

Abstract
Next‐generation sequencing (NGS) and digital PCR technologies allow analysis of the mutational profile of circulating cell‐free DNA (cfDNA) in individuals with advanced lung cancer. We have now evaluated the feasibility of cfDNA sequencing for mutation detection in patients with non‐small cell lung cancer at earlier stages. A total of 150 matched tumor and serum samples were collected from non‐small cell lung cancer patients at stages IA–IIIA. Amplicon sequencing with DNA extracted from tumor tissue detected frequent mutations in EGFR (37% of patients), TP53 (39%), and KRAS (10%), consistent with previous findings. In contrast, NGS of cfDNA identified only EGFR,TP53, and PIK3CA mutations in three, five, and one patient, respectively, even though adequate amounts of cfDNA were extracted (median of 4936 copies/mL serum). Next‐generation sequencing showed a high accuracy (98.8%) compared with droplet digital PCR for cfDNA mutation detection, suggesting that the low frequency of mutations in cfDNA was not due to a low assay sensitivity. Whereas the yield of cfDNA did not differ among tumor stages, the cfDNA mutations were detected in seven patients at stages IIA–IIIA and at T2b or T3. Tumor volume was significantly higher in the cfDNA mutation‐positive patients than in the negative patients at stages T2b–T4 (159.1 ± 58.0 vs. 52.5 ± 9.9 cm3, P = 0.014). Our results thus suggest that tumor volume is a determinant of the feasibility of mutation detection with cfDNA as the analyte.
Keywords: Cell‐free DNA, mutation, non‐small cell lung cancer, sequencing, tumor volume
Lung cancer is the leading cause of death related to cancer worldwide, with non‐small cell lung cancer (NSCLC) being the most common form of this disease.1 Targeted therapeutics such as tyrosine kinase inhibitors of the epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) have recently been introduced into clinical practice for individuals with NSCLC positive for actionable mutations of EGFR or ALK fusions, respectively. Molecular profiling that is able to predict the response to such drugs has thus become an important therapeutic strategy, allowing selection of the most appropriate treatment for individual patients.2 This strategy is limited, however, by the difficulty of obtaining tumor specimens, the collection of which often requires invasive procedures.
Sequencing of circulating cell‐free DNA (cfDNA), a non‐invasive approach to the detection of aberrant tumor‐derived DNA in blood, has the potential to allow early identification and management of solid tumors as well as prediction of drug sensitivity or resistance. Several studies have evaluated cfDNA as a potential biomarker in NSCLC patients, who tend to have a higher plasma cfDNA concentration than healthy individuals.3, 4, 5
Tumors at an advanced stage often shed cfDNA into the circulation, and mutations in this cfDNA can be detected with PCR‐based6, 7, 8, 9, 10 or sequencing‐based5, 11, 12, 13 assays. Deep sequencing of amplicons has proved feasible for the detection of somatic mutations in cfDNA if the total number of reads exceeds 300 000.13 Digital PCR is also a highly sensitive technology that allows the detection of mutations in cfDNA with a high accuracy relative to those in tumor cell DNA in individuals with advanced lung cancer.10 The clinicopathologic factors that are associated with the feasibility of mutation identification in cfDNA remain unknown, however. We have now compared the mutation profiles of surgically resected tumor specimens from patients with NSCLC of stage IA to IIIA with those of matched serum samples in order to ascertain the feasibility of mutation detection in cfDNA at such early disease stages as well as its determinant factors.
Materials and Methods
Patients and specimen collection
Matched lung cancer tissue and serum specimens were collected from 150 patients who underwent surgery for NSCLC at Tokyo Medical University Hospital (Tokyo, Japan) from January 2013 to July 2014. All tissue samples were immediately flash‐frozen in liquid nitrogen and stored at −80°C until analysis. Tumor volume was calculated as length × width × height during surgery. Blood samples were also collected during surgery and were centrifuged at 1400 g for 10 min, with serum being stored at −80°C until analysis. All patients provided written informed consent to participate in the study, including the collection of tumor and serum specimens for analysis. The study protocol was approved by the institutional ethics committees of Kindai University Faculty of Medicine (Osaka‐Sayama, Japan; approval no. 25‐135) and Tokyo Medical University Hospital (approval no. 2541).
DNA was isolated from the frozen tumor tissue with the use of an AllPrep DNA/RNA Mini Kit (Qiagen, Valencia, CA, USA). The quality and quantity of the DNA were verified with the use of a NanoDrop 2000 device (Thermo Fisher Scientific, Waltham, MA, USA) and PicoGreen dsDNA Assay Kit (Life Technologies, Foster City, CA, USA). Cell‐free DNA was purified from 0.52 to 1.0 mL serum with the use of a QIAamp Circulating Nucleic Acid Kit (Qiagen), and its copy number was determined with an RNaseP copy number assay (Life Technologies).
Sample processing
Sequencing analysis
Tumor DNA and cfDNA samples were subjected to analysis with next‐generation sequencing (NGS) panels for mutation detection. For library preparation, tumor DNA (10 ng) and cfDNA (maximum of 3000 copies) were subjected to multiplex PCR amplification with the use of an Ion AmpliSeq Library Kit 2.0 (Life Technologies) and Ion AmpliSeq Colon and Lung Cancer Panel version 2 (Life Technologies), the latter of which targets mutational hotspot regions of 22 cancer‐associated genes: AKT1, ALK, BRAF, CTNNB1, DDR2, EGFR, ERBB2, ERBB4, FBXW7, FGFR1, FGFR2, FGFR3, KRAS, MAP2K1, MET, NOTCH1, NRAS, PIK3CA, PTEN, SMAD4, STK11, and TP53. The PCR products were then ligated to Ion Xpress Barcode Adapters (Life Technologies) and purified with the use of Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA). The purified libraries were pooled and then sequenced with an Ion Torrent Proton instrument, Ion Proton Hi‐Q Sequencing Kit, and Ion PI version 3 Chip (all from Life Technologies). DNA sequencing data were accessed through the Torrent Suite version 4.4 program (Life Technologies). Reads were aligned with the hg19 human reference genome, and variants were called with the use of Variant Call Format version 4.4. Samples with a median coverage of <1000 per amplicon were not accepted for evaluation. For detection of mutations in tumor tissue DNA, raw variant calls were filtered with the following annotations: quality score of <100 and depth of coverage of <19. Germline mutations were excluded with the use of the Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB).14 For cfDNA mutation detection, the nucleotide count for each target base position from each allele count file in the variant calling report was used. The presence of mutant nucleotides was determined with the use of Poisson distribution statistics. The nucleotide count for each base position was applied to the Poisson function with the use of Microsoft Excel 2013 (Redmond, WA, USA). The value of the baseline error rate (non‐reference nucleotide count divided by the total nucleotide count) plus three standard deviations calculated from 10 normal blood DNA samples was used for the Poisson parameter lambda (λ) ranging from 0.00045 to 0.11859. If the nucleotide count of the mutant was significantly higher (α = 2 × 10−5) than that of the Poisson probability, the nucleotide was judged to have a mutation‐positive status. We established the α value with reference to a previous study.11 The minimum detection limit was calculated for a read number per site of 30 000 and α of 0.00002, with all values being shown in Table S1. The Ion AmpliSeq Colon and Lung Cancer Panel version 2 is designed to amplify 92 amplicons covering 1205 hot spot mutations. The theoretical minimum detection limit ranged from 12.7% to 0.103% and was <0.5% for 84.4% (1017/1205) of hot spot sites.
Digital PCR analysis
Digital PCR was carried out with a QX100 Droplet Digital PCR System (Bio‐Rad, Hercules, CA, USA). The primers and probes for detection of the EGFR L858R, KRAS G12C, and PIK3CA E545K and H1047R mutations were obtained from Bio‐Rad. The cycling conditions included an initial incubation at 95°C for 10 min, 40 cycles of 94°C for 30 s and 55°C for 60 s, and enzyme inactivation at 98°C for 10 min. After thermal cycling, the plates were transferred to a Droplet reader (Bio‐Rad), and the digital PCR data were analyzed with the Quanta Soft analytic software package (Bio‐Rad). The cut‐off values for mutations were determined with the use of plasma cfDNA and normal blood DNA derived from 10 healthy volunteers. Each cut‐off was set at 3 copies per one reaction because no background noise (0 copies per one reaction) was detected for any of the mutants in plasma cfDNA and normal blood DNA derived from the 10 healthy volunteers.10
Statistical analysis
Statistical analysis was carried out using the two‐tailed Student's t‐test (Prism software; GraphPad Software, San Diego, CA, USA). A P‐value of < 0.05 was considered statistically significant.
Results
Somatic mutations detected in lung cancer tissue by NGS
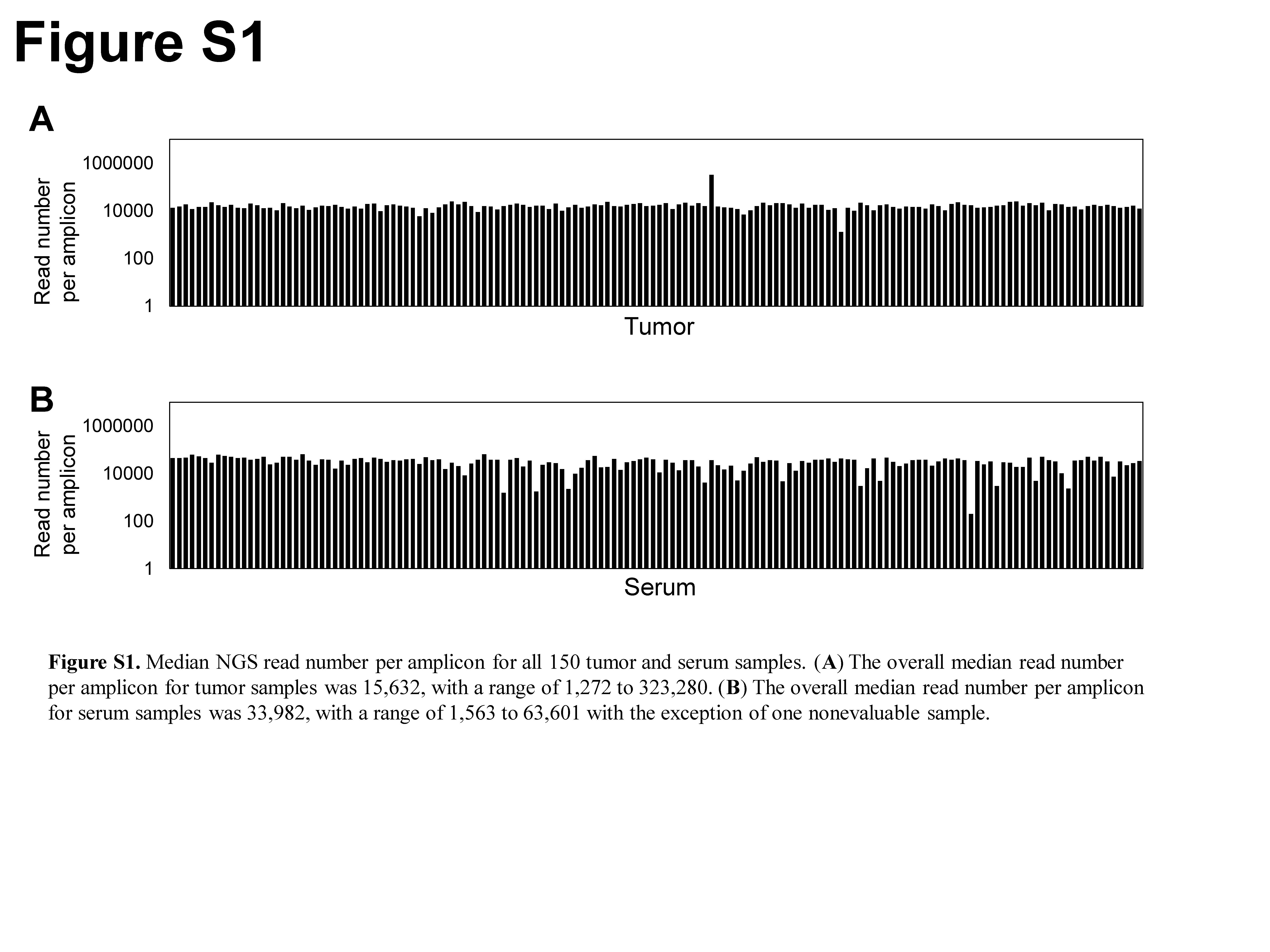
Frozen surgically resected tumor tissue and matched serum samples were obtained from 150 NSCLC patients (Table 1). All tumor DNA samples were successfully sequenced with the Ion Proton platform. The median read number per amplicon was 15 632, with a range of 1272 to 323 280 (Fig. S1). We identified TP53 mutations in 58 cases (38.7%), EGFR mutations in 56 (37.3%), KRAS mutations in 15 (10.0%), CTNNB1 mutations in 7 (4.7%), ERBB2, PIK3CA, BRAF, and PTEN mutations in 3 each (2.0%), and ERBB4, MET, ALK, FGFR2, NRAS, AKT1, and FBXW7 mutations in 1 each (0.7%) (Fig. 1a). No mutation was detected in 22.0% (33/150) of the samples. Mutation profiles for patients harboring at least one mutation are shown in Figure 1(b).
Table 1.
Characteristics of patients with non‐small cell lung carcinoma (n = 150) who participated in this study
| Characteristic | Classification | No. (%) |
|---|---|---|
| Age, years | Median (range) | 69 (23–85) |
| <65 | 51 (34.0) | |
| ≥65 | 99 (66.0) | |
| Sex | Male | 80 (53.3) |
| Female | 70 (46.7) | |
| Smoking status | Yes | 97 (64.7) |
| No | 48 (32.0) | |
| Unknown | 5 (3.3) | |
| Pathological stage | IA | 63 (42.0) |
| IB | 32 (21.3) | |
| IIA | 20 (13.3) | |
| IIB | 10 (6.7) | |
| IIIA | 25 (16.7) | |
| Tumor size (T stage) | T1a | 39 (26.0) |
| T1b | 36 (24.0) | |
| T2a | 44 (29.3) | |
| T2b | 10 (6.7) | |
| T3 | 20 (13.3) | |
| T4 | 1 (0.7) | |
| Lymph node metastasis | N0 | 104 (69.3) |
| (N stage) | N1 | 19 (12.7) |
| N2 | 21 (14.0) | |
| NX | 6 (4.0) |
Figure 1.
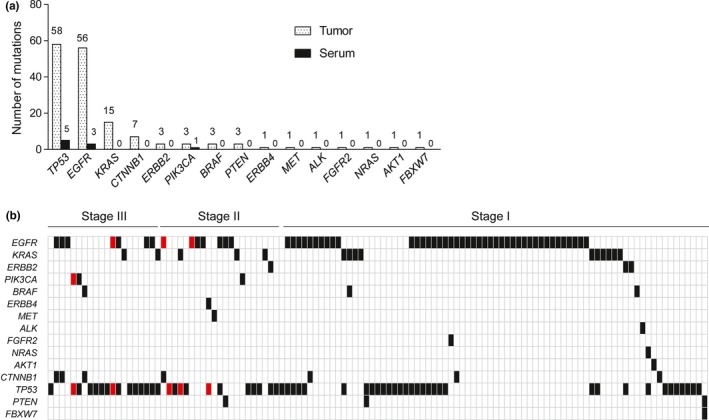
Mutation profiles for matched tumor and serum specimens of patients with non‐small cell lung carcinoma who participated in this study. (a) Incidence of mutations by gene in the study cohort as determined by next‐generation sequencing with tumor DNA and serum cfDNA samples. (b) Mutation profiles for the patients harboring at least one mutation. Black and red boxes indicate mutations detected in tumor samples alone or in both tumor and serum samples, respectively.
Somatic mutations detected in cfDNA by NGS
Serum cfDNA was extracted for all 150 patients, with a median yield (copy number) of 4936 (range, 572–373 658) (Fig. S2). A total of 149 of the 150 (99.3%) cfDNA samples were successfully sequenced with the Ion Proton platform, with sequencing failure being due to an insufficient read number per amplicon (case no. 200, 197 reads) in the one unsuccessful case. The median read number per amplicon for the 149 successfully sequenced cfDNA samples was 33 982, with a range of 1563–63 601 (Fig. S1).
We identified only a small number of non‐synonymous somatic mutations in cfDNA, including TP53 mutations in 5 cases (3.3%), EGFR mutations in 3 cases (2.0%), and a PIK3CA mutation in 1 case (0.7%) (Fig. 1a). The mutation profiles for patients harboring cfDNA mutations are shown in Figure 1(b). Compared with tumor tissue samples, the sensitivity and specificity for mutation detection in serum were 5.8% (9/155) and 100% (33/33), respectively, with an accuracy of 22.3% (42/188).
Accuracy of somatic mutation detection by NGS and digital PCR
To exclude the possibility of false negative results generated by NGS, we applied digital PCR to confirm mutations detected with NGS. The L858R mutation of EGFR, G12C of KRAS, and E545K and H1047R of PIK3CA accounted for a substantial proportion of the mutations detected in the tumor samples with NGS. The 42 tumor samples determined to be positive for these mutations by NGS and the paired serum samples were therefore analyzed for the same mutations with digital PCR (Table 2). For the 42 tumor samples, all of the mutations detected by NGS were also detected by digital PCR. For the corresponding serum samples, the two mutations (EGFR L858R in case no. 79 and PIK3CA E545K in case no. 65) detected by NGS were confirmed with digital PCR; only one mutation (KRAS G12C in case no. 215) was detected in the remaining 40 serum samples by digital PCR. Compared with digital PCR, the sensitivity and specificity of NGS for mutation detection were 97.8% (44/45) and 100% (39/39), respectively, with an accuracy of 98.8% (83/84).
Table 2.
Concordance for detection of major somatic mutations in tumor tissue and serum samples from patients with non‐small cell lung carcinoma using next‐generation sequencing (NGS) and digital PCR
| Mutation | Case no. | NGS (% of variant reads) | Digital PCR (copy number) | ||
|---|---|---|---|---|---|
| Tumor | Serum | Tumor | Serum | ||
| EGFR L858R | 55 | 16.3 | n.d. | 2520 | n.d. |
| 66 | 45.8 | n.d. | 4880 | n.d. | |
| 71 | 24.2 | n.d. | 2280 | n.d. | |
| 74 | 33.4 | n.d. | 2240 | n.d. | |
| 79 | 100.0 | 0.6 | 16660 | 74 | |
| 82 | 18.5 | n.d. | 1208 | n.d. | |
| 86 | 35.7 | n.d. | 4160 | n.d. | |
| 103 | 43.6 | n.d. | 3720 | n.d. | |
| 105 | 15.1 | n.d. | 1150 | n.d. | |
| 114 | 26.8 | n.d. | 1740 | n.d. | |
| 117 | 23.6 | n.d. | 3040 | n.d. | |
| 118 | 15.6 | n.d. | 1064 | n.d. | |
| 126 | 41.8 | n.d. | 4000 | n.d. | |
| 135 | 26.2 | n.d. | 1900 | n.d. | |
| 142 | 10.4 | n.d. | 680 | n.d. | |
| 143 | 7.8 | n.d. | 566 | n.d. | |
| 155 | 25.6 | n.d. | 2340 | n.d. | |
| 156 | 28.7 | n.d. | 2340 | n.d. | |
| 161 | 19.9 | n.d. | 2000 | n.d. | |
| 164 | 18.0 | n.d. | 1286 | n.d. | |
| 168 | 33.5 | n.d. | 6240 | n.d. | |
| 171 | 27.9 | n.d. | 2280 | n.d. | |
| 176 | 32.2 | n.d. | 3320 | n.d. | |
| 177 | 57.1 | n.d. | 8720 | n.d. | |
| 192 | 4.3 | n.d. | 186 | n.d. | |
| 204 | 49.0 | n.d. | 5460 | n.d. | |
| 208 | 35.7 | n.d. | 3240 | n.d. | |
| 209 | 25.7 | n.d. | 1580 | n.d. | |
| 212 | 11.8 | n.d. | 1100 | n.d. | |
| 220 | 4.5 | n.d. | 238 | n.d. | |
| KRAS G12C | 73 | 4.9 | n.d. | 278 | n.d. |
| 78 | 24.2 | n.d. | 2200 | n.d. | |
| 120 | 6.6 | n.d. | 858 | n.d. | |
| 123 | 15.8 | n.d. | 1528 | n.d. | |
| 149 | 4.2 | n.d. | 352 | n.d. | |
| 162 | 12.8 | n.d. | 1228 | n.d. | |
| 194 | 30.9 | n.d. | 2540 | n.d. | |
| 206 | 12.0 | n.d. | 882 | n.d. | |
| 215 | 32.4 | n.d. | 2560 | 4 | |
| PIK3CA E545K | 65 | 58.6 | 3.5 | 11260 | 43 |
| 184 | 31.2 | n.d. | 2660 | n.d. | |
| PIK3CA H1047R | 138 | 24.9 | n.d. | 1499 | n.d. |
n.d., not detected.
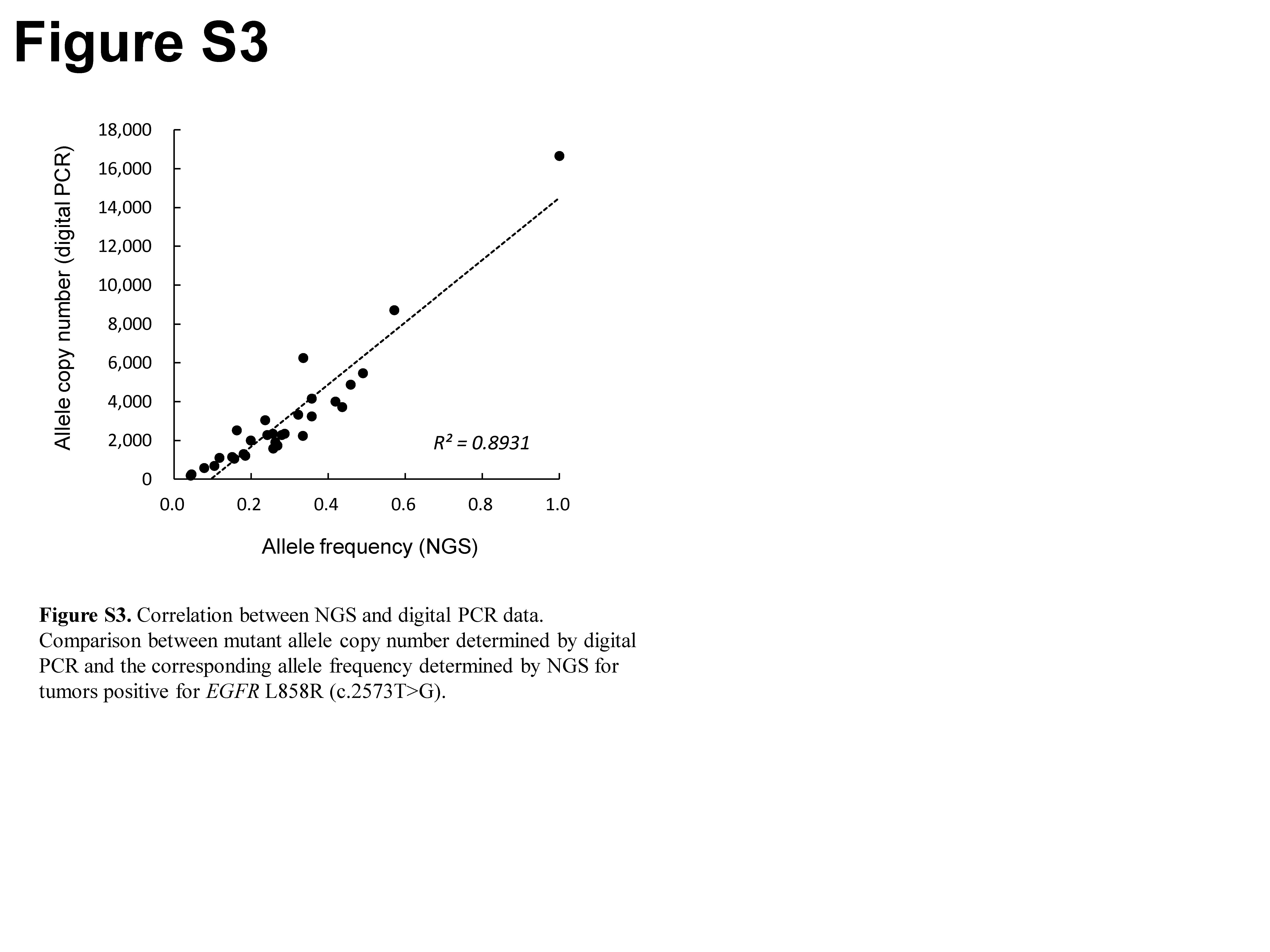
A comparison of variant allele frequency revealed a high correlation (R 2 = 0.992) between NGS and digital PCR for 30 tumor specimens positive for EGFR L858R (Fig. 2). We further examined the ability of NGS to detect mutant alleles by comparing the fraction of variant reads for these 30 tumor specimens positive for EGFR L858R with the corresponding allele copy number determined by digital PCR. The two parameters showed a good linear correlation (R 2 = 0.893) (Fig. S3), suggesting that the power of NGS for mutation detection was equivalent to that of digital PCR.
Figure 2.
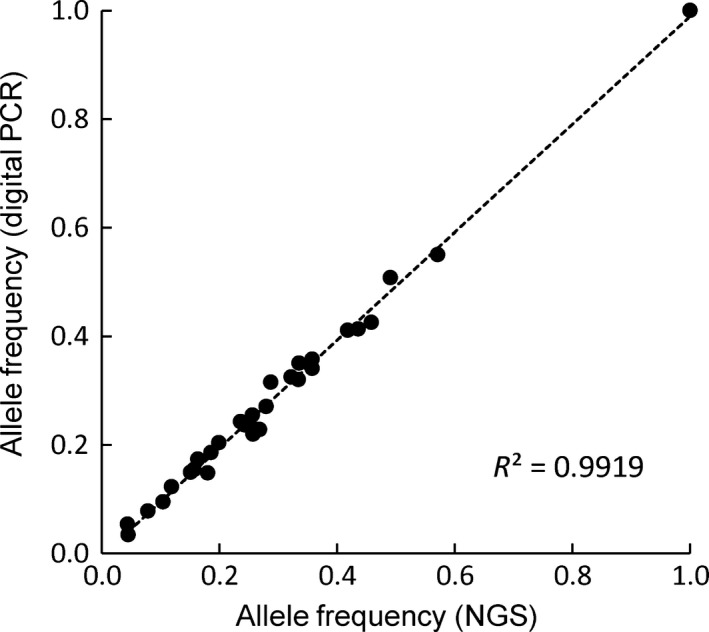
Correlation between next‐generation sequencing (NGS) and digital PCR data in a comparison between mutant allele frequency in EGFR L858R (c.2573T>G) mutation positive tumors. The correlation coefficient (R 2) is indicated.
To clear the baseline error rate in NGS assay, we measured the rate of two sample cohorts: 36 cfDNA samples in lung cancer patients with KRAS or EGFR exon 19 deletion mutation (KRAS/EGFRex19del) in tumor and 32 cfDNA samples in patients without any mutation in the tumor. Median allele frequency of cfDNA samples at position EGFR L858R (chr7:55259515) of NGS assay was 0.00631 (range, 0–0.11177) and 0.01046 (range, 0–0.04392) in tumor KRAS/EGFRex19del‐positive and no mutation groups, respectively. In addition, we examined the EGFR L858R mutation in cfDNA samples by digital PCR assay in these two groups. Allele frequency of EGFR L858R detected by digital PCR assay was 0.00 in all but one cfDNA sample of patient with no mutations in tumor (Fig. S4). These results indicate that the allele frequency was lower than the baseline error rate of NGS analysis.
Together, these results indicated that NGS performed well for mutation detection with both tumor tissue and serum samples.
Clinicopathologic features of cfDNA mutation‐positive patients
The study subjects included patients with surgically resectable NSCLC of stage IA–IIIA (Table 1), with more than half of the patients having a disease stage of IA or IB (95/150, 63.3%). We next examined the possible association of clinicopathologic features with serum cfDNA status. Determination of DNA copy number for cfDNA by quantitative PCR analysis revealed no statistically significant association between cfDNA yield and either disease stage (Fig. S2b) or T factor (Fig. S2c). The seven cases for which mutations were detected in cfDNA by NGS included two patients at stage IIA (T2b/N0/M0), three at stage IIB (two T3/N0/M0 and one T2b/N1/M0), and two at stage IIIA (T2b/N2/M0 and T3/N2/M0) (Fig. 3). Among patients with mutation‐positive tumors, mutations were detected in cfDNA for 2 of 17 patients at stage IIA, 3 of 5 patients at stage IIB, and 2 of 20 patients at stage IIIA, with no mutations being detected in cfDNA of any patient at stage IA or IB (Fig. 4a). With regard to T factor of the TNM classification, no mutation was detected in cfDNA of patients at T1a–T2a, with all such mutations being found in those at T2b or T3 (Fig. 4b). Mutations in cfDNA were detected for patients at N0, N1, or N2 of the TNM classification (Fig. 4c). These results thus suggested that detection of mutations in cfDNA of patients with disease at stage IA or IB or at T2a or lower is difficult, and that the feasibility of mutation detection with cfDNA may depend on the T factor rather than the N factor. We therefore compared tumor volume determined pathologically between patients positive or negative for cfDNA mutation among the subpopulation of patients with mutation‐positive tumors and a T factor of T2b–T4. Tumor volume in the cfDNA mutation‐positive group was significantly greater than that in the cfDNA mutation‐negative group (159.1 ± 58.0 vs. 52.5 ± 9.9 cm3, P = 0.014) (Fig. 4d). The maximum tumor diameter calculated at diagnosis was also larger in the cfDNA mutation‐positive group than in the cfDNA mutation‐negative group (5.3 ± 0.7 vs. 4.1 ± 0.3 cm, P = 0.050) (Fig. 4e). These results suggested that tumor volume is a determining factor for the feasibility of mutation detection with cfDNA.
Figure 3.
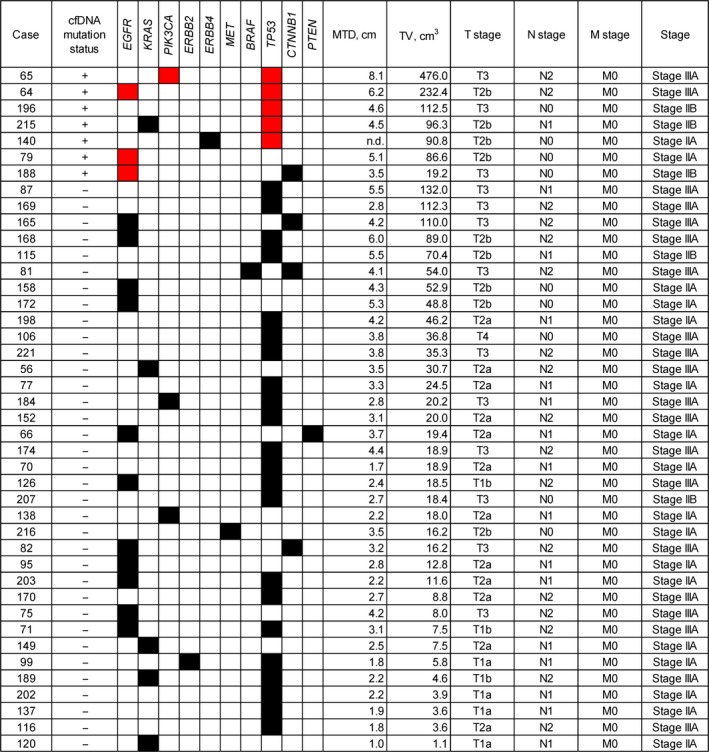
Mutation profiles and clinicopathologic characteristics for 42 mutation‐positive patients with non‐small cell lung carcinoma with a pathological stage of IIA–IIIA. Black and red boxes indicate mutations detected in tumor samples alone or in both tumor and serum samples, respectively. cfDNA, cell‐free DNA; MTD, maximum tumor diameter evaluated by diagnostic imaging; TV; tumor volume calculated during pathological examination.
Figure 4.
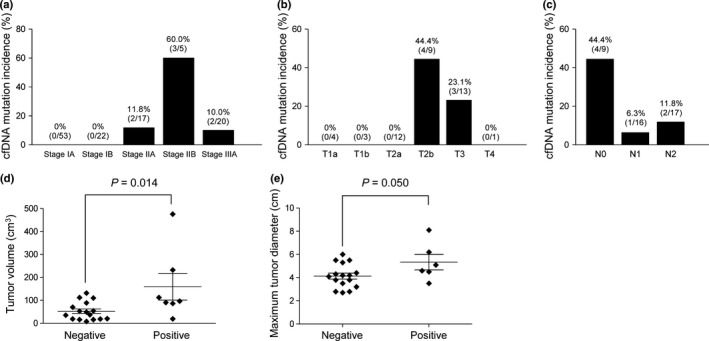
Relation between cfDNA mutation incidence and clinicopathologic features in patients with non‐small cell lung carcinoma. (a) Incidence of cfDNA mutations at each pathological stage for patients with mutation‐positive tumors. (b) Incidence of cfDNA mutations for each T factor among patients with mutation‐positive tumors and a pathological stage of IIA–IIIA. (c) Incidence of cfDNA mutations for each N factor among patients with mutation‐positive tumors and a pathological stage of IIA–IIIA. (d) Dot plot for tumor volume in cfDNA mutation‐positive (n = 7) or cfDNA mutation‐negative (n = 16) patients with mutation‐positive tumors and a T factor of T2b–T4. (E) Dot plot for maximum tumor diameter at diagnosis in cfDNA mutation‐positive (n = 6; unknown for one patient) and cfDNA mutation‐negative (n = 16) patients with mutation‐positive tumors and a T factor of T2b–T4. Mean ± SD values as well as P‐values determined by Student's t‐test are also shown in (d,e).
Discussion
Mutation profiling by NGS with tumor biopsy specimens has recently become established, with EGFR, TP53, and KRAS genes having been found to be frequently mutated in lung cancer specimens from Japanese patients.2 In the present study, mutations of EGFR, TP53, and KRAS were also frequently detected in NSCLC specimens, with those in KRAS and EGFR being mutually exclusive. Mutations in the genes analyzed were detected in 78.0% of tumor samples, with the mutation profiles being consistent with those observed previously.15, 16
The purpose of the present study was to investigate the mutation status for cfDNA isolated from serum of patients with NSCLC at an early stage. In comparison with matched tumor tissue, the sensitivity for mutation detection in serum by NGS was low (5.8%), consistent with previous results.17, 18 It thus appears to be difficult to detect mutations in cfDNA for patients with NSCLC at an early stage. In contrast, previous studies have reported a sensitivity of 70–80% for detection of EGFR mutations in plasma DNA compared with matched tumor DNA of patients with advanced NSCLC by beaming or digital PCR.5, 8, 9 We previously found the sensitivity for detection of activating mutations of EGFR in plasma cfDNA of such patients by digital PCR to be 66.7%.10
The feasibility of cfDNA sequencing is thought to be limited by the copy number of the DNA extracted from serum or plasma samples. The median copy number for the total of 150 samples in the present study was 4936, and library preparation and NGS were successful for 149 of these samples (99.3%). It is therefore unlikely that an insufficient amount of cfDNA was responsible for the low mutation frequency of cfDNA in our cohort. However, amplicons for NGS are derived from wild‐type as well as variant alleles, with a low frequency of mutant alleles compared with the wild‐type allele making it difficult to detect the former. The amplicon sequencing system was thus a possible limiting factor in our NGS assay. We therefore applied digital PCR to exclude this technical issue. The mutational status of cfDNA determined by NGS was confirmed with digital PCR, with the two approaches showing a high concordance. We therefore conclude that the NGS assay had sufficient power to detect a low frequency of mutations in serum, and that the discordance between the results obtained with tumor DNA and serum cfDNA was not due to the sensitivity of the NGS technology. The proportion of serum cfDNA that is derived from tumor cells thus appears to be much lower for patients with NSCLC at the resectable stage than for those with such tumors at an advanced stage. Our findings are consistent with those reported for different types of cancer including primary brain, renal, prostate, and thyroid tumors.19
We examined whether clinicopathologic features of the study patients might have influenced the mutation detection rate with cfDNA. The seven cfDNA mutation‐positive cases detected by NGS were all stage T2b or higher, suggesting that tumor volume is related to the rate of mutation detection in serum. The relation between the T factor and mutation detection in cfDNA, including the clinical relevance of mutation detection in cfDNA derived from patients with an advanced T factor, even at an early pathological stage, requires further validation. Lymph node metastasis (N stage) did not appear to be related to the rate of cfDNA mutation detection, suggesting that the lymphatic system might not play a major role in the spread of tumor cells into the circulation.
In conclusion, we found that 78.0% of resected NSCLC tumors harbored at least one mutation as detected by NGS. The incidence of cfDNA mutations in patients with early stage NSCLC was extremely low, however. Next‐generation sequencing showed a high accuracy compared with digital PCR for detection of mutations, indicating that NGS technology is similarly useful for the monitoring of mutant alleles. Given the multiplex nature of NGS, it is likely to become more widely adopted for monitoring of tumor progression and recurrence with cfDNA as the analyte.
Disclosure Statement
M.H. is an employee of SRL Inc. (Tokyo, Japan). K.N. has received lecture fees from Chugai Pharmaceutical, Daiichi Sankyo, and Sumitomo Bakelite. N.I. has received research grants from Ono Pharmaceutical, Japan Mediphysics, Eli Lilly Japan, Pfizer, Taiho Pharmaceutical, Chugai Pharmaceutical, and Bayer. The other authors have no conflict of interest.
Supporting information
Fig. S1. Median next‐generation sequencing read number per amplicon for all 150 non‐small cell lung carcinoma tumor and serum samples.
{kind=link}
Fig. S2. Yield of cell‐free DNA (cfDNA) for all 150 serum samples from patients with non‐small cell lung carcinoma.
{kind=link}
Fig. S3. Correlation between next‐generation sequencing (NGS) and digital PCR data.
{kind=link}
Fig. S4. Allele frequency at EGFR L858R position using next‐generation sequencing and digital PCR assay.
{kind=link}
Table S1. Poisson parameter (λ) and theoretical minimum detection limit (MDL) for next‐generation sequencing with the Ion AmpliSeq Colon and Lung Cancer Panel version 2 (Life Technologies, Foster City, CA, USA).
Acknowledgments
We thank K. Nagase, Y. Hosono, T. Miyazaki, and A. Kurumatani for cooperation and assistance. This work was supported by Applied Research for Innovative Treatment of Cancer (subject no. 14525177 to K.N.) from the Ministry of Health, Labor, and Welfare of Japan.
Cancer Sci 107 (2016) 1660–1666
Funding Information
Applied Research for Innovative Treatment of Cancer (14525177) Ministry of Health, Labor, and Welfare of Japan.
References
- 1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64: 9–29. [DOI] [PubMed] [Google Scholar]
- 2. Takeda M, Sakai K, Terashima M et al Clinical application of amplicon‐based next‐generation sequencing to therapeutic decision making in lung cancer. Ann Oncol 2015; 26: 2477–82. [DOI] [PubMed] [Google Scholar]
- 3. van der Drift MA, Hol BE, Klaassen CH et al Circulating DNA is a non‐invasive prognostic factor for survival in non‐small cell lung cancer. Lung Cancer 2010; 68: 283–7. [DOI] [PubMed] [Google Scholar]
- 4. Lee YJ, Yoon KA, Han JY et al Circulating cell‐free DNA in plasma of never smokers with advanced lung adenocarcinoma receiving gefitinib or standard chemotherapy as first‐line therapy. Clin Cancer Res 2011; 17: 5179–87. [DOI] [PubMed] [Google Scholar]
- 5. Couraud S, Vaca‐Paniagua F, Villar S et al Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never‐smokers: a proof‐of‐concept study from BioCAST/IFCT‐1002. Clin Cancer Res 2014; 20: 4613–24. [DOI] [PubMed] [Google Scholar]
- 6. Kimura H, Fujiwara Y, Sone T et al EGFR mutation status in tumour‐derived DNA from pleural effusion fluid is a practical basis for predicting the response to gefitinib. Br J Cancer 2006; 95: 1390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taniguchi K, Uchida J, Nishino K et al Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res 2011; 17: 7808–15. [DOI] [PubMed] [Google Scholar]
- 8. Douillard JY, Ostoros G, Cobo M et al First‐line gefitinib in Caucasian EGFR mutation‐positive NSCLC patients: a phase‐IV, open‐label, single‐arm study. Br J Cancer 2014; 110: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oxnard GR, Paweletz CP, Kuang Y et al Noninvasive detection of response and resistance in EGFR‐mutant lung cancer using quantitative next‐generation genotyping of cell‐free plasma DNA. Clin Cancer Res 2014; 20: 1698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ishii H, Azuma K, Sakai K et al Digital PCR analysis of plasma cell‐free DNA for non‐invasive detection of drug resistance mechanisms in EGFR mutant NSCLC: correlation with paired tumor samples. Oncotarget 2015; 6: 30850–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kukita Y, Uchida J, Oba S et al Quantitative identification of mutant alleles derived from lung cancer in plasma cell‐free DNA via anomaly detection using deep sequencing data. PLoS ONE 2013; 8: e81468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paweletz CP, Sacher A, Raymond CK et al Bias‐corrected targeted next‐generation sequencing for rapid, multiplexed detection of actionable alterations in cell‐free DNA from advanced lung cancer patients. Clin Cancer Res 2015; 22: 915–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sakai K, Tsurutani J, Yamanaka T et al Extended RAS and BRAF mutation analysis using next‐generation sequencing. PLoS ONE 2015; 10: e0121891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Narahara M, Higasa K, Nakamura S et al Large‐scale East‐Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS ONE 2014; 9: e100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Okamoto I, Sakai K, Morita S et al Multiplex genomic profiling of non‐small cell lung cancers from the LETS phase III trial of first‐line S‐1/carboplatin versus paclitaxel/carboplatin: results of a West Japan Oncology Group study. Oncotarget 2014; 5: 2293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sholl LM, Aisner DL, Varella‐Garcia M et al Multi‐institutional oncogenic driver mutation analysis in lung adenocarcinoma: the Lung Cancer Mutation Consortium experience. J Thorac Oncol 2015; 10: 768–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao X, Han RB, Zhao J et al Comparison of epidermal growth factor receptor mutation statuses in tissue and plasma in stage I‐IV non‐small cell lung cancer patients. Respiration 2013; 85: 119–25. [DOI] [PubMed] [Google Scholar]
- 18. Guo K, Zhang Z, Han L et al Detection of epidermal growth factor receptor mutation in plasma as a biomarker in Chinese patients with early‐stage non‐small cell lung cancer. Onco Targets Ther 2015; 8: 3289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bettegowda C, Sausen M, Leary RJ et al Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 2014; 6: 224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Median next‐generation sequencing read number per amplicon for all 150 non‐small cell lung carcinoma tumor and serum samples.
Fig. S2. Yield of cell‐free DNA (cfDNA) for all 150 serum samples from patients with non‐small cell lung carcinoma.
Fig. S3. Correlation between next‐generation sequencing (NGS) and digital PCR data.
Fig. S4. Allele frequency at EGFR L858R position using next‐generation sequencing and digital PCR assay.
Table S1. Poisson parameter (λ) and theoretical minimum detection limit (MDL) for next‐generation sequencing with the Ion AmpliSeq Colon and Lung Cancer Panel version 2 (Life Technologies, Foster City, CA, USA).