

Abstract
Diffuse large B‐cell lymphoma (DLBCL) is the most common subtype of malignant lymphoma; it derives from germinal center B cells. Although DLBCL harbors many genetic alterations, synergistic roles between such alterations in the development of lymphoma are largely undefined. We previously established a mouse model of lymphoma by transplanting gene‐transduced germinal center B cells into mice. Here, we chose one of the frequently mutated genes in DLBCL, Card11 mutant, to explore its possible synergy with other genes, using our lymphoma model. Given that BCL6 and BCL2 expression and/or function are often deregulated in human lymphoma, we examined the possible synergy between Card11, Bcl6, and Bcl2. Germinal center B cells were induced in vitro, transduced with Card11 mutant, Bcl6, and Bcl2, and transplanted. Mice rapidly developed lymphomas, with exogenously transduced Bcl2 being dispensable. Although some mice developed lymphoma in the absence of transduced Bcl6, the absence was compensated by elevated expression of endogenous Bcl6. Additionally, the synergy between Card11 mutant and Bcl6 in the development of lymphoma was confirmed by the fact that the combination of Card11 mutant and Bcl6 caused lymphoma or death significantly earlier and with higher penetrance than Card11 mutant or Bcl6 alone. Lymphoma cells expressed interferon regulatory factor 4 and PR domain 1, indicating their differentiation toward plasmablasts, which characterize activated B cell‐like DLBCL that represents a clinically aggressive subtype in humans. Thus, our mouse model provides a versatile tool for studying the synergistic roles of altered genes underlying lymphoma development.
Keywords: Bcl6, Card11, diffuse large B cell lymphoma, germinal center, mouse model
Diffuse large B‐cell lymphoma is the most common subtype of malignant lymphoma. Genetic abnormalities are heterogeneous between DLBCL clinical samples1, 2, 3 and a single patient harbors multiple genetic abnormalities, making it difficult to determine the genetic abnormalities specifically required for the development of lymphoma.
A number of transgenic and knockin/knockout mouse models have been generated to clarify the functional roles of the given genetic abnormalities in lymphomagenesis,4, 5, 6 yet many remain unexplored. To explore the combinatorial effects of multiple genetic abnormalities in lymphomagenesis, such genetically modified mice need to be crossed to give rise to mice harboring the genetic abnormalities of interest in combination. However, this strategy is laborious and time‐consuming. In addition, B cells of such genetically modified mice undergo SHM in genes encoding immunoglobulin as well as many other genes involved in lymphomagenesis, which provides an additional layer of difficulty in clarifying the roles of the genetic alterations of interest. In an effort to circumvent these difficulties, we previously established a mouse model of lymphoma using in vitro‐induced mouse GC B cells. The GC B cells were induced in vitro, retrovirally transduced with genes or mutated genes of interest in combination, and transplanted into immunodeficient mice.7, 8 In this model, the combinatorial effects of multiple genetic abnormalities can be assessed easily by infecting their corresponding retroviral vectors at once. Somatic hypermutation did not occur in the in vitro‐induced GC B cells, therefore, the role of the genes or mutated genes of interest in the development of the lymphoma could be assessed.7, 8, 9
Diffuse large B cell lymphomas are largely classified into two subtypes based on gene expression profile, the GCB and ABC types.10 ) Genetic abnormalities that deregulate functions and/or the expression of BCL6 and BCL2 frequently occur in both subtypes of human DLBCL. Chromosomal translocations involving BCL6 that result in the constitutive expression of BCL6 in B cells are exclusively found in ABC‐DLBCL.6 Interestingly however, BCL6 is transcriptionally upregulated by somatic mutations of MEF2B 11 or evades EP300/CREBBP‐mediated inactivation due to mutations in EP300/CREBBP genes,12, 13 in some GCB‐DLBCL cases. Likewise, although chromosomal translocations involving BCL2 that constitutively elevate BCL2 expression are found almost exclusively in GCB‐DLBCL, gene amplification of BCL2 is observed in ABC‐DLBCL.14, 15 Moreover, BCL6 and BCL2 play critical roles in the development and maintenance of DLBCL. For example, DLBCL cell lines and BCL6‐expressing patient‐derived DLBCL cells often depend on BCL6 transcription activity for survival.16 Elevated BCL2 expression promotes clonogenicity of lymphoblastoid cell lines17 and elicits lymphoma in some, if not all, mouse lymphoma models.18 However, enhanced activity of BCL6 or BCL2 per se is not sufficient to elicit lymphoma. Transgenic mice carrying a BCL6 (IμHABCL6 mice)4 or BCL2 transgene under the control of the IGH genes enhancer take almost 1 year to develop lymphomas.18, 19 Moreover, lymphomas that developed in these mice presented additional genetic abnormalities4, 20 such as translocation of Myc.18 Lymphoma development was significantly delayed in IμHABCL6 mice by genetic inactivation of activation‐induced cytidine deaminase21 or uracil–DNA glycosylase,22 both of which induce SHM in cancer‐related genes in addition to immunoglobulin genes.23 Together, these findings suggest that functionally enhanced BCL6 or BCL2 requires additional genetic “hits” to elicit lymphoma.
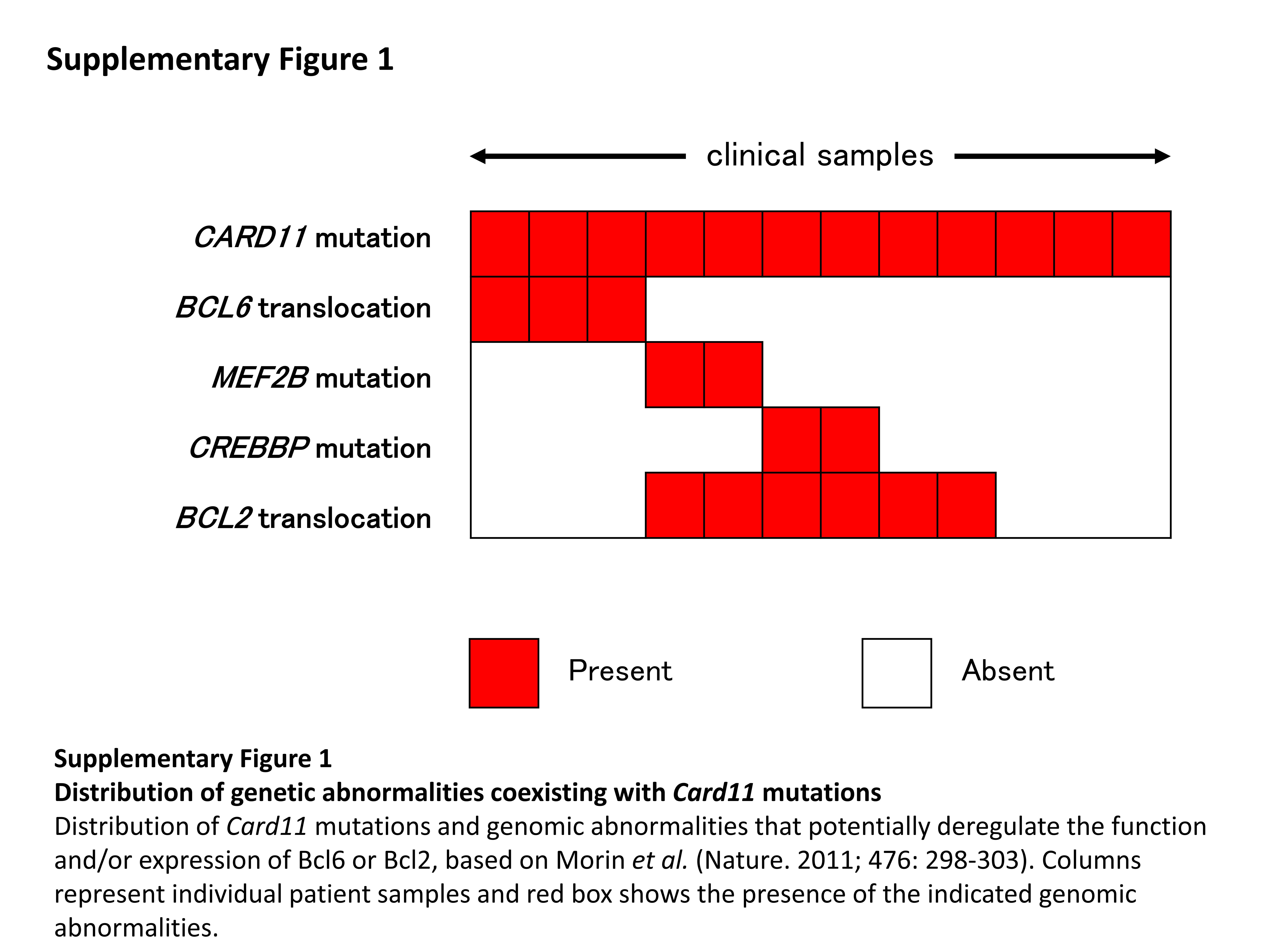
One such candidate “hit” is a somatic mutation of CARD11. CARD11 is mutated in approximately 10% of DLBCL cases,24 being more prevalent in ABC‐DLBCL, but also occurring in GCB‐DLBCL.1, 24 Card11 mutations occur during the process of lymphoma development in a subset of IμHABCL6 mice.20 In clinical lymphoma samples, CARD11 mutations are often accompanied by chromosomal translocations, gene amplifications, or mutations that lead to elevated activity of BCL6 or BCL2 (Fig. S1).13 CARD11 is a critical component of the NF‐κB pathway, transmitting B‐cell receptor signal to induce transcriptional activation of NF‐κB target genes. CARD11 mutations occurring in DLBCL activate the NF‐κB pathway even in the absence of B‐cell receptor input, providing survival signals, especially in ABC‐DLBCL.24 However, mutations of CARD11 seem insufficient for the development of lymphoma in humans. Only a limited number of affected persons within a family with a germline CARD11 mutation develop lymphoma.25
In this study, we investigated the possible synergy between Card11 mutation, Bcl6, and Bcl2, using our mouse model.
Materials and Methods
Induction of GC B cells, retroviral infection, and transplantation
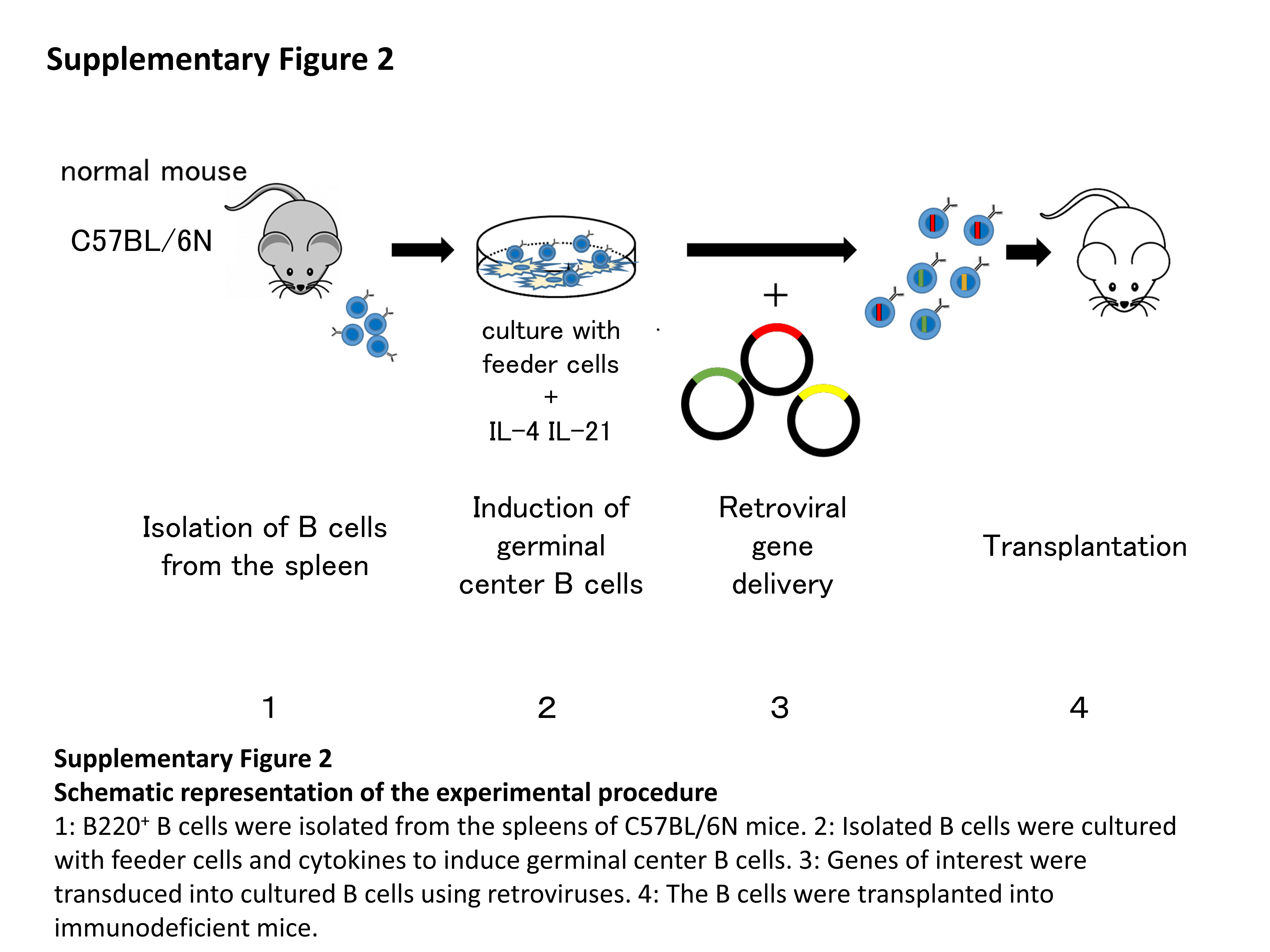
Induction of GC B cells was carried out as described previously.7, 9 Briefly, B220+ B cells were isolated from the spleens of C57BL/6N mice using antibodies for B220 (RA3‐6B2; eBioscience, San Diego, CA, USA) in combination with MACS columns (Miltenyi Biotec, Bergisch Gladbach, Germany). Isolated B cells were co‐cultured with X‐irradiated (150 Gy) NIH3T3 cells engineered to express mouse CD40 ligand and mouse Baff (3T3/CD40L/Baff cells) in RPMI‐1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% FCS, 5 × 10−5 M 2‐mercaptoethanol, 1 mM sodium pyruvate, 100 units/mL each of penicillin and streptomycin (Gibco), and recombinant interleukin‐4 (1 ng/mL; PeproTech, Rocky Hill, NJ, USA). Four days later, the B cells were transferred onto a new feeder layer and cultured with recombinant interleukin‐21 (10 ng/mL; PeproTech) for 4 days to induce GC B cells. The iGCB cells were filtered through 45‐μm nylon mesh to remove feeder cells and spin‐infected with the respective virus using RetroNectin (Takara Bio, Shiga, Japan) coated plates twice a day on days 3–5 after initiation of the culture. Cells were seeded back onto the feeder layer after infection. On day 8, gene‐transduced iGCB cells (1 × 107 cells) were transplanted i.p. into sublethally irradiated (2 or 2.5 Gy) NOD‐SCID mice (NOD.CB17‐Prkdc; Jackson Laboratory, Bar Harbor, ME, USA) (Fig. S2). All animal experiments were carried out according to protocols approved by the Institutional Animal Care and Use Committee at the Aichi Cancer Center (Nagoya, Japan).
Plasmids
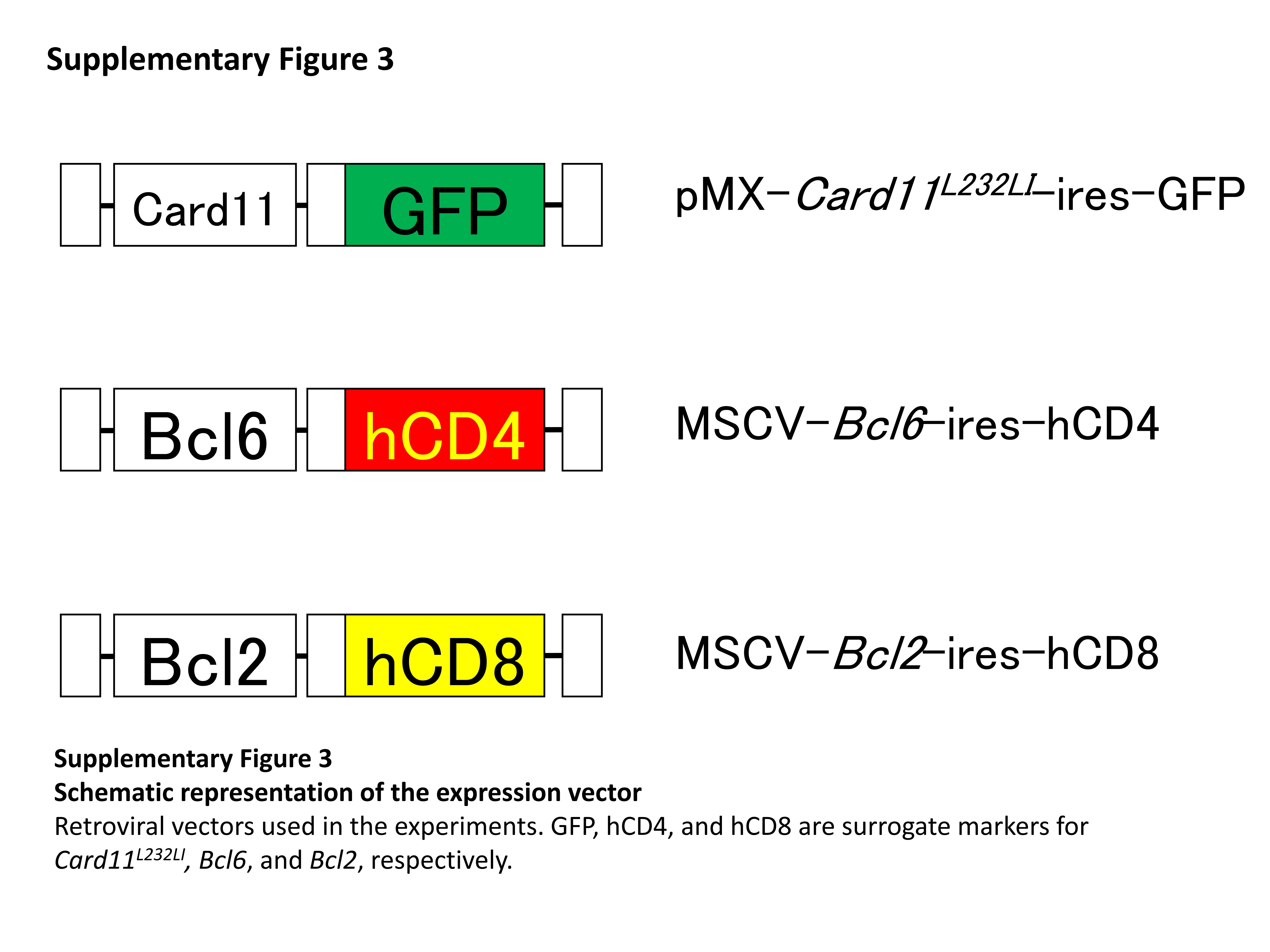
cDNA for mouse Bcl6 was amplified from mouse spleen cDNA by PCR and cloned into MSCV‐Bcl6‐ires‐human (h) CD4 retroviral vector. cDNA for mouse Bcl2 was cloned into MSCV‐Bcl2‐ires‐human (h) CD8, as previously described.26 pMX‐Card11 L232LI‐ires‐GFP was a generous gift from Dr. Keisuke Horikawa (The Australian National University, Canberra, Australia)27 (Fig. S3). These plasmids were co‐transfected with MCV‐Ecopac plasmid (a generous gift from Dr. Richard Van Etten, Tufts‐NEMC Cancer Center, Boston, MA, USA)28 into 293T cells to produce retroviruses.
Flow cytometry
Flow cytometric analysis was undertaken using antibodies for mouse B220 (RA3‐6B2), IgG1 (X‐56; Miltenyi Biotec), Fas (15A7), B7‐1/CD80 (16‐10‐A1), IgM (II/41), CD138 (281‐2), human CD4 (RPA‐T4), and human CD8 (RPA‐T8). Anti‐mouse IgG1 antibody (M1‐14D12) was used as an isotype‐matched control for anti‐human CD4 antibody. Antibodies were purchased from eBioscience, except for X‐56. Flow cytometric analysis was carried out using a FACSCalibur instrument (BD Biosciences, San Jose, CA, USA) and FlowJo software version 7.6.5 (Tree Star, Ashland, OR, USA).
Western blot analyses
Western blot analyses were carried out using anti‐BCL6 (D‐8; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐CARD11 (#4440; Cell Signaling Technology, Danvers, MA, USA), anti‐IRF4 (M‐17; Santa Cruz Biotechnology), anti‐PRDM1 (6D3; Santa Cruz Biotechnology), and anti‐actin (AC‐40; Sigma‐Aldrich, St. Louis, MO, USA), anti‐BCL2 (3F11; BD Pharmingen, San Diego, CA, USA), anti‐α‐tubulin (DM1A; Sigma‐Aldrich), anti‐Lamin B1 (L‐5; Thermo Fisher Scientific, Rockford, IL, USA), anti‐p65 (C‐20; Santa Cruz Biotechnology) antibodies. Cytosolic and nuclear extracts were obtained using the Nuclear Extract Kit (Active Motif, Carlsbad, CA, USA).
Analysis of cell clonality and SHM
To analyze the clonality and SHM of IGH genes in lymphoma cells, isolated genomic DNA was subjected to PCR using Ex Taq (Takara Bio) or KOD‐plus2 (Toyobo, Osaka, Japan) along with primer sets that amplify the VDJ regions of IGH genes, as described previously.29, 30 Combinations of forward (VHA, VHE, or VHG) and reverse (JH4 intron) primers were used. The resulting PCR products were gel‐purified, cloned into the pCR4‐TOPO vector (Invitrogen, Carlsbad, CA, USA), and sequenced. Every single plasmid was deemed to represent a distinct molecularly cloned sequence and, here, defined as a molecular clone. VDJ gene usage and somatic mutations were analyzed using igblast (http://www.ncbi.nlm.nih.gov/igblast/). To determine the presence of intraclonal heterogeneity in lymphoma cells, confirmed mutations and unconfirmed mutations were defined as follows:31 confirmed mutation, substitution mutations observed in more than one molecular clone sharing identical VDJ‐rearranged sequence from the same tumor sample; unconfirmed mutation, substitution mutations observed in a single molecular clone from the same tumor sample. Only the confirmed mutations were used for the analysis of intraclonal heterogeneity, or ongoing SHM. The unconfirmed mutations were disregarded because they can be caused by PCR error.
Results
Tumorigenicity of Card11 L232LI, Bcl6, and Bcl2‐transduced cells
To explore genes or mutated genes that collaborate with CARD11 mutant in lymphomagenesis, we reviewed published results of next generation sequencing of clinical samples, with special reference to those potentially enhancing the function of BCL6 or BCL2.13 Of 12 lymphoma cases harboring CARD11 mutation, chromosomal translocations involving BCL6 and BCL2 existed in three and six cases, respectively, and mutations of CREBBP or MEF2B existed in two cases each (Fig. S1). The expression of BCL6 and BCL2 is under the control of heterotopic enhancer through chromosomal translocations. CREBBP mutant lost its ability to inactivate BCL6 by acetylation,12 and MEF2B mutant can enhance BCL6 expression.11 Notably, BCL6 translocations and mutations of CREBBP or MEF2B are mutually exclusive, suggesting that they collaborate with CARD11 mutant in a non‐redundant manner in the development of lymphoma. Taken together, CARD11 mutations often co‐exist with functionally enhanced BCL6 or BCL2.
Therefore, we first investigated the possible collaboration between CARD11 mutant, BCL6, and BCL2 in lymphoma development. To this end, we used iGCB cells as a target for the transduction of Card11 mutant, Bcl6, and Bcl2 genes, given that DLBCL originates in GC B cells. B220+ murine B cells were isolated from the spleen of C57BL/6N mice, induced into GC B cells in culture, and retrovirally transduced with Card11 L232LI, Bcl6, and Bcl2. Mouse Card11 L232LI corresponds to the human CARD11 L225LI, an activating mutant. Card11 L232LI, Bcl6, and Bcl2 were programmed to co‐express GFP and the extracellular domains of human CD4 and CD8, respectively, as surrogate markers enabling the identification of the transduced cells by flow cytometry (Fig. S3). Gene‐transduced iGCB cells were then transplanted into immunodeficient mice, which were monitored for the development of lymphoma (Fig. S2). Experiments were independently carried out three times, using one (exp. 1), three (exp. 2), and two (exp. 3) mice, respectively. From a total of six mice transplanted with Card11 L232LI/Bcl6/Bcl2 triply transduced iGCB cells (referred to as “Card11mut+Bcl6 + Bcl2 #1 to #6”), three mice were dissected while alive when disease signs (either tumor mass coupled with ascites or hepatomegaly) manifested (Card11mut+Bcl6 + Bcl2 #1 to #3, Table S1). The remaining three mice were found dead, with two mice showing histologically evident lymphomas (Card11mut+Bcl6 + Bcl2 #5 and #6); histological analysis could not be carried out on one mouse (Card11mut+Bcl6 + Bcl2 #4). In total, five mice developed lymphomas and one died of unknown cause within 2 months of transplantation (Fig. 1a). Mice that developed lymphomas manifested apparent tumor masses (Card11mut+Bcl6 + Bcl2 #1 and #2) and/or hepatic involvement (Card11mut+Bcl6 + Bcl2 #2, #3, #5, and #6), which were accompanied by infiltration of atypical lymphoid cells morphologically consistent with DLBCL (Fig. S4).
Figure 1.
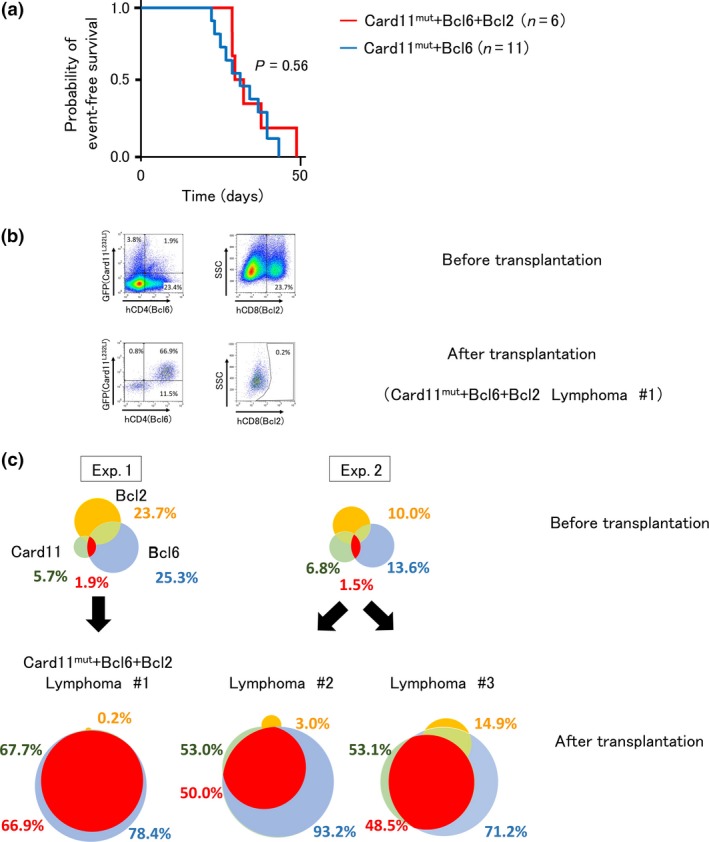
Tumorigenicity of Card11 L232 LI and Bcl6 with/without Bcl2‐transduced germinal center B cells. (a) Kaplan–Meier analysis of the probability of event‐free survival of mice transplanted with induced germinal center B (iGCB) cells transduced with Card11 L232 LI, Bcl6, and Bcl2 (red line). An event is defined as the presence of lymphoma on dissection or death. The probability of event‐free survival of the mice transplanted with iGCB cells transduced with Card11 L232 LI and Bcl6 is shown by the blue line. The difference was not statistically significant (P = 0.56), as determined by the log–rank test. (b) Flow cytometric analysis of gene‐transduced iGCB cells before transplantation (upper panels; experiment [Exp.] 1) and cells in lymphoma that developed after transplantation in a mouse (lower panels). Percentages of cells expressing GFP (representing Card11 L232 LI), hCD4 (Bcl6), and hCD8 (Bcl2) are depicted and truncated after the decimal point. (c) Venn diagrams showing the percentages of cells expressing exogenously transduced Card11 L232 LI (GFP, green circles), Bcl6 (hCD4, blue circles), and Bcl2 (hCD8, yellow circles) before (upper panel) and after transplantation, on lymphoma development (lower panel). One (#1) and two (#2 and #3) mice developed lymphoma in two independent experiments. Percentages of cells co‐expressing Card11 and Bcl6 are shown in red. The remaining three out six mice used in the experiment were found dead. Thus, viable cells were unavailable for flow cytometric analysis.
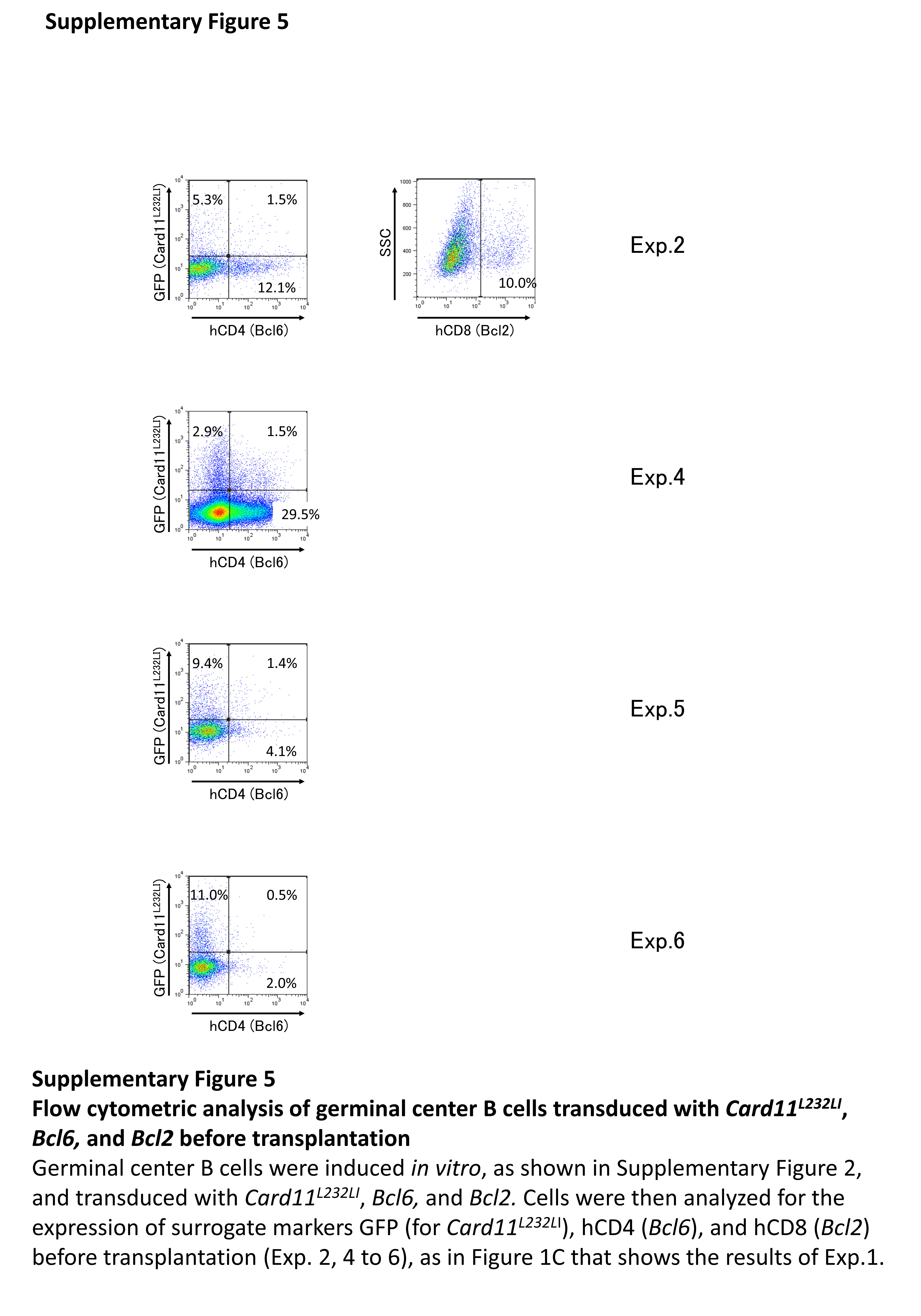
To determine which of the three genes (Card11 L232LI, Bcl6, and Bcl2) contributed to the development of lymphomas in mice, we examined the expression of surrogate markers, GFP for CARD11 L232LI, hCD4 for Bcl6, and hCD8 for Bcl2, on the cells by flow cytometry. As viable tumor cells were only available from the three mice dissected while alive (see description above), our analysis was necessarily confined to them (Card11mut+Bcl6 + Bcl2 #1–#3). Before transplantation, the percentages of cells expressing surrogate markers for the three transduced genes were variable and only a small fraction of the cells co‐expressed hCD4 or hCD8 along with GFP. In contrast, almost all cells obtained from the developed lymphomas co‐expressed both GFP and hCD4, with only a small fraction expressing hCD8, as exemplified in Card11mut+Bcl6 + Bcl2 #1 (Fig. 1b). A similar analysis was conducted in Card11mut+Bcl6 + Bcl2 #2 and #3 before (Fig. S5, exp. 2) and after lymphoma development (Fig. S6a) and the results are summarized using a Venn diagram (Fig. 1c). These findings suggest that the cells transduced with Card11 L232LI and Bcl6 grew selectively among the cells transduced with any possible combinations of the three genes. Bcl2 seemingly played only a limited role, if at all, in the development of lymphomas in this model.
Tumorigenicity of Card11 L232LI and Bcl6 ‐transduced cells
We next tested if a combination of Card11 mutant and Bcl6 is sufficient for lymphoma development in separate experiments. Induced GCB cells were transduced with the two genes and transplanted. All mice (n = 11) developed lymphoma or died within 2 months of transplantation with rapidity and penetrance comparable with those of mice transplanted with cells transduced with Card11 L232LI, Bcl2, and Bcl6 (referred to as “Card11mut+Bcl6 #1 to #11”; Fig. 2a, Table S1). Data were combined from four experiments carried out independently, using one (exp. 4), three (exp. 5), four (exp. 6), and three (exp. 7) mice. Seven mice were dissected while alive when disease signs manifested (Table S1, Fig. S7) and tumor cells were isolated and analyzed by flow cytometry (Card11mut+Bcl6 #1–#3, #5–#8; Figs. 2a, S6b). Of the remaining four mice dissected after death, one had histologically evident lymphoma (Card11mut+Bcl6 #11; Fig. S4) and one had no histologically detectable tumor (Card11mut+Bcl6 #10). Detailed analysis could not be undertaken on the remaining two mice (Card11mut+Bcl6 #4 and #9). Flow cytometric analysis revealed that Card11 L232LI‐transduced cells always constituted the major component of lymphomas (Fig. 2a), although the protein expression level of CARD11 was variable (Fig. 2b) between developed lymphoma cells. In contrast, the proportion of Bcl6‐transduced cells in the developed lymphomas varied between developed lymphomas. Only a small fraction of cells co‐expressed both Card11 L232LI and Bcl6 before transplantation. Cells co‐expressing Card11 L232LI and Bcl6 became dominant in four of seven mice (Card11 mut+Bcl6 #1, #2, #5, and #6) (Fig. 2a). These findings suggest that exogenously transduced Card11 L232LI and Bcl6 synergistically induced lymphomas in these mice, as was the case in mice transplanted with Card11 L232LI /Bcl6/Bcl2 transduced cells. Nevertheless, in the remaining three mice (Card11mut+Bcl6 #3, #7, and #8), lymphomas mainly consisted of cells expressing only the surrogate marker for Card11 L232LI, suggesting that Bcl6 was not required for the development of these lymphomas. However, Western blot analysis revealed that, in two lymphomas (Card11mut+Bcl6 #3 and #7), endogenous BCL6 was expressed at a level comparable to those of lymphoma cells expressing the exogenous Bcl6 gene with its surrogate marker and in a human GCB‐DLBCL cell line, OCI‐Ly8, which harbors BCL6 rearrangement (Fig. 2c).32 This finding implies that endogenous BCL6 was somehow upregulated, or rare clones with high endogenous BCL6 expression were selected during the course of lymphoma development in these recipient mice. Taken together, these findings suggest that Card11 mutant collaborates with exogenous or endogenous Bcl6 for the development of lymphoma.
Figure 2.
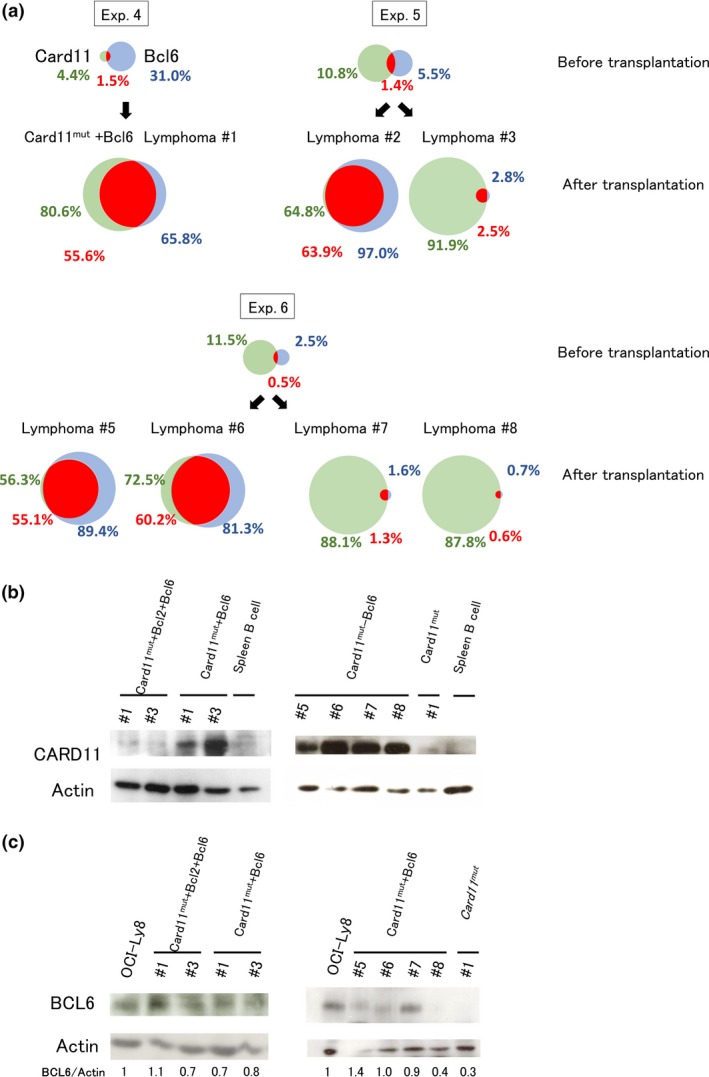
Synergistic activity of Card11 L232 LI and Bcl6. (a) Venn diagrams showing the percentages of cells expressing the surrogate markers for exogenously transduced Card11 L232 LI (GFP, green circles) and Bcl6 (hCD4, blue circles). Fractions of cells coexpressing GFP and hCD4 are shown in red. The remaining four mice, out of 11 mice used in the experiment (Exp.), were found dead. Thus, viable cells were unavailable for flow cytometric analysis. (b) Expression of CARD11 in the developed lymphoma. (c) BCL6 expression in the developed lymphoma. BCL6 protein levels are normalized to the levels of actin (protein loading control) and BCL6/actin ratios are presented and compared to that of OCI‐Ly8, a human lymphoma cell line, being set to 1.0.
Interestingly, in one (Card11mut+Bcl6 #8) of seven mice, BCL6 was undetectable in lymphoma cells (Fig. 2c), raising the possibility that transduced Card11 L232LI might induce lymphoma without the help of BCL6 in a limited number of cases. Indeed, in separate experiments using five mice transplanted with Card11 L232LI singly transduced cells, although lymphoma developed, the onset was significantly (P = 1.6 × 10−3) delayed and the penetrance was lower than that in mice transplanted with Card11 L232LI /Bcl6 co‐transduced cells. Only two of five mice receiving Card11 L232LI singly transduced cells developed lymphoma (referred to as “Card11mut #1 and #2”) (Fig. 3; see also Fig. S6c, Table S1). We therefore do not exclude the possibility that Card11 mutant is able to induce lymphoma by itself, but our results clearly show that Bcl6 facilitates Card11‐mediated lymphomagenesis. Three mice receiving Bcl6 only transduced iGCB cells died of unknown reasons (n = 1; 123 days after transplantation) or had no detectable lymphoma when killed (n = 2; 132 and 141 days after transplantation), suggesting that BCL6 alone is insufficient to induce lymphoma.
Figure 3.
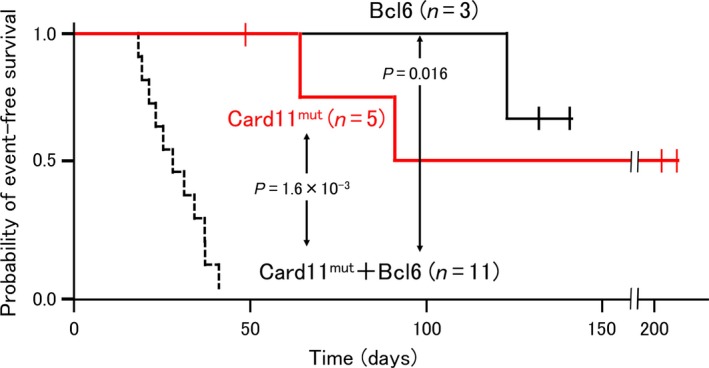
Tumorigenicity of induced germinal center B cells singly transduced with Card11 L232 LI or Bcl6. Kaplan–Meier analysis of the probability of event‐free survival of the mice receiving induced germinal center B cells transduced with Card11 L232 LI (red line) or Bcl6 alone (black line). An event is defined as the presence of lymphoma on dissection or death. The probability of event‐free survival was compared with that of mice transplanted with cells transduced with Card11 L232 LI and Bcl6 (dashed line). The difference in the probability of event‐free survival was statistically significant (P = 1.17 × 10−4), as determined by the log–rank test. Post‐hoc pairwise comparison was undertaken using Bonferroni correction. All P‐values <0.05 are shown in the figure.
Clonality and properties of lymphomas developed in mice
We next analyzed the clonality of lymphomas. The VDJ regions of IGH genes were PCR amplified, cloned into plasmids, and sequenced. The results revealed that these tumors were oligoclonal (Card11mut+Bcl6 #1) or monoclonal (Card11mut+Bcl6 #2 and #3) (Fig. 4a), suggesting that additional genetic or epigenetic factors, in addition to transduced genes, were needed for the development of lymphoma in our mouse model. Card11mut+Bcl6 #2 and #3 lymphoma had only one VDJ rearranged sequence without confirmed mutations through analysis of multiple plasmid clones, whereas Card11mut+Bcl6 #1 showed three distinct VDJ rearrangements, namely (IGHV1‐26*01, IGHD1‐1*01, IGHJ2*01), (IGHV1‐26*01, IGHD2‐3*01, IGHJ3*01), and (IGHV1‐80*01, IGHD1‐1*01, IGHJ3*02) (Table S2). Among these, only molecular clones using (V: 1‐26*01, D: 2‐3*01, J: 3*01) presented confirmed mutations, while the others did not. Ten molecular clones using (V: 1‐26*01, D: 2‐3*01, J: 3*01) presented identical confirmed mutations with no evidence of occurrence of ongoing SHM. The confirmed mutations were unlikely to be induced by PCR error, as they were identified in two independent experiments using different enzymes (Ex Taq and KOD‐plus2). These findings indicate that, in our model, lymphomas lacked ongoing SHM.
Figure 4.
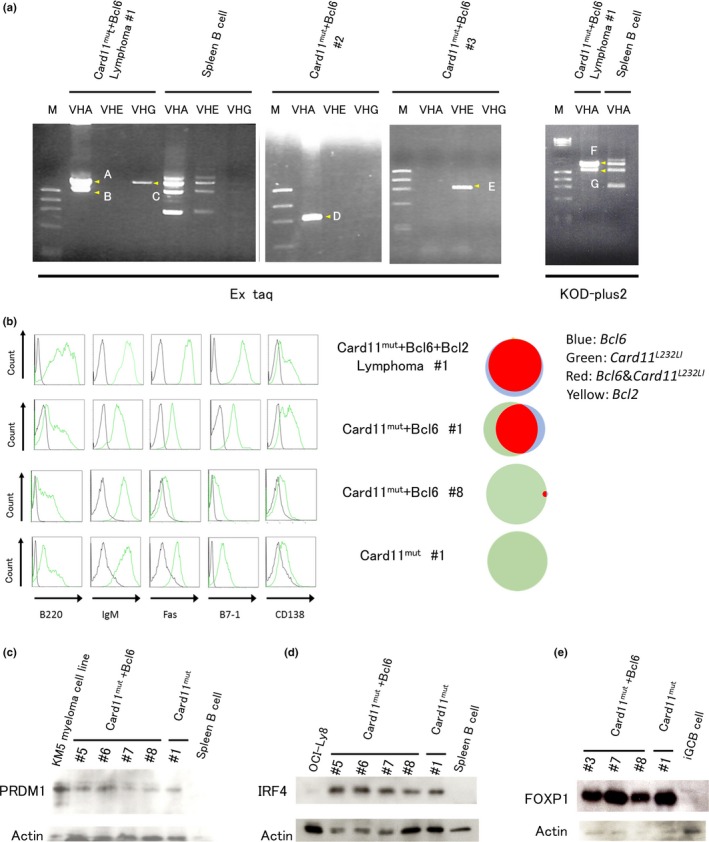
Clonality and properties of lymphomas. (a) Immunoglobulin heavy chain genes amplified by PCR from genomic DNA of lymphoma cells using combinations of VHA/VHE/VHG (forward) and JH4 intron (reverse) primers. PCR products amplified by Ex Taq or by KOD‐plus2 were fractionated by gel electrophoresis and bands were marked by A–E and F–G, respectively (yellow arrowheads). These amplified products were cloned into a plasmid vector and sequenced. Amplified products from genomic DNA of normal, non‐transduced spleen B cells were also electrophoresed as a control, to show their polyclonal status. M, markers. (b) Phenotype of the developed lymphoma cells as assessed by flow cytometry. Cells in the developed lymphoma were analyzed for the expression of the indicated molecules by flow cytometry (left). Control samples are represented by black lines. The fractions of cells that expressed each surrogate marker of transduced genes (GFP, Card11 L232 LI; hCD4, Bcl6; hCD8, Bcl2) of the corresponding tumor samples are represented by the Venn diagram (right). (c–e) Expression of PR domain 1 (PRDM1) (c), interferon regulatory factor 4 (IRF4) (d), and forkhead box P1 (FOXP1) (e) in developed lymphoma, as detected by Western blotting.
The tumor cells were positive for B220 (a pan‐B‐cell marker),33 IgM, and B7‐1 (an activated B‐cell marker) (Fig. 4b).34 They also expressed GC markers such as Fas,35 although the expression levels were variable between lymphomas. Intriguingly, these tumor cells also expressed CD138 (a plasma cell marker), albeit weakly, and PRDM1 (a plasma cell differentiating transcription factor), suggesting that they differentiated toward plasmablasts (Fig. 4c).36
Both IRF4 (Fig. 4d) and BCL2 (Fig. S8), known transcriptional targets of the NF‐κB pathway, were highly expressed, consistent with nuclear translocation of p65 (RelA) that shows NF‐κB activation (Fig. S9). Together with high expression of FOXP1 (Fig. 4e), a widely used classifier of ABC‐DLBCL in humans,37 these findings show that lymphoma cells developed in mice are consistent with DLBCL. Notably, all four secondary recipient mice transplanted with lymphoma cells derived from the primarily transplanted mice rapidly developed lymphoma within 20 days (Fig. S10), indicating the tumor‐propagating activity of the lymphomas; therefore, they are bona fide tumors.
Discussion
In this study, we showed the synergistic activity of Card11 L232LI and Bcl6 in lymphomagenesis in a mouse model. Evidence supporting the synergy between the two genes in the development of lymphoma are threefold: (i) iGCB cells doubly transduced with Card11 L232LI and Bcl6 grew selectively over singly transduced cells in 7 out of 10 mice receiving cells transduced with Card11 L232LI /Bcl6/Bcl2 (n = 3, Fig. 1c) or Card11 L232LI /Bcl6 (n = 4, Fig. 2a) and these mice rapidly succumbed to lymphoma; (ii) although some lymphomas (in two of the remaining three mice) did not express the surrogate marker of exogenously transduced Bcl6, the expression of intrinsic BCL6 was elevated (Fig. 2c); and (iii) mice transplanted with cells transduced with Card11 L232LI and Bcl6 developed lymphoma with significantly higher incidence and shorter latency than those transplanted with cells transduced with either one of the genes (Fig. 3).
Although synergistic, a combination of Card11 mutant and Bcl6 seems insufficient for lymphoma development, based on the following observations. First, lymphomas developed in mice that had oligoclonal or monoclonal IGH rearrangements, suggesting that not every single clone but a small number of clones contributed to the development of lymphoma (Fig. 4a). Second, our preliminary use of immunocompetent mice as recipients of Card11 L232LI /Bcl6 transduced iGCB cells reduced the incidence of lymphoma compared with immunodeficient mice. Although the reasons for the difference are not clear, evasion from immunosurveillance may facilitate lymphoma development, as suggested by the findings that clinical lymphomas often harbor mutations in immunosurveillance‐associated genes.38
Lymphomas developed in our model were consistent with ABC‐DLBCL, among others, in that they expressed IRF410, 39 and FOXP1,37 molecular markers of ABC‐DLBCL, and PRDM1, critically involved in post‐GC B cell differentiation.40, 41 They also showed evidence of NF‐κB activation.42 Lack of ongoing SHM, the hallmark of GCB‐DLBCL43 further supports the identity as ABC‐DLBCL.
The mechanism by which Bcl6 and Card11 L232LI might collaborate in lymphomagenesis remains unclear. Mutant CARD11 forms a complex with BCL10 and MALT1 and activates the NF‐κB pathway, independently of B‐cell receptor stimulation.44 Although Card11 L232LI activates NF‐κB signaling as such, the active NF‐κB signaling per se is insufficient for lymphoma development. Expression of constitutive active IKK2ca in GC B cells in mice activates NF‐κB, leading to enhanced cell proliferation, however, at the same time, provokes differentiation into plasma cells, failing to induce lymphoma development. The constitutive activation of NF‐κB and the blockade of terminal differentiation into plasma cells therefore appear to be required for the development of ABC‐DLBCL.45 The activated canonical NF‐κB pathway phosphorylates p65, which translocates into the nucleus, resulting in transcription of its target genes, including IRF4,41, 46 which, in turn, induces expression of PRDM1,47, 48 a transcription factor, which triggers the terminal differentiation into plasma cells. PRDM1 also functions as a tumor suppressor gene,6 as the encoded protein inhibits the survival and proliferation of B cells.49 When combined with conditional knockout of Prdm1, IKK2ca expressed in GC B cells elicits ABC‐DLBCL.5 The fact that PRDM1 is biallelically inactivated in human DLBCL6 supports the idea that complete, rather than partial repression of PRDM1 is required for malignant transformation of B cells. Given that BCL6 represses Prdm1 transcription in cell lines50 and that BCL6 translocation and PRDM1 inactivation are almost mutually exclusive in human DLBCL cases,6 expression of BCL6 is anticipated to collaborate with the NF‐κB signaling activated by CARD11 mutant in lymphomagenesis, but this possibility has not been tested in animal models. Thus, our study provides an experimental evidence for the collaboration of the two genes. Although functions of BCL6 are partially mediated by the repression of Prdm1, lymphoma cells in our mouse model expressed PRDM1, albeit at lower levels than a myeloma cell line, suggesting incompleteness of the BCL6‐mediated suppression of PRDM1, in stark contrast with PRDM1 knockout mice that recapitulates the biallelic inactivation of PRDM1 in human DLBCL.6 Therefore, our findings raise the possibility that BCL6 may have other functions besides repressing PRDM1. Mutant Card11 activates JNK,27 in addition to the NF‐κB pathway, increasing the amount of phosphorylated c‐Jun.51 BCL6 modulates the transcriptional activity of c‐Jun through physical interaction,52 which may represent an additional mechanism underlying the collaboration between CARD11 and BCL6. However, further studies are warranted to investigate the role of this pathway in the development of lymphoma.
Although exogenously expressed BCL2 was dispensable for lymphomagenesis in our model, endogenous BCL2 expression was elevated in lymphoma cells induced by co‐transduction of Card11 L232LI and Bcl6. Given that activated NF‐κB directly drives BCL2 expression,53, 54, 55 elevated BCL2 expression in our lymphoma, even without exogenous Bcl2, may reflect NF‐κB activation owing to Card11 L232LI transduction. This may at least partly explain elevated BCL2 expression in some human ABC‐DLBCL cases without BCL2 translocation.15 Although some DLBCL cases harbor both CARD11 mutation and BCL2 translocation, the sequence of these two events are not always known. In contrast, the two events happened almost simultaneously when induced in our experiments.
A mouse model of lymphoma, in which a mutant CARD11 is expressed from the embryonic stage in entire B cell fractions, has recently been reported. However, the animals developed lymphoma‐like disease very rapidly after birth and with 100% penetrance,51 in contrast to the low incidence of lymphoma (only two out of five mice) in our mice transplanted with Card11 L232LI only transduced iGCB cells. The difference in penetrance and rapidity of lymphoma development may reflect the difference in fractions of B cells (pan‐B cells vs GC B cells) and the developmental stage (embryonic vs adult) in which CARD11 is expressed. Human lymphomas usually develop in advanced age and, indeed, even in patients with germline CARD11 mutation, the development of monoclonal B‐cell disease culminates only in their 50s, despite polyclonal B‐cell lymphocytosis coupled with splenomegaly and follicular hyperplasia being observed during their childhood.25, 56 Together, these findings support the notion that CARD11 mutant needs additional genetic abnormalities to induce lymphoma.
CARD11 mutations are prevalent in ABC‐DLBCL,1, 24 a subtype of DLBCL, with poor prognosis.37, 57 Although ibrutinib, a Bruton tyrosine kinase inhibitor, is a promising drug for ABC‐DLBCL, it is ineffective for ABC‐DLBCL cases with CARD11 mutations,58 emphasizing the need for the development of new drugs. Our mouse model may help elucidate the molecular mechanisms underlying the development of DLBCL with CARD11 mutations to develop effective therapeutic agents.
Disclosure Statement
Shinobu Tsuzuki received research grant from the Japan Society for the Promotion of Science (JSPS KAKENHI Grant Number JP15K09492).
Abbreviations
- ABC
activated B cell‐like
- BCL
B‐cell lymphoma
- CARD11
caspase recruitment domain family member 11
- CREBBP
CREB binding protein
- DLBCL
diffuse large B‐cell lymphoma
- exp.
experiment
- FOXP1
forkhead box P1
- GC
germinal center
- GCB
germinal center B cell
- iGCB
induced germinal center B cell
- IGH
immunoglobulin heavy chain
- IRF
interferon regulatory factor
- NF‐κB
nuclear factor‐κB
- PRDM1
PR domain 1
- SHM
somatic hypermutation
Supporting information
Fig. S1. Distribution of genetic abnormalities coexisting with Card11 mutations.
{kind=link}
Fig. S2. Schematic representation of the experimental procedure.
{kind=link}
Fig. S3. Schematic representation of the expression vector.
{kind=link}
Fig. S4. Histology of developed lymphomas.
{kind=link}
Fig. S5. Flow cytometric analysis of germinal center B cells transduced with Card11 L232LI, Bcl6, and Bcl2 before transplantation.
{kind=link}
Fig. S6. Flow cytometric analysis of lymphoma cells.
{kind=link}
Fig. S7. Gross anatomy of a mouse with Card11 L232LI/Bcl6 doubly transduced lymphoma (Card11mut+Bcl6 #1).
{kind=link}
Fig. S8. Expression of BCL2 in developed lymphoma.
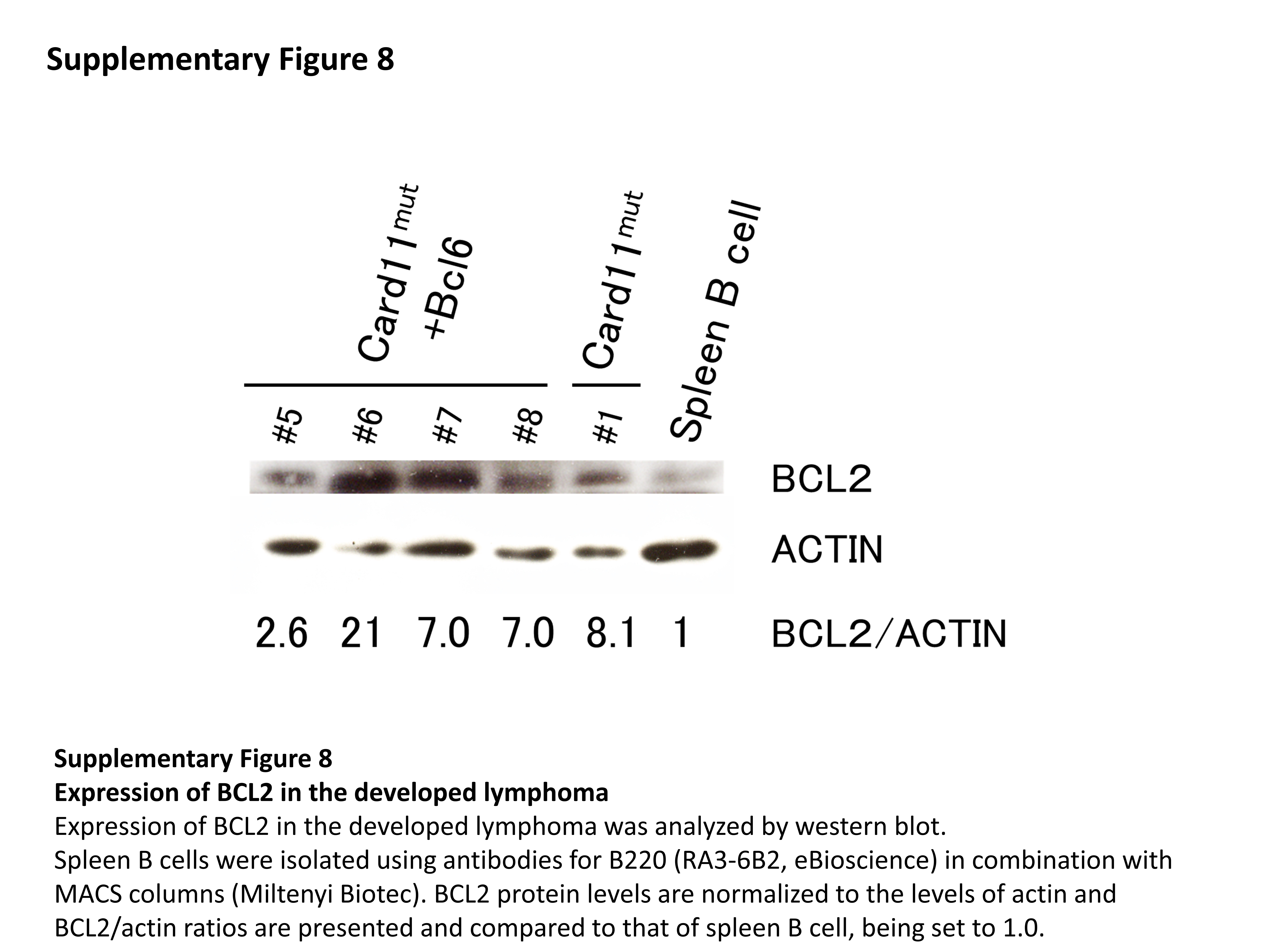{kind=link}
Fig. S9. Nuclear translocation of p65.
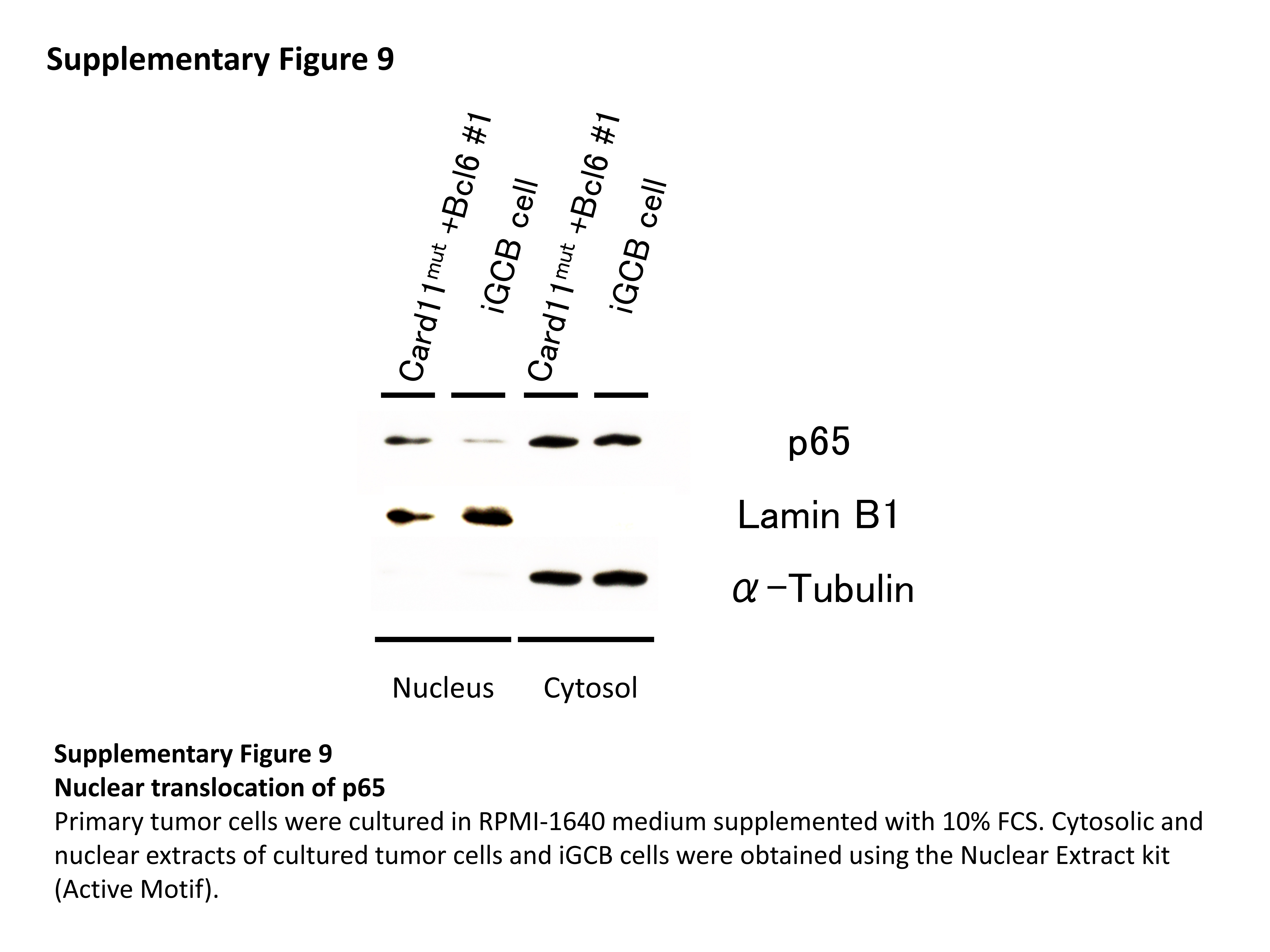{kind=link}
Fig. S10. Analysis of secondary transplantation of primary lymphoma cells.
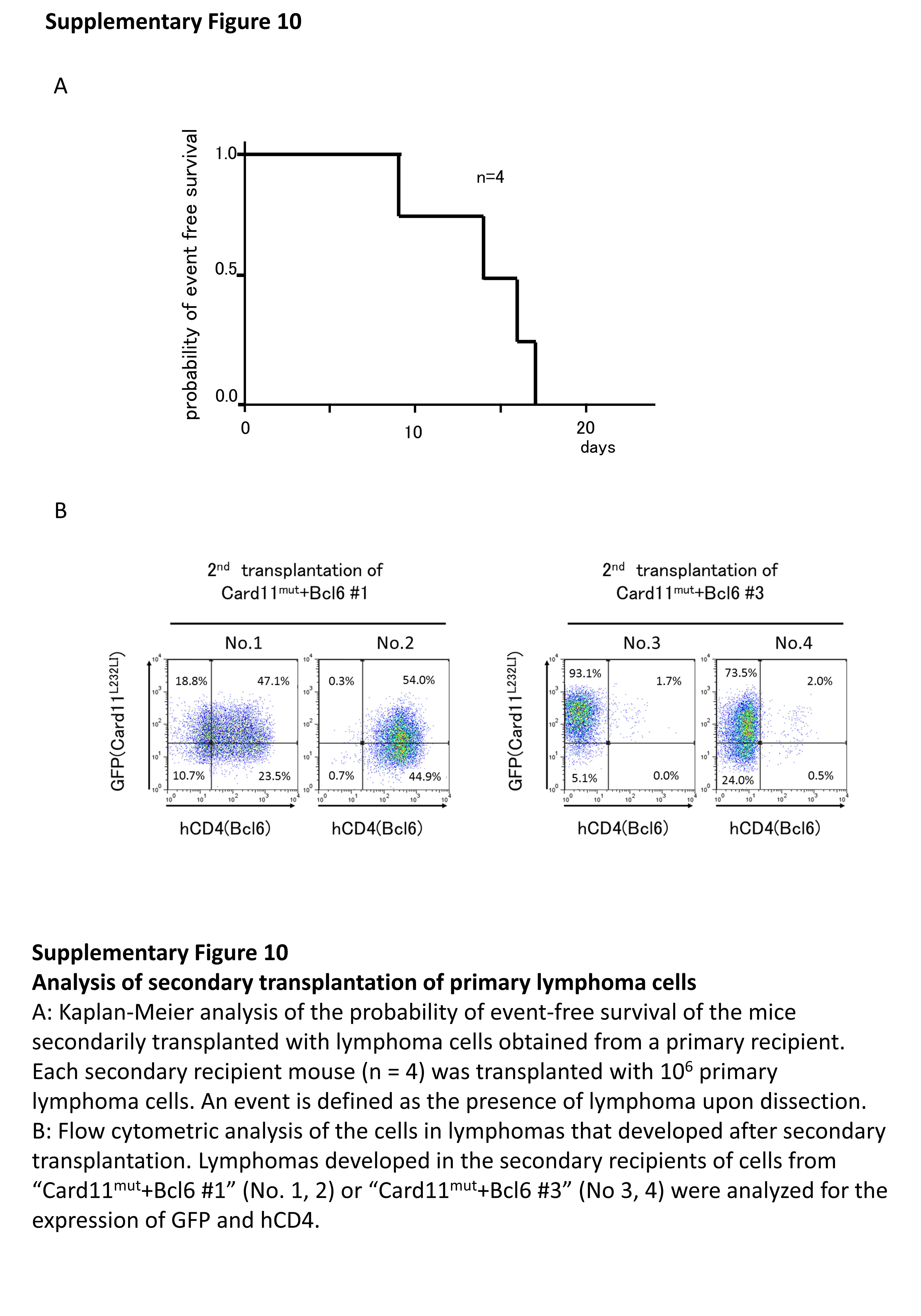{kind=link}
Table S1. Summary of the mice analyzed in this study
{kind=link}
Table S2. VDJ gene usage and analysis of ongoing somatic hypermutation of lymphoma cells
{kind=link}
Acknowledgments
We thank Seiko Sato for help with animal husbandry.
Cancer Sci 107 (2016) 1572–1580
Funding Information
Japan Society for the Promotion of Science (JP15K09492).
References
- 1. Pasqualucci L, Trifonov V, Fabbri G et al Analysis of the coding genome of diffuse large B‐cell lymphoma. Nat Genet 2011; 43: 830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang J, Grubor V, Love CL et al Genetic heterogeneity of diffuse large B‐cell lymphoma. Proc Natl Acad Sci USA 2013; 110: 1398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morin RD, Mungall K, Pleasance E et al Mutational and structural analysis of diffuse large B‐cell lymphoma using whole‐genome sequencing. Blood 2013; 122: 1256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cattoretti G, Pasqualucci L, Ballon G et al Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell 2005; 7: 445–55. [DOI] [PubMed] [Google Scholar]
- 5. Calado DP, Zhang B, Srinivasan L et al Constitutive canonical NF‐kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell‐like diffuse large cell lymphoma. Cancer Cell 2010; 18: 580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mandelbaum J, Bhagat G, Tang H et al BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell‐like diffuse large B cell lymphoma. Cancer Cell 2010; 18: 568–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arita K, Maeda‐Kasugai Y, Ohshima K et al Generation of mouse models of lymphoid neoplasm using retroviral gene transduction of in vitro‐induced germinal center B and T cells. Exp Hematol 2013; 41: 731–41 e9. [DOI] [PubMed] [Google Scholar]
- 8. Arita K, Tsuzuki S, Ohshima K, Sugiyama T, Seto M. Synergy of Myc, cell cycle regulators and the Akt pathway in the development of aggressive B‐cell lymphoma in a mouse model. Leukemia 2014; 28: 2270–2. [DOI] [PubMed] [Google Scholar]
- 9. Nojima T, Haniuda K, Moutai T et al In‐vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat Commun 2011; 2: 465. [DOI] [PubMed] [Google Scholar]
- 10. Alizadeh AA, Eisen MB, Davis RE et al Distinct types of diffuse large B‐cell lymphoma identified by gene expression profiling. Nature 2000; 403: 503–11. [DOI] [PubMed] [Google Scholar]
- 11. Ying CY, Dominguez‐Sola D, Fabi M et al MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol 2013; 14: 1084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pasqualucci L, Dominguez‐Sola D, Chiarenza A et al Inactivating mutations of acetyltransferase genes in B‐cell lymphoma. Nature 2011; 471: 189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morin RD, Mendez‐Lago M, Mungall AJ et al Frequent mutation of histone‐modifying genes in non‐Hodgkin lymphoma. Nature 2011; 476: 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iqbal J, Sanger WG, Horsman DE et al BCL2 translocation defines a unique tumor subset within the germinal center B‐cell‐like diffuse large B‐cell lymphoma. Am J Pathol 2004; 165: 159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iqbal J, Neppalli VT, Wright G et al BCL2 expression is a prognostic marker for the activated B‐cell‐like type of diffuse large B‐cell lymphoma. J Clin Oncol 2006; 24: 961–8. [DOI] [PubMed] [Google Scholar]
- 16. Cerchietti LC, Ghetu AF, Zhu X et al A small‐molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 2010; 17: 400–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nunez G, Seto M, Seremetis S et al Growth‐ and tumor‐promoting effects of deregulated BCL2 in human B‐lymphoblastoid cells. Proc Natl Acad Sci USA 1989; 86: 4589–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high‐grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 1991; 349: 254–6. [DOI] [PubMed] [Google Scholar]
- 19. Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl‐2. Nature 1990; 348: 331–3. [DOI] [PubMed] [Google Scholar]
- 20. Zhang B, Calado DP, Wang Z et al An oncogenic role for alternative NF‐kappaB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Rep 2015; 11: 715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pasqualucci L, Bhagat G, Jankovic M et al AID is required for germinal center‐derived lymphomagenesis. Nat Genet 2008; 40: 108–12. [DOI] [PubMed] [Google Scholar]
- 22. Gu X, Booth CJ, Liu Z, Strout MP. AID‐associated DNA repair pathways regulate malignant transformation in a murine model of BCL6‐driven diffuse large B‐cell lymphoma. Blood 2016; 127: 102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu M, Duke JL, Richter DJ et al Two levels of protection for the B cell genome during somatic hypermutation. Nature 2008; 451: 841–5. [DOI] [PubMed] [Google Scholar]
- 24. Lenz G, Davis RE, Ngo VN et al Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008; 319: 1676–9. [DOI] [PubMed] [Google Scholar]
- 25. Snow AL, Xiao W, Stinson JR et al Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med 2012; 209: 2247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakagawa M, Tsuzuki S, Honma K, Taguchi O, Seto M. Synergistic effect of Bcl2, Myc and Ccnd1 transforms mouse primary B cells into malignant cells. Haematologica 2011; 96: 1318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jeelall YS, Wang JQ, Law HD et al Human lymphoma mutations reveal CARD11 as the switch between self‐antigen‐induced B cell death or proliferation and autoantibody production. J Exp Med 2012; 209: 1907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li S, Ilaria RL Jr, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia‐like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med 1999; 189: 1399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ehlich A, Martin V, Muller W, Rajewsky K. Analysis of the B‐cell progenitor compartment at the level of single cells. Curr Biol 1994; 4: 573–83. [DOI] [PubMed] [Google Scholar]
- 30. Sander S, Calado DP, Srinivasan L et al Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 2012; 22: 167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lossos IS, Okada CY, Tibshirani R et al Molecular analysis of immunoglobulin genes in diffuse large B‐cell lymphomas. Blood 2000; 95: 1797–803. [PubMed] [Google Scholar]
- 32. Sanchez‐Izquierdo D, Siebert R, Harder L et al Detection of translocations affecting the BCL6 locus in B cell non‐Hodgkin's lymphoma by interphase fluorescence in situ hybridization. Leukemia 2001; 15: 1475–84. [DOI] [PubMed] [Google Scholar]
- 33. Coffman RL, Weissman IL. A monoclonal antibody that recognizes B cells and B cell precursors in mice. J Exp Med 1981; 153: 269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter JA. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med 1991; 173: 721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shinall SM, Gonzalez‐Fernandez M, Noelle RJ, Waldschmidt TJ. Identification of murine germinal center B cell subsets defined by the expression of surface isotypes and differentiation antigens. J Immunol 2000; 164: 5729–38. [DOI] [PubMed] [Google Scholar]
- 36. Shaffer AL, Lin KI, Kuo TC et al Blimp‐1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 2002; 17: 51–62. [DOI] [PubMed] [Google Scholar]
- 37. Choi WW, Weisenburger DD, Greiner TC et al A new immunostain algorithm classifies diffuse large B‐cell lymphoma into molecular subtypes with high accuracy. Clin Cancer Res 2009; 15: 5494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Challa‐Malladi M, Lieu YK, Califano O et al Combined genetic inactivation of beta2‐Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 2011; 20: 728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hans CP, Weisenburger DD, Greiner TC et al Confirmation of the molecular classification of diffuse large B‐cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004; 103: 275–82. [DOI] [PubMed] [Google Scholar]
- 40. Reljic R, Wagner SD, Peakman LJ, Fearon DT. Suppression of signal transducer and activator of transcription 3‐dependent B lymphocyte terminal differentiation by BCL‐6. J Exp Med 2000; 192: 1841–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saito M, Gao J, Basso K et al A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell 2007; 12: 280–92. [DOI] [PubMed] [Google Scholar]
- 42. Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell‐like diffuse large B cell lymphoma cells. J Exp Med 2001; 194: 1861–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lossos IS, Alizadeh AA, Eisen MB et al Ongoing immunoglobulin somatic mutation in germinal center B cell‐like but not in activated B cell‐like diffuse large cell lymphomas. Proc Natl Acad Sci USA 2000; 97: 10209–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lamason RL, McCully RR, Lew SM, Pomerantz JL. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC‐responsive inhibitory domain. Biochemistry 2010; 49: 8240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Basso K, Dalla‐Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol 2015; 15: 172–84. [DOI] [PubMed] [Google Scholar]
- 46. Chen LF, Greene WC. Shaping the nuclear action of NF‐kappaB. Nat Rev Mol Cell Biol 2004; 5: 392–401. [DOI] [PubMed] [Google Scholar]
- 47. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor‐4 coordinates isotype switching with plasma cell differentiation. Immunity 2006; 25: 225–36. [DOI] [PubMed] [Google Scholar]
- 48. Kwon H, Thierry‐Mieg D, Thierry‐Mieg J et al Analysis of interleukin‐21‐induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 2009; 31: 941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shapiro‐Shelef M, Lin KI, McHeyzer‐Williams LJ, Liao J, McHeyzer‐Williams MG, Calame K. Blimp‐1 is required for the formation of immunoglobulin secreting plasma cells and pre‐plasma memory B cells. Immunity 2003; 19: 607–20. [DOI] [PubMed] [Google Scholar]
- 50. Tunyaplin C, Shaffer AL, Angelin‐Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl‐6 inhibits plasmacytic differentiation. J Immunol 2004; 173: 1158–65. [DOI] [PubMed] [Google Scholar]
- 51. Knies N, Alankus B, Weilemann A et al Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant B‐cell proliferation via cooperative NF‐kappaB and JNK activation. Proc Natl Acad Sci USA 2015; 112: E7230–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vasanwala FH, Kusam S, Toney LM, Dent AL. Repression of AP‐1 function: a mechanism for the regulation of Blimp‐1 expression and B lymphocyte differentiation by the B cell lymphoma‐6 protooncogene. J Immunol 2002; 169: 1922–9. [DOI] [PubMed] [Google Scholar]
- 53. Catz SD, Johnson JL. Transcriptional regulation of bcl‐2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 2001; 20: 7342–51. [DOI] [PubMed] [Google Scholar]
- 54. Zhao B, Barrera LA, Ersing I et al The NF‐kappaB genomic landscape in lymphoblastoid B cells. Cell Rep 2014; 8: 1595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Viatour P, Bentires‐Alj M, Chariot A et al NF‐ kappa B2/p100 induces Bcl‐2 expression. Leukemia 2003; 17: 1349–56. [DOI] [PubMed] [Google Scholar]
- 56. Brohl AS, Stinson JR, Su HC et al Germline CARD11 mutation in a patient with severe congenital B cell lymphocytosis. J Clin Immunol 2015; 35(1): 32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Visco C, Li Y, Xu‐Monette ZY et al Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B‐cell lymphoma: a report from the International DLBCL Rituximab‐CHOP Consortium Program Study. Leukemia 2012; 26: 2103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilson WH, Young RM, Schmitz R et al Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 2015; 21: 922–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Distribution of genetic abnormalities coexisting with Card11 mutations.
Fig. S2. Schematic representation of the experimental procedure.
Fig. S3. Schematic representation of the expression vector.
Fig. S4. Histology of developed lymphomas.
Fig. S5. Flow cytometric analysis of germinal center B cells transduced with Card11 L232LI, Bcl6, and Bcl2 before transplantation.
Fig. S6. Flow cytometric analysis of lymphoma cells.
Fig. S7. Gross anatomy of a mouse with Card11 L232LI/Bcl6 doubly transduced lymphoma (Card11mut+Bcl6 #1).
Fig. S8. Expression of BCL2 in developed lymphoma.
Fig. S9. Nuclear translocation of p65.
Fig. S10. Analysis of secondary transplantation of primary lymphoma cells.
Table S1. Summary of the mice analyzed in this study
Table S2. VDJ gene usage and analysis of ongoing somatic hypermutation of lymphoma cells