl‐2 cell survival regulatory family member proteins, as well as cyclin D1, cyclin E, and p‐Rb, which are important for progression through G1 cell cycle phase. MM cells exposed to both inhibitors revealed synergy contributing to the diminished survival, compared to single‐drug therapy. Blots of cell lysates were prepared from cells treated for 48 h.
Western blot analyses revealed that both drugs were capable of tilting the balance of pro‐ vs. anti‐apoptosis Bcl‐2 family proteins in MM. Combination therapy increased expression of pro‐apoptotic member Bim in most cell lines (although less so than did GSK1120212 alone), and reduced pro‐survival factors including Bcl‐2, Bcl‐xl, and Mcl‐1 as well as the phosphorylated form of Bad (Figure 4D). NVP‐BEZ235 treatment inhibited the expression of cyclins D1 and E (Figure 4D), contributing to the G0/G1 cell cycle arrest (Figure S4). GSK1120212 treatment reduced the expression of cyclin D1 to various and typically lessor degrees than did NVP‐BEZ235, and did not result in similar apparent G0/G1 cell cycle arrest in the panels of canine MM cells. Both drugs combined, however, reduced basal expression of the cyclins D1 and E as well as the level of p‐Rb, an inactive form of the cell cycle suppressor. Overall, when the two drugs were used together, it appeared that they facilitated each other's antiproliferative effect and collaborated in increased cytotoxicity.
Combined inhibitor mediated arrest of solid MM tumor growth
We next tested whether the targeted cytotoxicity of the inhibitors observed in vitro could be translated into in vivo therapy. Female nude mice with established canine melanoma M5 xenografts (75–150 mm3) were treated with GSK1120212 (1.5 mg/kg) and NVP‐BEZ235 (30 mg/kg), as single agents or in combination. GSK1120212 alone at 1.5 mg/kg had a suboptimal therapeutic effect, as MM tumor sizes were similar to vehicle‐treated control mice (P > 0.5) (Figure 5A, B). NVP‐BEZ235 delayed tumor growth (P = 0.005 on day 10 of administration) compared to vehicle‐treated control mice (Figure 5B). By combining the two drugs, the tumor growth was inhibited to a greater extent than with either drug alone (P < 0.0005, treatment day 10) (Figure 5A, B). The doses used in this study are safe and well tolerated as no significant body weight loss was observed during the treatments (Figure S5).
Figure 5.
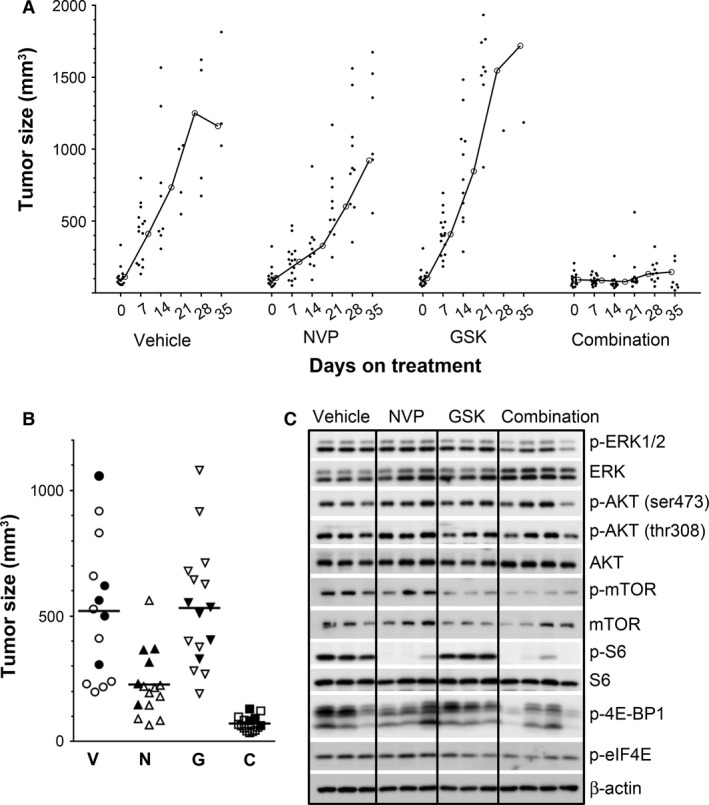
The combination of NVP‐BEZ235 and GSK1120212 inhibited the growth of MM xenografts. Nude mice bearing well‐established M5 melanoma cell xenografts (75–150 mm3) were randomized to receive daily oral doses of NVP‐BEZ235 at 30 mg/kg (N), GSK1120212 at 1.5 mg/kg (G), these two doses combined of the two agents (C), or drug vehicles as control (V), for up to 39 days, at which time tumor size in the majority of the control and individual inhibitor‐treated mice reached experimental end point. Tumor volumes were measured longitudinally. All data are from a single experiment and all mice were evaluated concurrently. (A) MM growth kinetics during treatment; data points represent individual mouse tumor volumes at day of treatment. Line tracings represent treatment group mean tumor volume over time of treatment. (B) Tumor volume measured 10 days after initiation of treatment showed significantly less growth of MM in NVP‐BEZ235‐treated mice (P = 0.005), and in mice receiving combination therapy (P < 0.0005). Data points represent individual mouse tumors. Bars represent treatment group mean tumor volume. (C) Five tumor bearing mice [indicated by solid symbols in (B)] were electively removed from each treatment group at 10 days following the initiation of treatment to analyze for target modulation efficacy. Representative Western blot analyses revealed inhibition of signaling nodes in tumors from mice treated for 10 days.
GSK11220212 treatment resulted in no noticeable reduction of p‐ERK in tumors harvested 10 days after treatment was initiated, as assessed by WB (Figure 5C). However, reduced p‐ERK level was detected in tumors from mice treated with the drug combination (Figure 5C), and the timing of this finding was associated with clear evidence of in vivo tumor growth inhibition (Figure 5A, B). NVP‐BEZ235 administered alone, or in combination with GSK11220212, inhibited mTOR phosphorylation in MM xenograft tumors, but had more modest effect on p‐AKT levels. The combination inhibitory effect on p‐AKT in vivo was more efficacious than NVP‐BEZ235 alone. Furthermore, downstream effectors S6, eIF4E, and 4E‐BP1 were diminished, particularly in drug combination‐treated mice, corroborating the target inhibition in MM cells treated in vitro (Figure 5C).
The suboptimal growth inhibition produced in vivo by the MEK inhibitor alone, compared to its efficacy in vitro, was investigated further by relating plasma GSK1120212 levels to p‐ERK in the melanoma xenografts from the cohort of mice examined at 10 days post‐treatment. Assessment of p‐ERK in the nuclear compartment of melanomas was undertaken by IHC for further insight into target modulation in solid tumors and served as a surrogate for MAPK‐stimulated proliferative potential. Plasma inhibitor concentrations were determined to be a reasonable proxy for tissue level of drug in other experiments (unpublished data). Significantly diminished nuclear p‐ERK in combination inhibitor‐treated mice, compared to mice treated with GSK1120212 alone, or vehicle control (Figure 6A–E), occurred in association with significantly greater plasma GSK1120212 in combination inhibitor‐treated mice (Figure 6F). Therefore, while GSK1120212 monotherapy at 1.5 mg/kg was suboptimal in treating MM in a mouse model, its combination with NVP‐BEZ235 was able to modulate oncogenic signaling events and inhibit solid tumor growth, thereby promoting the survival of animals. These findings help to support evidence for synergistic combination inhibitor therapy in vivo.
Figure 6.
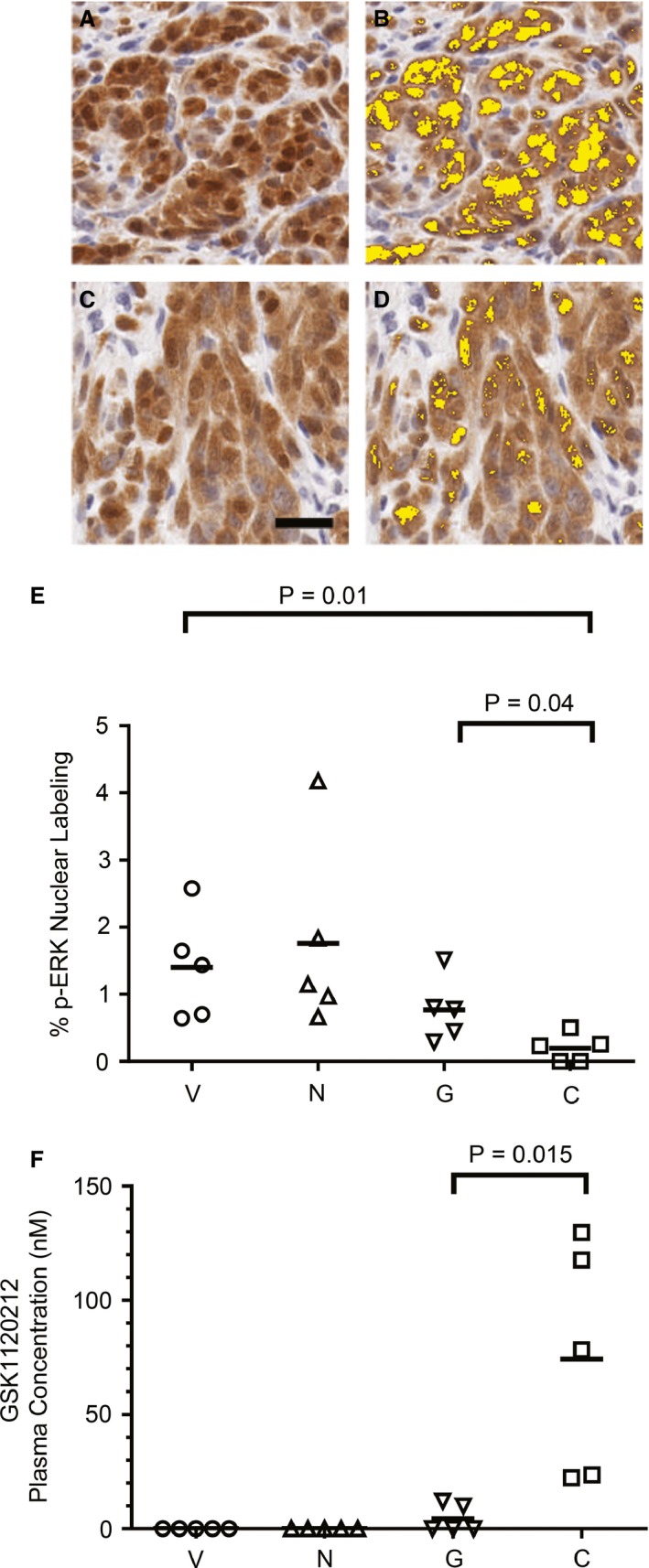
Melanomas from mice treated with the combination treatment had less nuclear p‐ERK in association with greater plasma GSK1120212 concentration. Data are derived from the same animal experiment as in Figure 5 from mice examined at 10 days of therapy. Photomicrographs of p‐ERK IHC in a melanoma from representative (A) vehicle‐only and (C) both GSK1120212 and NVP‐BEZ235 combination‐treated mice. Brown chromogen, hematoxylin counterstain. Bar = 10 um. (B) and (D) are images of the same microscopic field of view as in A and C, respectively, with post‐image analysis processing pseudo‐color markup. Nuclear p‐ERK expression is shown as yellow. (E) Nuclear expression of ERK activation quantified on a tumor area basis by image analysis for all treatment groups [V (vehicle), N (BEZ235), G (GSK1120212), and C (combination)]. Data points represent individual mouse tumors; bars represent mean of the treatment group. Percentage nuclear p‐ERK was significantly less in combination‐treated mice (C) than single‐agent treatment groups (N, G), or vehicle (V). (F) Plasma level of GSK1120212 was significantly greater in mice treated with combination GSK1120212 and NVP‐BEZ235 (C) than in mice treated with GSK1120212 alone (G) (P = 0.015). Data points represent individual mouse plasma drug concentrations. Bars represent treatment group mean GSK1120212 concentration. Values at zero represent findings below the quantifiable limit of the assay.
Discussion
Cancer therapies designed to concurrently inhibit multiple signaling pathways serve to address innate and acquired resistance to targeted therapies. Rational selection of agents dictates that valid model systems be engaged early in development for potential clinical translation. Spontaneous canine melanoma has been considered an optimal preclinical animal model for developing suitable strategies, particularly for MM (Fowles et al., 2015; Gillard et al., 2014; Simpson et al., 2014). Our findings amplify the validity of using spontaneous canine MM as a human cancer preclinical, or surrogate‐clinical model, and provide evidence of synergistic therapeutic efficacy in melanoma when simultaneously targeting multiple nodes in RAS/MEK and PI3K/mTOR pathways.
Animal models that present the entire spectrum of cancer from benign neoplasms to primary tumors and metastases are rare. There are distinct advantages to piloting therapeutics for both human and canine patients in clinically affected dogs with MM. Dogs with spontaneous melanoma are immune‐competent, more phylogenetically similar to humans than are rodents, and have a shorter natural life span and relatively brief disease course. It is feasible to enroll small veterinary patient cohorts amenable to frequent monitoring and sampling. These cohorts may be used for assessing target modulation, performing comparative genomics discovery, and for evaluating PK/PD parameters as well as for assessing patient outcomes. In addition, an absence of established standard of care for most canine cancers permits obtaining owner/client consent for implementing combination drug targeting approaches in treatment‐naive settings (Gordon et al., 2009; Paoloni and Vail, 2013). Distinct from genetically engineered mouse models, dogs represent a unique opportunity to investigate certain subtypes of naturally occurring spontaneous melanoma formation, progression, metastasis, and response to disease intervention in a large mammal species featuring analogous genetic and intertumoral diversity (Simpson et al., 2014). Canine preclinical models hold potential for early identification of more precise efficacy benefits for human patients.
The genetic mechanisms underlying RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathway activations in canine and human MM remain obscure. V600E BRAF mutation is common in human cutaneous melanomas but scarce in human MM (Curtin et al., 2005; Fowles et al., 2015; Gillard et al., 2014; Shelly et al., 2005; Tacastacas et al., 2014). BRAF mutations were absent in 46 primary canine MM evaluated by sequence analysis in this study. Correspondingly, the lack of BRAF mutations has been reported previously (Fowles et al., 2015; Gillard et al., 2014; Shelly et al., 2005), although more recent evidence suggests that canine melanomas may have an orthologous BRAF mutation in low frequency (3 of 54 cases) (Mochizuki et al., 2015). The relative frequency of the predicted glutamic acid substitution for valine in BRAF was conspicuously greater in cases of benign cutaneous melanocytomas, compared to the malignant melanomas reported (Mochizuki et al., 2015). Cutaneous melanocytic neoplasms arising from the haired‐skin in the dog are often benign and are not believed to be associated with ultraviolet solar radiation injury, in contrast to its association with BRAF mutation in human cutaneous melanomagenesis (Bergman et al., 2013; Besaratinia and Pfeifer, 2008; Goldschmidt and Hendrick, 2002). N‐RAS Q61 mutations in canine melanoma tumor tissues do appear more often than mutations in BRAF. The low incidence of N‐RAS mutation in this and other studies (Fowles et al., 2015; Gillard et al., 2014; Mayr et al., 2003), however, does not seem to account for the frequent ERK activation documented in canine melanoma. Notably, we did not find BRAF or N‐RAS mutations in the benign melanocytic proliferative lesions of the oral mucosa in dogs (analogous to blue nevi), which have low malignant potential (Esplin, 2008) and lack AKT and ERK activation (Simpson et al., 2014). While activation of RAS/ERK and PI3K/AKT/mTOR signaling would appear to strongly influence malignant behavior of both species’ melanocytes, connections between pathway activation and cancer genetics, including orthologous BRAF and N‐RAS mutations, remain yet to be defined.
Activation of one or both RAS/ERK and PI3K/AKT/mTOR pathways in the majority of MM from both humans and dogs examined overlaps with the pattern of similar activation in well‐documented cutaneous melanomas and other cancers (Bogenrieder and Herlyn, 2010; Fowles et al., 2015; Grazia et al., 2014; Karbowniczek et al., 2008; Margolin et al., 2012). The RAS/ERK and the PI3K/AKT/mTOR pathways are known to intersect with variable feedback and regulatory influences overlapping downstream (Ersahin et al., 2015; Mendoza et al., 2011; Shi et al., 2011; Van Dort et al., 2015). For example, mTOR inhibition has been shown to result in reciprocal increase in p‐ERK in human cancer (Bailey et al., 2014; Carracedo et al., 2008; Zhu et al., 2015). Corresponding findings in the current study revealed coordinate increased ERK activation in canine MM cell lines upon mTOR inhibition by the dual PI3K/mTOR inhibitor NVP‐BEZ235 in vitro. Likewise, AKT activation in melanoma has been shown to play a key role in acquired resistance to BRAF or MEK inhibitors (Atefi et al., 2011; Greger et al., 2012). Such interplay provides evidence for possible sources of drug resistance that may be circumvented by simultaneously targeting intersecting pathways, such as RAS/ERK and PI3K/AKT/mTOR. Combined inhibitor approaches in dogs with melanoma, such as the one detailed herein, may constitute opportunity for piloting combined targeted therapeutic development for human melanoma patients, as well as for enhancing therapeutic response in affected animals.
Further basis supporting the concept that dogs are ideal candidates for modeling human cancers was obtained by indications that the inhibitors used in these studies exerted mechanisms of actions similar to those in human cancer (Gilmartin et al., 2011; Maira et al., 2008). Individually, the GSK1120212 MEK inhibitor induced apoptosis in canine melanoma cells, while the NVP‐BEZ235 dual PI3K/mTOR inhibitor primarily led to G0/G1 cell cycle arrest at the IC50 concentration, consistent with known drug actions in human cells. Furthermore, both agents when combined not only maintained their own inhibitory effects on signaling effectors, but also potentiated each compound's cytotoxic effect, suggesting a cross‐inhibitory effect on melanoma when the two drugs are applied simultaneously. Downstream‐activated mediators in both pathways such as ERK, mTOR, S6, and 4E‐BP1 were appropriately inhibited by the combination of GSK1120212 and NVP‐BEZ235. Synergistic cytotoxicity and diminished cell titer were observed for all canine MM cell lines tested with the combination in vitro, and the responses due to inhibition of MEK and PI3K/mTOR activities appeared to be manifest regardless of the intrinsic activation level of RAS/ERK or AKT/mTOR. In addition, basal increased expression or activation of Bcl‐2 family member proteins in the treatment‐naive canine MM cell lines is also similar to human melanoma, and can be factors contributing to resistance against cancer treatment (Hartman and Czyz, 2013). Exposure of MM cell lines to the two inhibitors in combination was associated with decreased p‐Bad, Bcl‐2, Bcl‐xl, Mcl‐1, and increased Bim, an expression pattern of Bcl‐2 family proteins favoring apoptosis, which was evidenced in MM by caspase 3/7 activation and reduced cell survival. These findings help to elucidate the mechanism of successfully combined GSK1120212 and NVP‐BEZ235 therapeutic targeting of RAS/ERK and PI3K/AKT/mTOR in melanoma. The combination furthermore led to melanoma tumor stasis in xenograft experiments, where the modulation of some mediators was evident in both signal transduction pathways at 10 days following the start of treatment in mice.
Combining pathway inhibitors that are able to exert synergistic therapeutic effects with potential to minimize some consequences of acquired drug resistance is an attractive therapeutic strategy in clinical oncology. MM's typical presentation at more advanced stage, already in a vertical phase of growth and disseminated, coupled with the poor prognosis and limited therapeutic responses in both human and dogs, makes a continued search for more effective means to treat the disease warranted. Even though early clinical trials simultaneously targeting MAPK and PI3K/AKT/mTOR in human patients have been disappointing due to lack of demonstrable responses coupled with high toxicities (Tolcher et al., 2015a, b), newer generations of inhibitors may be proven to be safer with less adverse side effects. Combining two drugs may beneficially minimize side effects of any one inhibitor, although if similarly dosed as monotherapy might limit effectiveness. Despite some suboptimal efficacy when employed as single agents, we showed in this study that the combination of GSK1120212 and NVP‐BEZ235 is well tolerated and highly effective in a mouse xenograft model. Further studies about the nature of synergism observed in vivo are warranted, including investigations into drug–drug interactions and altered pharmacokinetic factors such as bioavailability, saturation, and clearance. For example, formulations used for delivery may influence bioavailability (Williams et al., 2011), and protein binding of one agent may alter distribution of another (Wanke et al., 2013); however, the precise mechanisms for the apparent biological synergy in vivo remain undefined. Moreover, this study provides substantial additional evidence of the benefits of preclinical, and likely surrogate‐clinical, development of combined pathway targeted melanoma therapies in dogs affected by naturally occurring MM, with potential application to human MM patients. As the combination was effective on a panel of MM cells regardless of the intrinsic MEK/ERK and PI3K/AKT/mTOR activities, this regimen may have application for a range of both human and canine MM patients. Definition of the underlying genetic alterations contributing to canine melanomagenesis should be investigated more thoroughly for this naturally occurring canine cancer to be most effectively combated and made most predictive as a human disease model. Evaluation could be accelerated in clinical trials of naturally occurring melanomas in dogs to facilitate greater understanding for human treatment (Rankin et al., 2012; Vail et al., 2009).
Methods
Canine melanoma specimens and cell lines
Canine melanoma cell lines were kindly provided by Dr. Michael Kent at University of California, Davis [UCDK9M1 (M1), UCDK9M2 (M2), UCDK9M3 (M3), UCDK9M4 (M4), UCDK9M5 (M5)], Drs. Jared Fowles and Dan Gustafson of Colorado State University, Fort Collins (Jones), and Dr. David Vail of University of Wisconsin, Madison (17Cm98CM). All cell lines are originally from oral mucosal melanomas. M2, M5, and 17Cm98CM lines are metastatic MMs derived from lymph nodes and M1 represents skin metastasis. Cells were cultured in DMEM with 10% FBS in 5% CO2 at 37°C. Frozen and formalin‐fixed paraffin‐embedded primary canine mucosal melanomas, sampled from pet dogs according to contemporary veterinary medical standards of care following owner/client informed consent, were obtained from the Pfizer‐Comparative Canine Oncology and Genomics Consortium (CCOGC) tissue specimen biorepository (http://ccogc.net/collection-process/). For inclusion in this study, tumor diagnoses of biorepository specimens were reconfirmed according to established consensus criteria (Simpson et al., 2014).
Antibodies and inhibitors
Antibodies against AKT, p‐AKTSer473, p‐AKTThr308, mTOR, p‐mTORSer2448, p‐4E‐BP1Thr37/46, eIF4E, p‐eIF4E, S6, p‐S6Ser235/236, ERK1/2, p‐ERK 1/2, p‐Rb, Bcl‐2 family proteins (including p‐BadSer112 (data shown) and Ser136), and cyclin E were obtained from Cell Signaling Technologies (Danvers, MA, USA). Anti‐cyclin D1 was from Abcam (Cambridge, MA, USA). Anti‐β‐actin was obtained from Sigma‐Aldrich (St. Louis, MO, USA). PI3K/mTOR dual inhibitor NVP‐BEZ235 (Dactolisib) was purchased from Selleck Chemical (Boston, MA, USA) and MEK inhibitor GSK1120212 (Trametinib) was purchased from ChemieTek (Indianapolis, IN, USA).
Western blotting
Snap‐frozen canine melanoma tumor specimens were homogenized in RIPA buffer (Cell Signaling) using a biovortexer (Bellco Glass, Inc., Vineland, NJ, USA). Cell lysates of cultured cell lines were generated by lysing cells in cell lysis buffer (Cell Signaling) for 20 min on ice. Tumor and cell lysates were cleared by centrifugation at 18 000 g for 10 min; supernatants were separated from cell debris for Western blot (WB) analyses. The cleared tumor and cell lysates were separated on 4–20% Tris‐glycine gels (Invitrogen) and transferred onto PVDF membranes (Bio‐Rad, Hercules, CA, USA). Membranes were probed with primary antibodies followed by respective horseradish peroxidase (HRP)‐conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA, USA). Immunoreactive bands were detected using chemiluminescence.
Polymerase Chain Reaction (PCR) amplification and sequencing
Genomic DNA was isolated from formalin‐fixed paraffin‐embedded (FFPE) tissues. The FFPE DNA kit (Qiagen, Valencia, CA, USA) was used to isolate genomic DNA from five scrolls of 7‐micrometer‐thick paraffin sections for each sample. PCR amplification focused on hotspot regions surrounding codon 600 in BRAF (forward 5′TGCTTGCTCTGATAGGAAAATG3′ and reverse5′CCACAAAATGGATCCAGACA3′), codon 12 of N‐RAS (forward 5′GACTGAGTACAAACTGGTGG3′ and reverse 5′GGGCCTCACCTCTATGGTG3′), and codon 61 of N‐RAS (forward 5′TCTTACCGAAAACAGGTGGTTATAG3′ and reverse 5′GTCCTCATGTATTGGTCTCTCATGGCAC3′) (Karbowniczek et al., 2008; Murua Escobar et al., 2004). PCR was performed using 100 ng of genomic DNA, primers, and PCR master mix (Qiagen) with 35 cycles with denaturing at 95°C for 45 s, annealing at 56°C for 45 s, and extension at 72°C for 60 s. Sequence analyses were performed on PCR products using sense and antisense primers.
Drug inhibition studies
Five canine MM cell lines, Jones, M1, M2, M3, and M5 were used. Cells were plated in 96‐well plates. Drug inhibitors (in DMSO) were added once to wells 24 h after plating cells. For dose responses, GSK1120212 was dosed at serial 1:4 dilutions from 10 μM to 0.125 nM and NVP‐BEZ235 was dosed at 1:4 dilutions from 2 μM to 0.125 nM; GSK1120212 and NVP‐BEZ235 were combined at a 1:1 ratio (range, 0.125 nM–2 μM) for combination therapy. Cell viability was determined by MTS assay (Promega, Madison, WI, USA), 72 h after the addition of drugs. A dose near 50% inhibitory concentration (IC50) of each drug in each cell line was used in subsequent kinetic studies, in which cell viability was determined at 24, 48, and 72 h. Five to six wells per cell line, per drug concentration/combination were used for each treatment, and the experiment was repeated at least twice.
To analyze cell cycle, cells were plated onto 6‐well plates and drug‐treated accordingly for 48 h. Cells were then washed with PBS and fixed with 70% ethanol. RNA was removed by adding RNase A (Sigma), and DNA was stained with propidium iodide. Cell cycle was analyzed using FACScalibur (BD). Evidence of caspase 3/7 activation was tested to detect apoptosis using CellEvent Caspase 3/7 Detection Reagent (ThermoFisher, Waltham, MA, USA) following manufacturer's protocol. Briefly, cells were grown and treated on glass cover slips in 24‐well plates. At the end of drug treatment, 5 mM of CellEvent Caspase‐3/7 Green Detection Reagent was added to each well and the cells were incubated in 37°C for 30 min followed by fixation with 4% formaldehyde for 20 min. Cover slips were then mounted onto glass slides using Vectashield DAPI‐containing mounting medium (Vector Laboratories Inc, Burlingame, CA, USA). Images were acquired using Axio microscope equipped with AxioCam HRc camera system (Zeiss, Thornwood, NY, USA). Five fields of view for each treatment were chosen, and the number of green caspase 3/7 positive cells as well as total number of cells was counted to calculate percentages of apoptotic cells.
Immunohistochemistry and image analysis
A tissue microarray (TMA) composed of primary canine melanomas from the CCOGC biorepository was created by the NCI Tissue Array Research Program Laboratory (Sogabe et al., 2014). A human TMA containing 40 human mucosal melanoma tumors was purchased from US Biomax, Inc. (Gaithersburg, MD, USA). Portions of melanoma xenografts from five mice representing each treatment group (see below) collected 10 days after the initiation of treatments were processed for routine preparation of formalin‐fixed paraffin‐embedded histology glass slide‐mounted tissue sections. Immunohistochemistry (IHC) was performed as previously described (Simpson et al., 2014). Labeling signals were developed with red chromogen (Vector Laboratories or Dako) for TMA primary tumor slides, and peroxidase‐catalyzed brown chromogen 3,3′‐diaminobenzidine (DAB, Dako) for tumor xenografts. Negative assay control included slides processed for IHC by omitting primary antibody. Intensity and degree of IHC‐treated tissue immunolabeling on TMAs was assessed accounting for both relative lack, limited or concentrated immunolabeling intensity, and percentages of cells labeled (<10%, 10–30%, 30–100%), resulting in a negative, low, or high score, respectively.
p‐ERK IHC‐labeled melanoma xenograft tissue sections were digitally scanned at 20× magnification using an Aperio ScanScope AT2 digital slide scanner (Leica Biosystems, Buffalo Grove, IL, USA) to create whole‐slide image data files at 0.5 μm/pixel resolution. Images files were viewed and analyzed using HALO applications (Indica Labs, Inc., Corrales, NM, USA). Intact tumor was manually segmented as regions of interest for analyses. Minimum tumor area analyzed was 2.6 mm2. Relative percentage quantification of p‐ERK‐immunolabeling signal intensity (brown) in the nuclear compartment was analyzed among treatment groups on a tumor area basis by paired t test using color deconvolution.
Animal studies
All animal experiments were carried out under review by NCI animal care and use programs in compliance with current US Public Health Service Guide for the Care and Use of Laboratory Animals (8th edition as revised, 2015). Six‐ to eight‐week‐old female nu/nu athymic nude mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). 1 × 106 M5 cells were injected into the right flank of mice. Mice with well‐established tumors (75–150 mm3) were randomized into four groups (15 mice per group), 14 days after tumor cell inoculation. Mice were treated with (i) GSK1120212 in 0.5% hydroxypropyl methylcellulose (HPMC)/0.2% Tween at 1.5 mg/kg, (ii) NVP‐BEZ235 in N‐methyl‐2‐pyrrolidone/Polyethylene glycol 300 (NMP/PEG300) at 30 mg/kg, (iii) combinations of (i) and (ii), or treatment (iv) drug vehicles of (i) and (ii). Treatments (drugs and vehicles) were given by oral gavage; GSK1120212 was given daily and NVP‐BEZ235 five days a week. The dosing regimen was based upon preliminary experiments in mice aimed at piloting tolerable treatment levels. Two of four mice treated with the highest tested doses of 3 mg/kg for GSK1120212 and 45 mg/kg for NVP‐BEZ235 in combination for 1 week revealed evidence of mild centrilobular hepatocellular vacuolar degeneration. Hence, 1.5 mg/kg GSK1120212 and 30 mg/kg NVP‐BEZ235 were chosen for follow‐up study. In the subject efficacy study, melanoma tumor dimensions were measured twice a week in mice using a caliper, and tumor volumes were calculated using π/6 × larger dimension × (smaller dimension)2. Body weights were recorded twice a week to gauge possible toxicity of treatments. Approximately four hours after oral gavage, a cohort of five mice from each treatment group were bled for plasma specimens and then removed from study 10 days after initial treatment began. Tumors from this cohort were harvested and tissues were fixed in 10% neutral‐buffered formalin and routinely prepared for histology, and lysates were separately prepared for WB. Treatment continued on the remaining 10 mice per group. Mice were electively taken off study when tumors approached 2000 mm3 in volume, and oral dosing was discontinued when >80% of control animals had reached tumor‐size end point (approximately day 35 to 39 of oral dose administration).
Mass spectrometry
Trametinib and dactolisib were quantitatively measured using a validated ultra HPLC‐tandem mass spectrometry (MS/MS) assay, with a calibration range (for both compounds) of 5–2000 ng/ml. Briefly, 100 μl of each plasma or conditioned medium sample was transferred to a well of a Waters Ostro® phospholipid removal plate before addition of 300 μl of methanol. The contents of each well were aspirated to thoroughly mix and precipitate proteins before a positive pressure manifold pushed the clean supernatant through to a collection plate using compressed nitrogen gas. Five microliters of the resulting supernatant was injected onto a Waters ACQUITY BEH C18 column (2.1 × 50 mm, 1.7 μm) before tandem mass spectrometric detection in the positive MRM mode. The assay was validated per FDA guidelines (US Food and Drug Administration. Guidelines for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics. 2015).
Statistics
Statistical significance was determined by t test using prism 6 software (version 6.0b, GraphPad, San Diego, CA, USA). Assessment of synergy for the inhibitor combinations was performed using the chou‐talalay algorithm (Chou, 2010) using compusyn (ComboSyn Inc., Paramus, NJ, USA).
Supporting information
Figure S1. Western blot analysis of additional patient tumors indicates that canine melanomas exhibit a diverse range of ERK1/2 and AKT activities. Tumor lysates were prepared from snap‐frozen melanoma specimens. Lane numbers indicate unique canine patient primary tumor samples.
Figure S2. Inhibition of MEK by GSK1120212 (GSK, trametinib) and PI3K/mTOR by NVP‐BEZ235 (NVP, dactolisib) in MM was time‐ and dose‐dependent. Five canine MM cell lines were treated with GSK or NVP at 31, 125, 500, and 2000 nM concentrations in vitro for 4 and 24 h, and analyzed by Western blot with anti‐p‐ERK1/2 or anti‐p‐AKT. (D, DMSO Control).
Figure S3. Densitometric assessment of p‐AKT(serine 473) in Western blots, normalized to (A) beta actin or (B) native AKT as internal controls, using Image J, revealed variable levels of inhibition in MM cell lines due to exposure to NVP‐BEZ235 (N, dactolisib) separately, or in combination (C) with GSK1120212 (G, trametinib) for 48 h. Results expressed as a ratio to control DMSO‐treated cells. p‐AKT blot images are from Figure 3.
Figure S4. Cell cycle analysis of canine MM cell lines (A) M1, (B) M2, and (C) M5 and (D) Jones. Cells were treated with GSK1120212 (G), NVP‐BEZ235 (N), a combination of the two agents (C), or DMSO control (D) for 48 h. Cells were fixed and labeled with propidium iodide after 48 h treatment to analyze the distribution of cells in various phases of cell cycle. Ratios of cycle phases varied by treatment and cell line and typically included relative increased population in G0/G1 (N), and sub‐G fraction for (G) and (C).
Figure S5. Body weights of M5 MM xenograft bearing mice treated with GSK1120212, NVP‐BEZ235, a combination of the two agents, or vehicle control. Mice were treated for up to 39 days after tumors were established.
Acknowledgements
This research was supported by the Intramural Research Program, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland. Kind support is acknowledged from the Animal Cancer Foundation and from the Canine Comparative Oncology and Genomics Consortium Biorepository. HTM is a molecular pathology fellow in the NIH Comparative Biomedical Scientist Training Program, an NCI‐administered Graduate Partnership Program in partnership with the University of Maryland. CHCH is currently at Pfizer, Inc., Groton, CT. AA is currently at Meso Scale Discovery, LLC, Rockville, MD. We thank Amy K. LeBlanc for critical reading of the manuscript. We thank Terry Van Dyke for input into experimental design.
References
- Atefi, M. , Von Euw, E. , Attar, N. et al. (2011). Reversing melanoma cross‐resistance to BRAF and MEK inhibitors by co‐targeting the AKT/mTOR pathway. PLoS ONE 6, e28973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, S.T. , Zhou, B. , Damrauer, J.S. , Krishnan, B. , Wilson, H.L. , Smith, A.M. , Li, M. , Yeh, J.J. , and Kim, W.Y. (2014). mTOR inhibition induces compensatory, therapeutically targetable MEK activation in renal cell carcinoma. PLoS ONE 9, e104413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman, P.J. (2007). Canine oral melanoma. Clin. Tech. Small Anim. Pract. 22, 55–60. [DOI] [PubMed] [Google Scholar]
- Bergman, P.J. , Kent, M.S. , and Farese, J.P. (2013). Melanoma In Withrow and MacEwen's Small Animal Oncology. Withrow S.J., Vail D.M., and Page R.L., eds. (St. Louis, MO: Elsevier; ), pp. 321–334. [Google Scholar]
- Besaratinia, A. , and Pfeifer, G.P. (2008). Sunlight ultraviolet irradiation and BRAF V600 mutagenesis in human melanoma. Hum. Mutat. 29, 983–991. [DOI] [PubMed] [Google Scholar]
- Bogenrieder, T. , and Herlyn, M. (2010). The molecular pathology of cutaneous melanoma. Cancer Biomark. 9, 267–286. [DOI] [PubMed] [Google Scholar]
- Bosenberg, M. , Arnheiter, H. , and Kelsh, R. (2014). Melanoma in mankind's best friend. Pigment Cell Melanoma Res. 27, 1. [DOI] [PubMed] [Google Scholar]
- Carracedo, A. , Ma, L. , Teruya‐Feldstein, J. et al. (2008). Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K‐dependent feedback loop in human cancer. J. Clin. Investig. 118, 3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal, R.D. , Antonescu, C.R. , Wolchok, J.D. et al. (2011). KIT as a therapeutic target in metastatic melanoma. JAMA 305, 2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal, R.D. , Spencer, S.A. , and Lydiatt, W. (2012). Mucosal melanoma: a clinically and biologically unique disease entity. J. Natl. Compr. Canc. Netw. 10, 345–356. [DOI] [PubMed] [Google Scholar]
- Chang, A.E. , Karnell, L.H. , and Menck, H.R. (1998). The National Cancer Data Base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer 83, 1664–1678. [DOI] [PubMed] [Google Scholar]
- Chou, T.C. (2010). Drug combination studies and their synergy quantification using the Chou‐Talalay method. Cancer Res. 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Chu, P.Y. , Pan, S.L. , Liu, C.H. , Lee, J. , Yeh, L.S. , and Liao, A.T. (2013). KIT gene exon 11 mutations in canine malignant melanoma. Vet. J. 196, 226–230. [DOI] [PubMed] [Google Scholar]
- Curtin, J.A. , Fridlyand, J. , Kageshita, T. et al. (2005). Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 353, 2135–2147. [DOI] [PubMed] [Google Scholar]
- Curtin, J.A. , Busam, K. , Pinkel, D. , and Bastian, B.C. (2006). Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 24, 4340–4346. [DOI] [PubMed] [Google Scholar]
- Ersahin, T. , Tuncbag, N. , and Cetin‐Atalay, R. (2015). The PI3K/AKT/mTOR interactive pathway. Mol. BioSyst. 11, 1946–1954. [DOI] [PubMed] [Google Scholar]
- Esplin, D.G. (2008). Survival of dogs following surgical excision of histologically well‐differentiated melanocytic neoplasms of the mucous membranes of the lips and oral cavity. Vet. Pathol. 45, 889–896. [DOI] [PubMed] [Google Scholar]
- Flaherty, K.T. , Infante, J.R. , Daud, A. et al. (2012). Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 367, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowles, J.S. , Denton, C.L. , and Gustafson, D.L. (2015). Comparative analysis of MAPK and PI3K/AKT pathway activation and inhibition in human and canine melanoma. Vet. Comp. Oncol. 13, 288–304. [DOI] [PubMed] [Google Scholar]
- Gillard, M. , Cadieu, E. , De Brito, C. et al. (2014). Naturally occurring melanomas in dogs as models for non‐UV pathways of human melanomas. Pigment Cell Melanoma Res. 27, 90–102. [DOI] [PubMed] [Google Scholar]
- Gilmartin, A.G. , Bleam, M.R. , Groy, A. et al. (2011). GSK1120212 (JTP‐74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res. 17, 989–1000. [DOI] [PubMed] [Google Scholar]
- Goldschmidt, M.H. , and Hendrick, M.J. (2002). Melanocytic tumors In Tumors in Domestic Animals. Meuten D.J., ed. (Ames, Iowa: Iowa State University Press; ), pp. 78–84. [Google Scholar]
- Gomes, J. , Queiroga, F.L. , Prada, J. , and Pires, I. (2012). Study of c‐kit immunoexpression in canine cutaneous melanocytic tumors. Melanoma Res. 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Gordon, I. , Paoloni, M. , Mazcko, C. , and Khanna, C. (2009). The Comparative Oncology Trials Consortium: using spontaneously occurring cancers in dogs to inform the cancer drug development pathway. PLoS Med. 6, e1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grazia, G. , Penna, I. , Perotti, V. , Anichini, A. , and Tassi, E. (2014). Towards combinatorial targeted therapy in melanoma: from pre‐clinical evidence to clinical application (review). Int. J. Oncol. 45, 929–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger, J.G. , Eastman, S.D. , Zhang, V. , Bleam, M.R. , Hughes, A.M. , Smitheman, K.N. , Dickerson, S.H. , Laquerre, S.G. , Liu, L. , and Gilmer, T.M. (2012). Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 11, 909–920. [DOI] [PubMed] [Google Scholar]
- Hartman, M.L. , and Czyz, M. (2013). Anti‐apoptotic proteins on guard of melanoma cell survival. Cancer Lett. 331, 24–34. [DOI] [PubMed] [Google Scholar]
- Karbowniczek, M. , Spittle, C.S. , Morrison, T. , Wu, H. , and Henske, E.P. (2008). mTOR is activated in the majority of malignant melanomas. J. Invest. Dermatol. 128, 980–987. [DOI] [PubMed] [Google Scholar]
- Kwei, K.A. , Baker, J.B. , and Pelham, R.J. (2012). Modulators of sensitivity and resistance to inhibition of PI3K identified in a pharmacogenomic screen of the NCI‐60 human tumor cell line collection. PLoS ONE 7, e46518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maira, S.M. , Stauffer, F. , Brueggen, J. et al. (2008). Identification and characterization of NVP‐BEZ235, a new orally available dual phosphatidylinositol 3‐kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 7, 1851–1863. [DOI] [PubMed] [Google Scholar]
- Margolin, K.A. , Moon, J. , Flaherty, L.E. , Lao, C.D. , Akerley, W.L. 3rd , Othus, M. , Sosman, J.A. , Kirkwood, J.M. , and Sondak, V.K. (2012). Randomized phase II trial of sorafenib with temsirolimus or tipifarnib in untreated metastatic melanoma (S0438). Clin. Cancer Res. 18, 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr, B. , Schaffner, G. , Reifinger, M. , Zwetkoff, S. , and Prodinger, B. (2003). N‐ras mutations in canine malignant melanomas. Vet. J. 165, 169–171. [DOI] [PubMed] [Google Scholar]
- Mclaughlin, C.C. , Wu, X.C. , Jemal, A. , Martin, H.J. , Roche, L.M. , and Chen, V.W. (2005). Incidence of noncutaneous melanomas in the U.S. Cancer 103, 1000–1007. [DOI] [PubMed] [Google Scholar]
- Mendoza, M.C. , Er, E.E. , and Blenis, J. (2011). The Ras‐ERK and PI3K‐mTOR pathways: cross‐talk and compensation. Trends Biochem. Sci. 36, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki, H. , Kennedy, K. , Shapiro, S.G. , and Breen, M. (2015). BRAF mutations in canine cancers. PLoS ONE 10, e0129534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, A. , Mori, T. , Sakai, H. , Murakami, M. , Yanai, T. , Hoshino, Y. , and Maruo, K. (2011). Analysis of KIT expression and KIT exon 11 mutations in canine oral malignant melanomas. Vet. Comp. Oncol. 9, 219–224. [DOI] [PubMed] [Google Scholar]
- Murua Escobar, H. , Gunther, K. , Richter, A. , Soller, J.T. , Winkler, S. , Nolte, I. , and Bullerdiek, J. (2004). Absence of ras‐gene hot‐spot mutations in canine fibrosarcomas and melanomas. Anticancer Res. 24, 3027–3028. [PubMed] [Google Scholar]
- Newman, S.J. , Jankovsky, J.M. , Rohrbach, B.W. , and Leblanc, A.K. (2012). C‐kit expression in canine mucosal melanomas. Vet. Pathol. 49, 760–765. [DOI] [PubMed] [Google Scholar]
- Paoloni, M. , and Vail, D.M. (2013). Clinical trials and developmental therapeutics In Small Animal Clinical Oncology. Withrow S.J., Vail D.M., and Page R.L., eds. (St. Louis, MO: Elsevier/Saunders; ), pp. 290–304. [Google Scholar]
- Rankin, K.S. , Starkey, M. , Lunec, J. , Gerrand, C.H. , Murphy, S. , and Biswas, S. (2012). Of dogs and men: comparative biology as a tool for the discovery of novel biomarkers and drug development targets in osteosarcoma. Pediatr. Blood Cancer 58, 327–333. [DOI] [PubMed] [Google Scholar]
- Shelly, S. , Chien, M.B. , Yip, B. , Kent, M.S. , Theon, A.P. , Mccallan, J.L. , and London, C.A. (2005). Exon 15 BRAF mutations are uncommon in canine oral malignant melanomas. Mamm. Genome 16, 211–217. [DOI] [PubMed] [Google Scholar]
- Shi, H. , Kong, X. , Ribas, A. , and Lo, R.S. (2011). Combinatorial treatments that overcome PDGFRbeta‐driven resistance of melanoma cells to V600EB‐RAF inhibition. Cancer Res. 71, 5067–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, R. , Naishadham, D. , and Jemal, A. (2012). Cancer statistics, 2012. CA Cancer J. Clin. 62, 10–29. [DOI] [PubMed] [Google Scholar]
- Simpson, R.M. , Bastian, B.C. , Michael, H.T. et al. (2014). Sporadic naturally occurring melanoma in dogs as a preclinical model for human melanoma. Pigment Cell Melanoma Res. 27, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley, R.C. , Spangler, W.L. , Esplin, D.G. , Kitchell, B.E. , Bergman, P.J. , Ho, H.Y. , Bergin, I.L. , and Kiupel, M. (2011). Prognostic markers for canine melanocytic neoplasms: a comparative review of the literature and goals for future investigation. Vet. Pathol. 48, 54–72. [DOI] [PubMed] [Google Scholar]
- Sogabe, S. , Togashi, Y. , Kato, H. et al. (2014). MEK inhibitor for gastric cancer with MEK1 gene mutations. Mol. Cancer Ther. 13, 3098–3106. [DOI] [PubMed] [Google Scholar]
- Tacastacas, J.D. , Bray, J. , Cohen, Y.K. , Arbesman, J. , Kim, J. , Koon, H.B. , Honda, K. , Cooper, K.D. , and Gerstenblith, M.R. (2014). Update on primary mucosal melanoma. J. Am. Acad. Dermatol. 71, 366–375. [DOI] [PubMed] [Google Scholar]
- Tolcher, A.W. , Bendell, J.C. , Papadopoulos, K.P. et al. (2015a). A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 26, 58–64. [DOI] [PubMed] [Google Scholar]
- Tolcher, A.W. , Patnaik, A. , Papadopoulos, K.P. et al. (2015b). Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 75, 183–189. [DOI] [PubMed] [Google Scholar]
- Vail, D.M. , Thamm, D.H. , Reiser, H. et al. (2009). Assessment of GS‐9219 in a pet dog model of non‐Hodgkin's lymphoma. Clin. Cancer Res. 15, 3503–3510. [DOI] [PubMed] [Google Scholar]
- Van Dort, M.E. , Galban, S. , Wang, H. , Sebolt‐Leopold, J. , Whitehead, C. , Hong, H. , Rehemtulla, A. , and Ross, B.D. (2015). Dual inhibition of allosteric mitogen‐activated protein kinase (MEK) and phosphatidylinositol 3‐kinase (PI3K) oncogenic targets with a bifunctional inhibitor. Bioorg. Med. Chem. 23, 1386–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanke, R. , Harjivan, S.G. , Pereira, S.A. , Marques, M.M. , and Antunes, A.M. (2013). The role of competitive binding to human serum albumin on efavirenz‐warfarin interaction: a nuclear magnetic resonance study. Int. J. Antimicrob. Agents 42, 443–446. [DOI] [PubMed] [Google Scholar]
- Williams, H.D. , Nott, K.P. , Barrett, D.A. , Ward, R. , Hardy, I.J. , and Melia, C.D. (2011). Drug release from HPMC matrices in milk and fat‐rich emulsions. J. Pharm. Sci. 100, 4823–4835. [DOI] [PubMed] [Google Scholar]
- Yasumizu, Y. , Miyajima, A. , Kosaka, T. , Miyazaki, Y. , Kikuchi, E. , and Oya, M. (2014). Dual PI3K/mTOR inhibitor NVP‐BEZ235 sensitizes docetaxel in castration resistant prostate cancer. J. Urol. 191, 227–234. [DOI] [PubMed] [Google Scholar]
- Zhu, Y.R. , Min, H. , Fang, J.F. , Zhou, F. , Deng, X.W. , and Zhang, Y.Q. (2015). Activity of the novel dual phosphatidylinositol 3‐kinase/mammalian target of rapamycin inhibitor NVP‐BEZ235 against osteosarcoma. Cancer Biol. Ther. 16, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Western blot analysis of additional patient tumors indicates that canine melanomas exhibit a diverse range of ERK1/2 and AKT activities. Tumor lysates were prepared from snap‐frozen melanoma specimens. Lane numbers indicate unique canine patient primary tumor samples.
Figure S2. Inhibition of MEK by GSK1120212 (GSK, trametinib) and PI3K/mTOR by NVP‐BEZ235 (NVP, dactolisib) in MM was time‐ and dose‐dependent. Five canine MM cell lines were treated with GSK or NVP at 31, 125, 500, and 2000 nM concentrations in vitro for 4 and 24 h, and analyzed by Western blot with anti‐p‐ERK1/2 or anti‐p‐AKT. (D, DMSO Control).
Figure S3. Densitometric assessment of p‐AKT(serine 473) in Western blots, normalized to (A) beta actin or (B) native AKT as internal controls, using Image J, revealed variable levels of inhibition in MM cell lines due to exposure to NVP‐BEZ235 (N, dactolisib) separately, or in combination (C) with GSK1120212 (G, trametinib) for 48 h. Results expressed as a ratio to control DMSO‐treated cells. p‐AKT blot images are from Figure 3.
Figure S4. Cell cycle analysis of canine MM cell lines (A) M1, (B) M2, and (C) M5 and (D) Jones. Cells were treated with GSK1120212 (G), NVP‐BEZ235 (N), a combination of the two agents (C), or DMSO control (D) for 48 h. Cells were fixed and labeled with propidium iodide after 48 h treatment to analyze the distribution of cells in various phases of cell cycle. Ratios of cycle phases varied by treatment and cell line and typically included relative increased population in G0/G1 (N), and sub‐G fraction for (G) and (C).
Figure S5. Body weights of M5 MM xenograft bearing mice treated with GSK1120212, NVP‐BEZ235, a combination of the two agents, or vehicle control. Mice were treated for up to 39 days after tumors were established.