

Abstract
Germline PMS2 gene mutations were detected by RT‐PCR/direct sequencing of total RNA extracted from puromycin‐treated peripheral blood lymphocytes (PBL) and multiplex ligation‐dependent probe amplification (MLPA) analyses of Japanese patients with colorectal cancer (CRC) fulfilling either the revised Bethesda Guidelines or being an age at disease onset of younger than 70 years, and screened by mismatch repair protein immunohistochemistry of formalin‐fixed paraffin embedded sections. Of the 501 subjects examined, 7 (1.40%) showed the downregulated expression of the PMS2 protein alone and were referred to the genetic counseling clinic. Germline PMS2 mutations were detected in 6 (85.7%), including 3 nonsense and 1 frameshift mutations by RT‐PCR/direct sequencing and 2 genomic deletions by MLPA. No mutations were identified in the other MMR genes (i.e. MSH2,MLH1 and MSH6). The prevalence of the downregulated expression of the PMS2 protein alone was 1.40% among the subjects examined and IHC results predicted the presence of PMS2 germline mutations. RT‐PCR from puromycin‐treated PBL and MLPA may be employed as the first screening step to detect PMS2 mutations without pseudogene interference, followed by the long‐range PCR/nested PCR validation using genomic DNA.
Keywords: Colorectal cancer, immunohistochemistry, Lynch syndrome, mismatch repair gene, PMS2
Lynch syndrome is an autosomal dominantly inherited hereditary cancer syndrome characterized by the familial accumulation of early‐onset cancers such as carcinomas of, for example, the colon, endometrium, ovary, urinary tract and stomach.1 Four responsible genes have been identified to date (MSH2, MLH1, MSH6 and PMS2), all of which participate in the mismatch repair (MMR) system in DNA synthesis. MSH2 or MLH1 mutation is most prevalent among germline mutations detected in Lynch kindred, followed by MSH6, while the prevalence of PMS2 germline mutations in Japanese patients has only been discussed in a few case reports.2, 3 An immunohistochemical (IHC) examination of formalin‐fixed paraffin‐embedded (FFPE) colon cancer tissues using a combination of specific antibodies directed to each of the four MMR proteins has been applied to screen for Lynch syndrome and this new modality has enabled the identification of the responsible gene.4 MLH1 and PMS2 form a MutLα heterodimer and the downregulated expression of the MLH1 protein is typically accompanied by the loss of PMS2 protein expression, implying that inactivated MLH1 fails to dimerize PMS2 in order to form MutLα. In contrast, the loss of PMS2 protein expression alone indicates the presence of a PMS2 gene mutation. In the present study, we report the diagnosis of germline PMS2 mutations in patients referred to the genetic counseling clinic due to the downregulated expression of the PMS2 protein in colon cancer tissues, in an attempt to clarify the prevalence of Lynch syndrome caused by PMS2 germline mutations among Japanese patients with colorectal cancer (CRC).
Subjects and Methods
Postoperative CRC patients were screened by IHC for the expression of the mismatch repair proteins. Between 31 January 2012 and 26 December 2015, 501 postoperative CRC patients undergoing endoscopic surveillance fulfilled the eligibility criteria; that is, either the revised Bethesda Guidelines or an age at disease onset of younger than 70 years.1, 5 The Institutional Review Board (IRB) approved the protocols for IHC and subsequent gene testing for MMR genes (Study Nos. 2011‐108, 2013‐190 and 2013‐303). Once written informed consent was provided, patients underwent an IHC examination for mismatch repair protein expression (MSH2, MLH1, MSH6 and PMS2). Subjects with the loss of MMR protein expression were recruited by the genetic counseling clinic to undergo genetic counseling and subsequent gene testing for germline mutations in MMR genes at the Department of Genetic Medicine and Services, the National Cancer Center Hospital, Tsukiji, Chuo‐ku, Tokyo. RT‐PCR/direct sequencing analysis, PCR/direct sequencing analysis and an Multiplex Ligation‐dependent Probe Amplification (MLPA) assay were performed at the Oncogene Research Unit/Cancer Prevention Unit, Tochigi Cancer Center Research Institute, Utsunomiya, Tochigi Prefecture according to the IRB approved protocols for multi‐institutional study of hereditary cancer (Study Nos. A‐019 and A‐291).
Immunohistochemistry
The protocol for IHC has been reported previously.6 Briefly, deparaffinized 4‐mm‐thick sections from each paraffin block were exposed to 0.3% hydrogen peroxide for 15 min to block endogenous peroxidase activity. Antigen retrieval was performed by autoclaving in a 10‐mM citrate buffer (pH 6.0) for 10 min. Anti‐MLH1 (G168–728; 1:200 dilution; BD Biosciences, Franklin Lakes, NJ, USA), anti‐MSH2 (FE11; 1:100 dilution; Biocare Medical, Concord, CA, USA), anti‐PMS2 (A16‐4; 1:200 dilution; BD Biosciences) and anti‐MSH6 antibodies (SP93;1:200 dilution; Spring Bioscience, Pleasanton, CA, USA) were used as the primary antibodies. We used an automated stainer (Dako, Tokyo, Japan) according to the vendor's protocol for staining. ChemMate EnVision (Dako, Tokyo, Japan) methods were used for detection. The IHC results were evaluated by two pathologists independently and the results coincided completely.
Microsatellite instability
Subjects showing downregulated expression of the PMS2 protein alone in IHC underwent microsatellite instability (MSI) testing according to the original National Cancer Institute (NCI) microsatellite panel, including BAT25, BAT26, D2S123, D5S346 and D17S250.7 The protocol for MSI analysis was as reported previously.8 Tumors were classified as high‐frequency MSI (MSI‐H) when two or more of the five NCI‐recommended panels of microsatellite markers showed MSI, in accordance with the recommendation of the National Cancer Institute Workshop.7
Gene testing for germline PMS2 mutations
An RT‐PCR was employed as the first screening step, in which puromycin‐treated peripheral blood lymphocytes (PBL) were used for RNA extraction.9 cDNA synthesis, RT‐PCR and direct sequencing were performed as described previously. Briefly, 7 mL of heparinized fresh blood was incubated with or without puromycin (Sigma Chemical, St. Louis MO, USA) at a concentration of 0.2 mg/mL at 37°C for 2 h. Leukocytes were isolated from peripheral blood using Lymphoprep (Alere Technologies AS, Oslo, Norway), mixed with 1 mL of ISOGEN (Nippon Gene, Tokyo, Japan) and stored frozen at −80°C. Total RNA was extracted using the acid guanidium phenol/chloroform method. Reverse transcription was performed with 200 units of MMLV reverse transcriptase SuperScript II (Life Technologies Inc., Carlsbad MD, USA). Direct sequencing was performed using a Bigdye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems, Fostercity CA, USA) and ABI 3100 Genetic Analyzer (PE Applied Biosystems). The primers used for RT‐PCR and direct sequencing are described in a list in the Supporting Information. The extraction of genomic DNA was performed by proteinase K digestion and phenol/chloroform extraction as described previously.10 When mutations were detected by the RT‐PCR/direct sequencing analysis, they were validated in genomic DNA using a PCR/direct sequencing analysis. Because the presence of pseudogenes may hamper the detection of PMS2‐specific mutations, we used a long‐range PCR/nested PCR approach to confirm mutations, as described previously by Clendenning et al.11 (Fig. 1 and Tables S1,S2).
Figure 1.
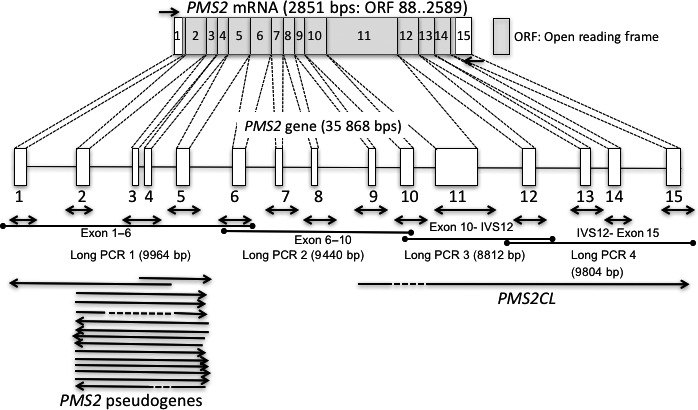
Genomic organization, mRNA of the PMS2 gene, and strategy for gene testing. Genomic organization of the PMS2 gene (35 868 bps) and PMS2 mRNA (2851 bps). Boxes indicate exons and the open reading frame (ORF) is indicated by the gray box. Short arrows indicate primer locations for RT‐PCR and amplicon locations for an exon‐by‐exon analysis in a long/nested PCR analysis. Long arrow lines indicate the location of pseudogenes.
Multiplex ligation‐dependent probe amplification
Multiplex ligation‐dependent probe amplification was employed in combination with the RT‐PCR/direct sequencing analysis in order to detect PMS2 gene rearrangements using a kit (P008‐B1 or P008‐C1) purchased from MRC‐Holland (Amsterdam, the Netherlands), according to the manufacturer's recommendations.12 A fragment analysis was performed using an ABI310 Prism Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The electropherograms of the subjects were compared with control samples in order to detect genomic rearrangements in the PMS2 gene. The PMS2 allele downstream of exon 12 reportedly has frequent gene conversions with the PMS2CL pseudogene, which contains exons 9 and 11–15 of the PMS2 gene. In the analysis of genomic regions from exons 11 to 15 of the PMS2 gene, three sets of probes were designed for each genomic region. Each Probe set hybridized to specific sequences for PMS2, PMS2CL or the consensus sequence for PMS2 and PMS2CL. As for the exon 12, probes specific for PMS2 or PMS2CL were designed to anneal the intron 12 (IVS12) sequences of the PMS2 and PMS2CL genes to avoid the pseudogene interference, whereas the universal probe set was designed to anneal the consensus sequence of the exon 12. Of the 96 subjects examined by MLPA, IHC was performed in 33 subjects and the other subjects were assayed for the diagnosis of Lynch syndrome as a part of the multi‐institutional study (Study Nos. A‐019 and A‐291). They are used as assay controls for depicting the profiles of copy number variation from IVS12 through exon 15 of the PMS2 and PMS2CL genes among Japanese. The numbers of deleterious mutations detected in the other MMR genes were as follows: 20 subjects had deleterious mutations for MSH2, 21 subjects for MLH1 and 8 subjects for MSH6, respectively. Forty samples were diagnosed as the wild type for MSH2, MLH1 and MSH6. Of the Seven subjects showing downregulated expression of the PMS2 protein alone, four deleterious mutations (3 nonsense and 1 frameshift mutations) of the PMS2 gene were detected by the RT‐PCR/direct sequencing analysis, as described in the results section.
Results
Of the 501 subjects examined, 7 (1.40%) showed the loss of PMS2 protein expression alone and referred to the clinic for genetic counseling and provided written informed consent to undergo genetic testing (Table 1). All subjects were male and a significant difference was noted in the reduced PMS2 expression status between genders (male 7/290: female 0/211, P = 0.0233). Genetic testing was completed on seven subjects and deleterious PMS2 germline mutations were detected in 6/7 (85.7%) of the index subjects (Table 1). Three nonsense and one frameshift mutations were detected by the RT‐PCR/direct sequencing analysis using puromycin‐treated PBL.
Table 1.
Clinical characteristics of subjects with the loss of PMS2 protein expression alone and the kindred undergoing genetic testing
| Subject number | Sex | Age (years) | Colorectal cancer location (age, years) | Other cancers (age, years) | Histological Dx | Mutation | Compatibility with the Revised Bethesda Guideline† | MSI status‡ |
|---|---|---|---|---|---|---|---|---|
| 1 | Male | 52 | T. Colon (50) |
25 × 20 mm, Type 2 well to moderately differentiated adenocarcinoma pMP,n0 (0/15) |
Exon 9 c.943C>T R315X |
Incompatible | MSI‐H(5/5) | |
| 2 | Male | 64 | S. Colon (61) |
Stomach (58) 9 × 6 mm poorly differentiated adenocarcinoma and signet ring cell adenocarcinoma, m,n0 (0/36) |
9 × 9 mm well differentiated adenocarcinoma |
Not identified | Compatible: 2) | MSI‐H(5/5) |
| 3 | Male | 63 | A. Colon (62) |
67 × 47 mm pSS, n0 (0/54) |
Exon 11 c.1882C>T R628X |
Compatible: 4) | MSI‐H(4/5) | |
| 4 | Male | 46 | S. Colon (40) |
50 × 55 mm moderate to poorly differentiated adenocarcinoma pSS, n0 (0/23) |
PMS2 genomic del Exon 12–15 |
Compatible: 1) | MSI‐H(5/5) | |
| 5 | Male | 59 | A. colon (58) |
25 × 22 mm pSM2, n0 (0/21), H0 |
PMS2 genomic del Exon 11–15 |
Incompatible | MSI‐H(5/5) | |
| 6 | Male | 55 | S. Colon (55) |
20 × 13 mm well differentiated adenocarcinoma pSM2, n0 (0/26) |
IVS14 c.2446‐1G>A, r.2446delG frameshift mutation |
Incompatible | MSI‐H(5/5) | |
| 7 | Male | 63 | Rectum (Rb) (63) |
35 × 25 mm moderately differentiated adenocarcinoma A, n0 (0/30) |
Exon 3 c.241G>T, p.Glu81Stop |
Incompatible | MSI‐H(4/5) |
†Compatibility with the Revised Bethesda Guidelines14 is indicated when the individual fulfills any of the items in the following: 1) Colorectal cancer diagnosed in a patient who is <50 years of age. 2) Presence of synchronous, metachronous colorectal, or other hereditary non‐polyposis colorectal cancer (HNPCC) associated tumors, §regardless of age. 3) Colorectal cancer with the MSI‐H histology¶ diagnosed in a patient who is <60 years of age. 4) Colorectal cancer diagnosed in one or more first‐degree relatives with an HNPCC‐related tumor, with one of the cancers being diagnosed under age 50 years. 5) Colorectal cancer diagnosed in two or more first‐ or second‐degree relatives with HNPCC‐related tumors, regardless of age. ‡Microsatellite instability (MSI) was examined according to the original National Cancer Institute (NCI) microsatellite panel including BAT25, BAT26, D2S123, D5S346 and D17S2507. Microsatellite instability–high (MSI‐H) in tumors refers to changes in two or more of the five NCI‐recommended panels of microsatellite markers. §HNPCC‐related tumors include colorectal, endometrial, stomach, ovarian, pancreas, ureter and renal pelvis, biliary tract, and brain (usually glioblastoma as seen in Turcot syndrome) tumors, sebaceous gland adenomas and keratoacanthomas in Muir–Torre syndrome, and carcinoma of the small bowel. ¶Presence of tumor infiltrating lymphocytes, Crohn's‐like lymphocytic reaction,mucinous/signet‐ring differentiation, or medullary growth pattern. There was no consensus among the Workshop participants on whether toinclude the age criteria in guideline 3 above; participants voted to keep <60 years of age in the guidelines.
In subjects nos. 1, 3 and 7, three nonsense mutations were detected; c.943C>T, p.Arg315Stop in exon 9, c.1882C>T, Arg628Stop in exon 11 and c.241G>T, p.Glu81Stop in exon 3 (Figs 2, 3, 4). In subjects nos. 1 and 3, signals from the mutated allele were enhanced in puromycin‐treated samples, showing the presence of nonsense‐mediated mRNA decay (NMD) in samples without the puromycin treatment (Figs 2,3).
Figure 2.
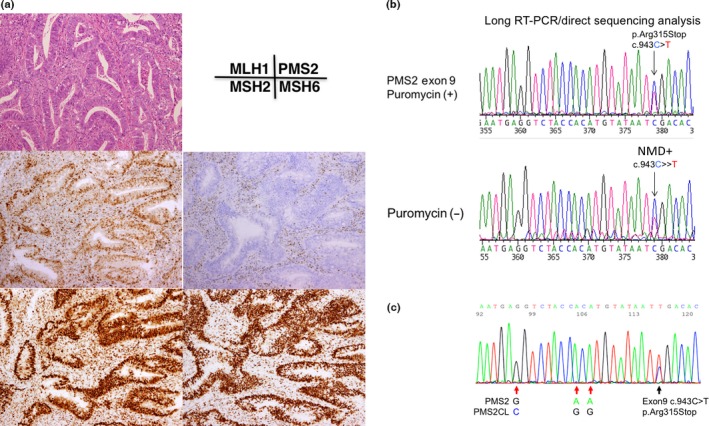
Immunochemical (IHC) and gene testing of subject no. 1. (a) IHC using four antibodies to mismatch repair (MMR) proteins. Note the loss of PMS2 protein expression in cancer cell nuclei. (b) RT‐PCR/direct sequencing analysis of the PMS2 gene in subject No. 1. The upper panel indicates the sequencing profile showing the c.943C>T (p.Arg315Stop) mutation from the puromycin‐treated total RNA sample and the lower panel shows the sequencing profile of the sample without the puromycin treatment. Note that the signal ratio of the mutated allele (T) to the wild‐type allele (C) is weaker in the sample without the puromycin treatment than in that with the puromycin treatment, indicating nonsense‐mediated mRNA decay (NMD). (c) Long/nested PCR/direct sequencing analysis of the PMS2 gene. The black arrow indicates that the same mutation (c.943C>T, pArg315Stop) was detected in an analysis using DNA by a long PCR/nested PCR/direct sequencing analysis. Red arrows indicate the nucleotide positions coding different DNA sequences between PMS2 and PMS2CL, showing that the sequences of the PMS2 paralogue are not contaminated in the PCR product.
Figure 3.
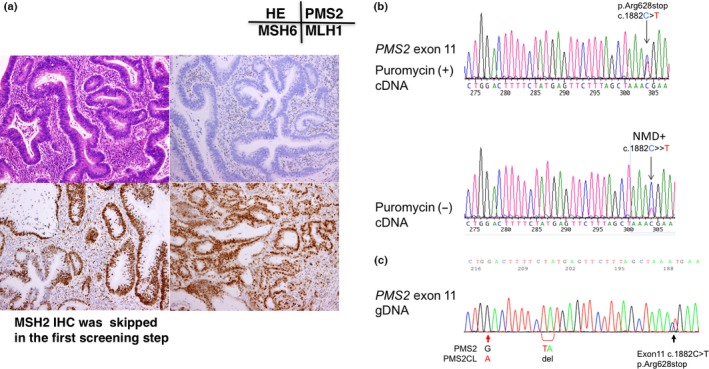
Immunochemical (IHC) and gene testing of subject No. 3. (a) IHC using four antibodies to mismatch repair (MMR) proteins. Note the loss of PMS2 protein expression in cancer cell nuclei. (b) RT‐PCR/direct sequencing analysis of the PMS2 gene in subject No. 3. The upper panel indicates the sequencing profile showing the c.1882C>T (p.Arg628Stop) mutation from the puromycin‐treated total RNA sample and the lower panel shows the sequencing profile without the puromycin treatment. Note that the signal ratio of the mutated allele (T) to the wild‐type allele (C) was weaker in the sample without the puromycin treatment than in that with the puromycin treatment, indicating nonsense‐mediated mRNA decay (NMD). (c) Long/nested PCR/direct sequencing analysis of the PMS2 gene. The black arrow indicates that the same mutation (c.1882C>T, pArg628Stop) was detected in the analysis using DNA by a long PCR/nested PCR/direct sequencing analysis. Red arrows indicate the nucleotide positions coding different DNA sequences between PMS2 and PMS2CL, showing that the sequences of the PMS2 paralogue were not contaminated in the PCR product.
Figure 4.
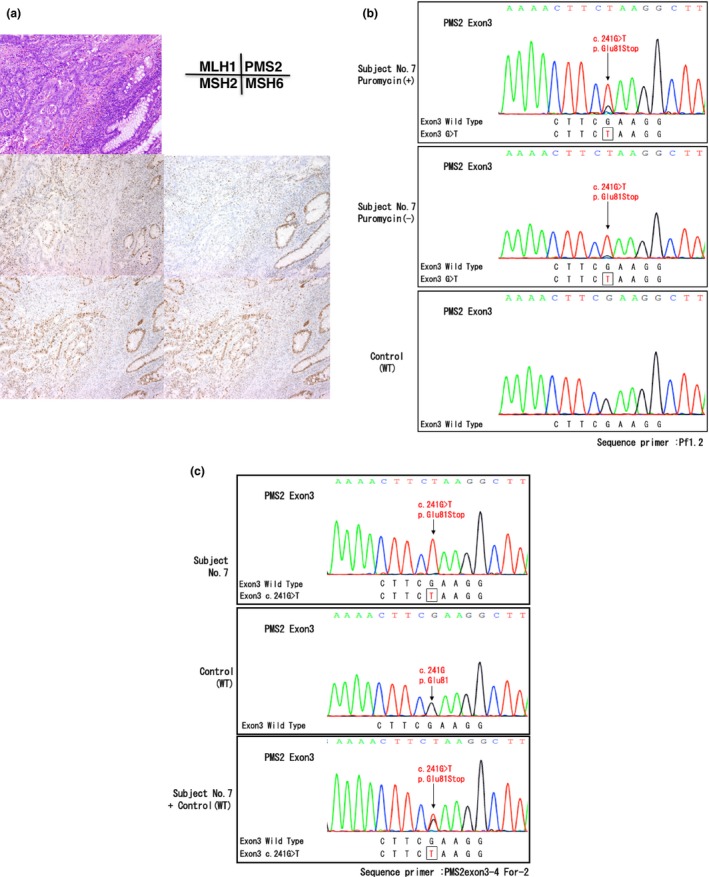
Gene testing of subject No. 7. (a) Immunochemistry (IHC) using four antibodies to mismatch repair (MMR) proteins. Note the loss of PMS2 protein expression in cancer cell nuclei. (b) RT‐PCR/direct sequencing analysis of the PMS2 gene in subject No. 7. The upper panel indicates the sequencing profile showing the c.241G>T (p.Glu81Stop) mutation from a puromycin‐treated total RNA sample and the middle panel shows the sequencing profile without the puromycin treatment. Note that the signal ratio of the mutated allele to the wild‐type allele increased in the absence of the puromycin treatment and the c.241G>T (p.Glu81Stop) mutation appeared to be a homozygous mutation. (c) Long/nested PCR/direct sequencing analysis of the PMS2 gene in subject No. 7. An analysis of the genomic DNA by a long PCR/nested PCR/direct sequencing analysis showed the c.241G>T mutation (upper panel) without the presence of the wild type allele as compared to that of the control (middle panel). Equal amounts of the genomic DNA from subject no. 7 and the healthy control was mixed and subjected to the long PCR/nested PCR/direct sequencing analysis. This experiment resulted in the DNA sequence looking like the heterozygous mutation (lower panel), supporting the reliability of the homozygous mutation detected in subject No. 7.
In subject no. 7, a nonsense mutation c.241G>T, p.Glu81Stop was detected in exon 3 of the PMS2 gene by RT‐PCR/direct sequencing (Fig. 4b). A signal from the wild type allele was reduced and modest as compared to the background noise, and the signal from the mutated allele looked enhanced in the sample without puromycin treatment, implying that the wild type allele has no mRNA transcript. This finding was compatible with the result of long PCR/nested PCR analysis using genomic DNA, in which the same mutation (p.Glu81Stop) in exon 3 of the PMS2 gene was detected as a homozygous mutation (Fig. 4c). To exclude the possibility of the PCR artifact, we made a compound DNA sample in which the equal amounts of the genomic DNA from the control and subject no. 7 were mixed and subjected to the long PCR/nested PCR/direct sequencing analysis. This assay resulted in the mixture of the wild type and the mutated sequences resembling the heterozyote and subject no. 7 was assumed to have a homozygous mutation (Fig. 4c).
In subject no. 6, a G>A mutation in the splicing acceptor site of the exon 15 (c.2446‐1G>A) resulted in a frameshift mutation. Because exon 15 is the last exon of the PMS2 gene, the G>A mutation in the splicing acceptor site resulted in the appearance of a new splicing site instead of exon skipping or NMD (Fig. 5b). All of these mutations were validated in the analysis using genomic DNA samples (Figs 2c,3c,4c,5c).
Figure 5.
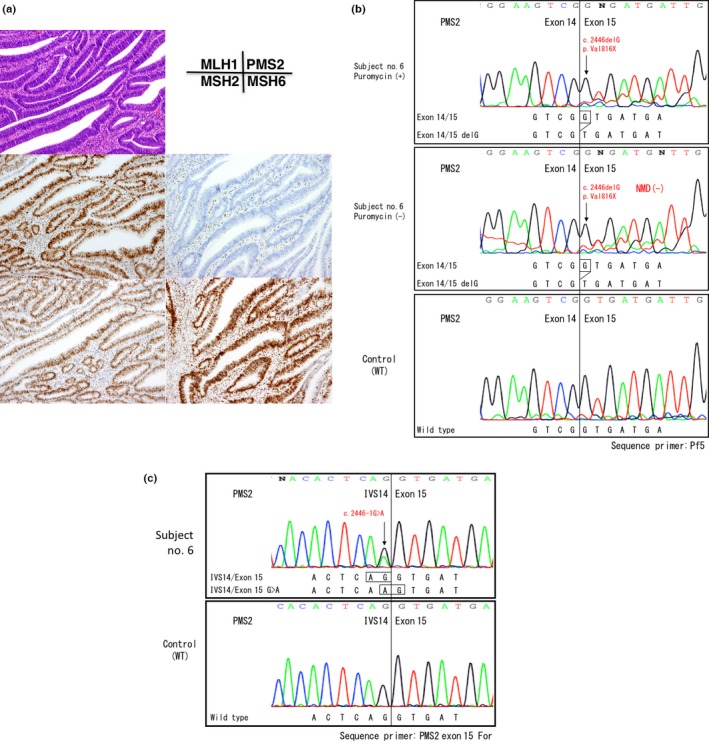
Gene testing of subject No. 6. (a) Immunochemistry (IHC) using four antibodies to mismatch repair (MMR) proteins. Note the loss of PMS2 protein expression in cancer cell nuclei. (b) RT‐PCR/direct sequencing analysis of the PMS2 gene in subject No. 6. The upper panel indicates the sequencing profile showing the c.2446delG (p.Val816Stop) frameshift mutation from a puromycin‐treated total RNA sample and the middle panel shows the sequencing profile without the puromycin treatment. Note that the signal ratios of the frameshift mutation to the wild‐type sequence were almost the same with or without the puromycin treatment, indicating the absence of nonsense‐mediated mRNA decay (NMD). (c) Long/nested PCR/direct sequencing analysis of the PMS2 gene. An analysis of genomic DNA by a long PCR/nested PCR/direct sequencing analysis showed the c.2446‐1G>A mutation in the splicing acceptor site of exon 15 (black arrow). Because exon 15 was the last exon of the PMS2 gene, a mutation in the exon–intron boundary activated cryptic splicing site and caused a 1‐bp‐deleted frameshift mutation in mRNA without NMD.
In the analysis using MLPA, we examined the total copy numbers of PMS2 and PMS2CL genes; the copy number detected by the probes hybridizing the consensus sequences in PMS2 or the PMS2CL genes from exons 11 to 15 in 96 subjects. In 93 subjects, the total copy number was 4 throughout exons 11 to 15, while the copy number of each exon in PMS2 and PMS2CL differed due to gene conversions. We summarized the combination of copy number alterations in each exon of PMS2 and PMS2CL (Fig. 6a). Two types of genomic deletions in the PMS2 gene were detected in three subjects from 2 index patients and 1 relative; that is, genomic deletions from exons 11 to 15 (subject number 5 and his relative) and from exons 12 to 15 (subject no. 4) (Fig. 6b,c). Gene conversion between PMS2 and PMS2CL was infrequent in exon 11 and IVS 12; therefore, the loss of the PMS2 gene copy number in exon 11 and IVS 12 implies a sign of the genomic deletion of the PMS2 gene. In the analysis of exons 13 and 14, 41/93 (44.1%) or 42/93 (45%) normal subjects had more than three copies of the PMS2 gene in exons 13 and 14, respectively, while 37/93 (39.8%) normal subjects had more than three copies of the PMS2CL gene in exon 15.
Figure 6.
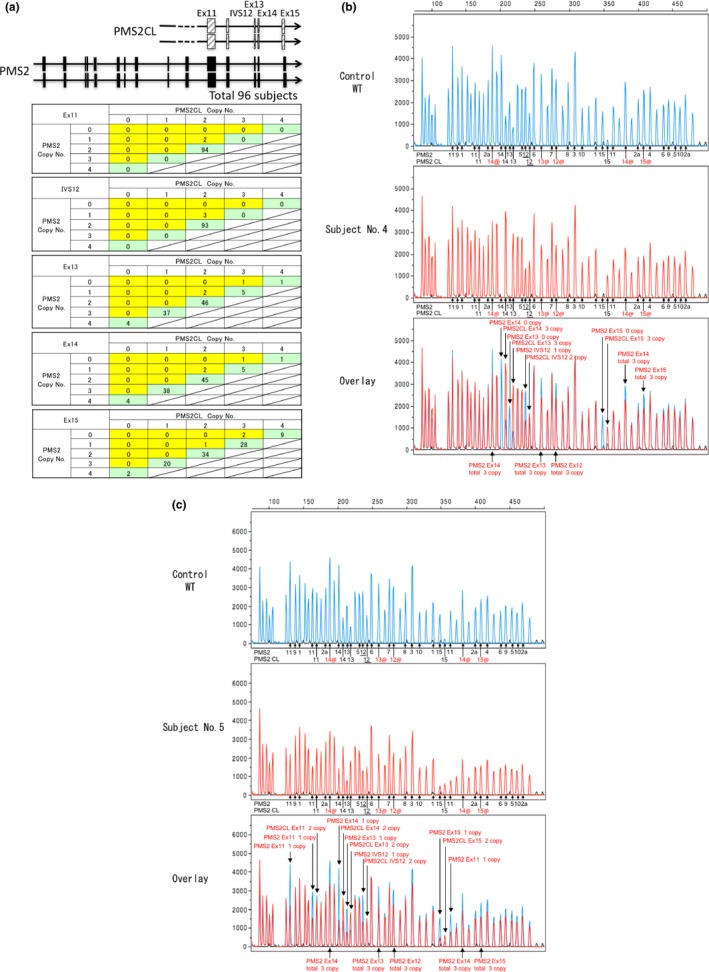
Multiplex ligation‐dependent probe amplification (MLPA) analysis of the PMS2 gene. In (b) and (c), the underlined number 12 indicates the IVS12 of the PMS2 or the PMS2CL gene, where the probes were designed to anneal. Numbers followed by@ and in red※ indicate the position of the probes hybridizing the consensus sequences of the PMS2 and PMS2CL genes and the signals indicate the total copy numbers of the PMS2 and PMS2CL genes in exons 12, 13, 14 and 15. (a) The PMS2 downstream exon 11 sequence has close homology with PMS2CL paralogues and gene conversion frequently occurred between PMS2 and PMS2CL. Ninety‐six subjects were analyzed by MLPA and the copy numbers of the PMS2,PMS2CL and consensus sequences between PMS2 and PMS2CL were evaluated from exons 11 to 15. In normal subjects, the copy numbers of the consensus sequences were 4 (green box) and, in the case of a genomic deletion, the total copy number decreased to 3 (yellow box). (b) The genomic deletion of PMS2 exons 12–15 was detected in subject No. 4. (c) The genomic deletion of PMS2 exons 11–15 was detected in subject No. 5.
Discussion
The classical phenotype of Lynch syndrome, also known as hereditary non‐polyposis colorectal cancer (HNPCC), was defined by the Amsterdam criteria in 1990,13 which were characterized by the “3‐2‐1” rule as follows:
At least three relatives have histologically confirmed CRC, 1 of whom is a first degree relative of the other 2; familial adenomatous polyposis must be excluded;
At least two successive generations are involved;
At least 1 of the cancers was diagnosed before the age of 50.
Patients meeting the Amsterdam criteria were eligible for the germline DNA testing of MMR genes, mostly giving rise to deleterious mutations in either the MSH2 or MLH1 gene. In this study, all subjects were postoperative CRC patients fulfilling either the revised Bethesda Guidelines14 or CRC patients younger than 70 years at the age of disease onset.5 The latter condition appears to be slightly too broad to screen for Lynch syndrome as the CRC younger than 70 years made up 43% (53357/124921) of all CRC cases in Japan in 2011,15 while those fulfilling the Amsterdam criteria in Japan comprised 0.2% of all CRC cases.16 In the present study, the loss of PMS2 protein expression alone was observed in seven subjects (1.39%), among which only three subjects were compatible with the revised Bethesda Guideline and none fulfilled the Amsterdam criteria, as indicated in the pedigrees (Table 1 and Fig. S1). Subjects with the loss of PMS2 protein expression alone in CRC tissues were all male and a significant difference was observed between the genders (P = 0.0233). A total of six subjects developed CRC between 50 and 63 years, and 1 subject developed CRC at 40 years. Ten Broeke et al.17 report that the cumulative risk of CRC for male PMS2 mutation carriers by the age of 70 years was 19% in their study on the familial cancer registry, while that among female carriers was 11% for CRC and 12% for endometrial cancer (EC). The mean age of CRC development was 52 years, and there was a significant difference in the mean age of CRC between the probands (mean, 47 years; range, 26–68 years) and other family members with a PMS2 mutation (mean, 58 years; range, 31–86 years; P < 0.001). They conclude that CRC and EC risks were markedly lower than those previously reported for the other MMR genes, not withstanding the influence of genetic modifiers and lifestyle factors on cancer risks.17
Gene testing was completed in seven subjects and deleterious PMS2 mutations were confirmed in 6 (85.7%), among which RT‐PCR detected four deleterious mutations and MLPA detected two genomic deletions. The diagnosis of PMS2 gene mutations still appears to be challenging due to the existing pseudogenes, the number of which is estimated to be at least 15.11 Clendenning et al.11 reported a method to avoid the interference of the pseudogenes using a two‐step amplification of the PMS2 gene, with the first step involving the amplification of the PMS2 gene in four large fragments, followed by exon‐specific nested PCR amplification. They analyzed 41 samples that were negative for the expression of PMS2 alone or that of PMS2/MLH1 without germline MLH1 mutations and identified 11 putative pathogenic mutations. In an analysis of subjects showing the loss of PMS2 protein expression alone, 10/16 (62.5%) subjects had putative pathogenic mutations, while missense variants were detected at the same time and some were pointed out to be artifacts caused by the pseudogene interference.18
An RT‐PCR/direct sequencing analysis using total RNA extracted from puromycin‐treated PBL may be a useful option for avoiding pseudogene interference. Messenger RNA harboring deleterious mutations is degradable due to NMD. Puromycin is a protein translation inhibitor that is known to suppress NMD. Total RNA extracted from puromycin‐treated PBL was subjected to an RT‐PCR/direct sequencing analysis. This method has been used as a routine step in our laboratory for the gene testing of Lynch syndrome‐related genes such as MSH2 and MLH1,9 and we applied this technique to detect PMS2 gene mutations.
Subject no. 2 showed a frameshift mutation skipping to exon 5 in the midst of exon 4 (r.301_353del) (data not shown). The mRNA abundance of this frameshift mutation was modest, but enhanced in the presence of puromycin treatment, so that we speculated this was likely to be a pathogenic mutation resulting NMD. In analysis using genomic DNA, however, no mutations were detected in sequences spanning exons 4 and 5. There seem to be several explanations for this discrepancy between RT‐PCR and PCR. First, a number of pseudogenes were reported, particularly in this region, rendering a difficulty in sequencing exons 4 and 5 of the PMS2 gene, even if examined using the long PCR/nested PCR approach (Fig. 1).18 Second, the modest signal of the frameshift mutation in RT‐PCR may imply the presence of a germline mosaicism in PBL, which could not be detected by the routine PCR/direct sequencing approach.
In subject no. 6, we detected r.2446delG mutation in the exon–intron boundary of the last coding exon of the PMS2 (c.2446‐1G>A). Reportedly, the nonsense mutation in the last exon does not result in NMD, as suggested by the fact that the stop codon is located in the last exon in the majority of genes (80%).19
In subject no. 7, c.241G>T, p.Glu81Stop mutation detected in exon 3 of the PMS2 gene appeared to be a homozygous mutation. Homozygous mutation of the PMS2 gene accounts for a constitutive mismatch repair deficiency syndrome (CMMR‐D) or biallelic mismatch repair deficiency (BMMR‐D), which is characterized as follows: (i) early onset gastrointestinal adenocarcinoma (<20 year‐old) or multiple adenomatous polyps; or (ii) childhood brain, lymphoma/leukemia with one of the following features such as consanguinity or neurofibromatosis type 1 (NF1) features.20 As abnormal IHC findings were observed in both normal and tumor tissues, IHC for the MMR proteins would be helpful in the diagnosis of CMMR‐D.21 In subject no. 7, the age at onset was late (i.e. 63 years old) and normal colonic gland and stromal lymphocytes expressed PMS2 protein. These findings were incompatible with clinical features of CMMR‐D. Another explanation may be a failure to amplify the wild‐type allele for unknown reason. No information was obtained about the consanguinity, but gene testing of the first‐degree relatives may be helpful to elucidate the possibility of homozygosity for the c.241G>T, p.Glu81Stop mutation.
In subject nos. 4 and 5, the genomic deletion spanning exon 15 of the PMS2 gene was detected by MLPA. Because the reverse primer for RT‐PCR was designed to anneal to the 3′ untranslated region in exon 15, this primer set did not amplify the cDNA sequence lacking exon 15. Therefore, the combined use of MLPA may be useful. The presence of the pseudogene PMS2CL also adds to the complexity of interpreting the data obtained using MLPA. The PMS2CL sequence shows close similarities with the PMS2 gene downstream of exon 12 and frequent gene conversion occurs between PMS2 and PMS2CL. The MLPA kit designed to analyze the PMS2 gene may be used to quantify the copy numbers of the PMS2, PMS2CL and consensus sequences from exons 11 to 15 in order to identify copy number aberrations in each exon. Because we initially had no control samples for this assay, we analyzed a total of 96 samples, including those harboring deleterious mutations in other MMR genes or normal control subjects to estimate the copy number variations from exons 11 to 15 of the PMS2 or the corresponding sequences of the PMS2CL genes (Fig. 6a). Only three subjects had one or zero copies in PMS2 and three copies in consensus sequences, indicating the loss of the PMS2 allele, and these results were consistent with the loss of the PMS2 protein expression shown by IHC. In the other subjects, the copy number of the consensus sequence was 4 from exons 11 to 15, while the ratio of PMS2 and PMS2CL genes differed between exons. In exons 11 and 12 (designated as IVS12), the copy number in PMS2 and PMS2CL was 2, indicating that gene conversion is an infrequent genetic event in this region, while the copy numbers of PMS2 and PMS2CL differed in exons 13, 14 and 15. There were more than three copies of PMS2 in exons 13 and 14 and more than three copies of PMS2CL in exon 15. Variable copy numbers in exons 13, 14 and 15 indicate the presence of the frequent gene conversions between PMS2 and PMS2CL in this region (Fig. 6a), and the decrease of the total copy numbers for PMS2 and PMS2CL suggests the presence of the genomic rearrangement either in the PMS2 or the PMS2CL gene, but it is difficult to confirm this without knowing the IHC results.
The prevalence of germline PMS2 mutations has been reported to vary between 2% and 8.7%,22, 23, 24 while only two mutations have been reported in Japanese patients to date.2, 3 In the present study, the prevalence of PMS2 mutations was estimated in Japanese CRC subjects with prior IHC screening for the expression of PMS2, in combination with the RT‐PCR/direct sequencing analysis using puromycin‐treated RNA samples and MLPA. This study is ongoing and will ultimately provide comprehensive information on the prevalence of other MMR gene mutations.
In conclusion, 7 (1.40%) out of the 501 subjects examined showed the loss of PMS2 protein expression alone and were referred to the genetic counseling clinic. Germline PMS2 mutations were detected in 6 (85.7%), including 3 nonsense and 1 frameshift mutations detected by RT‐PCR/direct sequencing and two genomic deletions detected by MLPA. IHC results predicted PMS2 germline mutations. RT‐PCR from puromycin‐treated PBL enabled the detection of PMS2 germline mutations by suppressing NMD. Similarly, the MLPA analysis effectively quantified copy numbers, particularly in exons 14 and 15 in PMS2 and its paralogous sequence, PMS2CL, showing the complexity due to gene conversion. As the pseudogene interference may hamper a diagnosis of the pathogenic PMS2 mutations, the IHC screening, RT‐PCR from puromycin‐treated RNA, the long‐range PCR/nested PCR approach and MLPA are all important diagnostic modalities, and their use in combination may achieve a high standard in diagnosing germline mutations of the PMS2 gene.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. Pedigrees corresponding to subjects No. 1 through No. 7.
Table S1. PCR primer and sequence primer sequences for RT‐PCR and RT‐PCR conditions.
Table S2. PCR primer sequences for a long PCR/nested PCR/direct sequencing analysis.
Acknowledgments
This research was partially supported by The National Cancer Center Research and Development Fund (25‐A‐1), the Practical Research for Innovative Cancer Control and the Program for Promoting Practical Applications of Genomic Medicine from the Japan Agency for Medical Research and Development, AMED (15ck0106097 h0102 and 15cK0106168 h0201). The author thanks Kyouko Takai, CGC for her assistance with the preparation of figures.
Cancer Sci 107 (2016) 1677–1686
Funding Information
This research was partially supported by The National Cancer Center Research and Development Fund (25‐A‐1), the Practical Research for Innovative Cancer Control and the Program for Promoting Practical Applications of Genomic Medicine from the Japan Agency for Medical Research and Development, AMED (15ck0106097 h0102 and 15cK0106168 h0201).
References
- 1. Vasen HF, Möslein G, Alonso A et al Guidelines for the clinical management of Lynch syndrome (hereditary non‐polyposis cancer). J Med Genet 2007; 44: 353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miyaki M, Nishio J, Konishi M et al Drastic genetic instability of tumors and normal tissues in Turcot syndrome. Oncogene 1997; 15: 2877–81. [DOI] [PubMed] [Google Scholar]
- 3. Nomura S, Fujimoto Y, Yamamoto N et al A case of early onset rectal cancer of Lynch syndrome with a novel deleterious PMS2 mutation. Jpn J Clin Oncol 2015; 45: 987–92. [DOI] [PubMed] [Google Scholar]
- 4. Mojtahed A, Schrijver I, Ford JM, Longacre TA, Pai1 RK. A two‐antibody mismatch repair protein immunohistochemistry screening approach for colorectal carcinomas, skin sebaceous tumors, and gynecologic tract carcinomas. Mod Pathol 2011; 24: 1004–14. [DOI] [PubMed] [Google Scholar]
- 5. Balmaña J, Balaguer F, Cervantes A, Arnold D, ESMO Guidelines Working Group . Familial risk‐colorectal cancer: ESMO Clinical Practice Guidelines. Ann Oncol 2013; 24(Suppl 6): 73–80. [DOI] [PubMed] [Google Scholar]
- 6. Sekine S, Ogawa R, Oshiro T et al Frequent lack of GNAS mutations in colorectal adenocarcinoma associated with GNAS‐mutated villous adenoma. Genes Chromosomes Cancer 2014; 53: 366–72. [DOI] [PubMed] [Google Scholar]
- 7. Boland CR, Thibodeau SN, Hamilton SR et al A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58: 5248–57. [PubMed] [Google Scholar]
- 8. Miyakura Y, Sugano K, Konishi F et al Extensive methylation of hMLH1 promoter region predominates in proximal colon cancer with microsatellite instability. Gastroenterology 2001; 121: 1300–9. [DOI] [PubMed] [Google Scholar]
- 9. Nomura S, Sugano K, Kashiwabara H et al Enhanced detection of deleterious and other germline mutations of hMSH2 and hMLH1 in Japanese hereditary nonpolyposis colorectal cancer kindreds. Biochem Biophys Res Commun 2000; 271: 120–9. [DOI] [PubMed] [Google Scholar]
- 10. Sugano K, Kyogoku A, Fukayama N et al Methods in laboratory investigation. Rapid and simple detection of c‐Ki‐ras2 gene codon 12 mutations by nonradioisotopic single‐strand conformation polymorphism analysis. Lab Invest 1993; 68: 361–6. [PubMed] [Google Scholar]
- 11. Clendenning M, Hampel H, LaJeunesse J et al Long‐range PCR facilitates the identification of PMS2‐specific mutations. Hum Mutat 2006; 27: 490–5. [DOI] [PubMed] [Google Scholar]
- 12. Wernstedt A, Valtorta E, Armelao F et al Improved multiplex ligation‐dependent probe amplification analysis identifies a deleterious PMS2 allele generated by recombination with crossover between PMS2 and PMS2CL. Genes Chromosomes Cancer 2012; 51: 819–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non‐Polyposis Colorectal Cancer (ICG‐HNPCC). Dis Colon Rectum 1991; 34: 424–5. [DOI] [PubMed] [Google Scholar]
- 14. Umar A, Boland CR, Terdiman JP et al Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96: 261–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsuda A, Matsuda T, Shibata A et al Cancer incidence and incidence rates in Japan in 2008: a study of 25 population‐based cancer registries for the Monitoring of Cancer Incidence in Japan (MCIJ) Project. Jpn J Clin Oncol 2013; 44: 388–96. [DOI] [PubMed] [Google Scholar]
- 16. Kawakami K, Baba S. Analysis of the HNPCC registries data reported at the 43rd Japanese society for cancer of the colon and rectum (ISCC) meeting. New strategies for treatment of hereditary colorectal cancer. Churchill Livingstone, Tokyo, 1996, pp. 229–33.
- 17. ten Broeke SW, Brohet RM, Tops CM et al Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol 2015; 33: 319–25. [DOI] [PubMed] [Google Scholar]
- 18. Niessen RC, Kleibeuker JH, Jager PO, Sijmons RH, Hofstra RM. Letter to the editor: Getting rid of the PMS2 pseudogenes: mission impossible? Hum Mutat 2007; 28: 414. [DOI] [PubMed] [Google Scholar]
- 19. Nagy E, Maquat LE. A rule for termination‐codon position within intron‐containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 1998; 23: 198–9. [DOI] [PubMed] [Google Scholar]
- 20. Durno CA, Sherman PM, Aronson M et al Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR‐D) syndrome. Eur J Cancer 2015; 51: 977–83. [DOI] [PubMed] [Google Scholar]
- 21. Bakry D, Aronson M, Durno C et al Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer 2014; 50: 987–96. [DOI] [PubMed] [Google Scholar]
- 22. Hampel H, Frankel WL, Martin E et al Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352: 1851–60. [DOI] [PubMed] [Google Scholar]
- 23. Hendriks YM, Jagmohan‐Changur S, van der Klift HM et al Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 2006; 130: 312–22. [DOI] [PubMed] [Google Scholar]
- 24. Niessen RC, Kleibeuker JH, Westers H et al PMS2 involvement in patients suspected of Lynch syndrome. Genes Chromosom Cancer 2009; 48: 322–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Pedigrees corresponding to subjects No. 1 through No. 7.
Table S1. PCR primer and sequence primer sequences for RT‐PCR and RT‐PCR conditions.
Table S2. PCR primer sequences for a long PCR/nested PCR/direct sequencing analysis.