

Abstract
Alcohol is the most socially accepted addictive drug. Alcohol consumption is associated with some health problems such as neurological, cognitive, behavioral deficits, cancer, heart and liver disease. Mechanisms of alcohol-induced toxicity are presently not yet clear. One of the mechanisms underlying alcohol toxicity has to do with its interaction with amino acid-homocysteine (Hcy), which has been linked with brain neurotoxicity. Elevated homocysteine (Hcy) impairs with various physiological mechanisms in the body, especially metabolic pathways. Hcy metabolism is predominantly controlled by epigenetic regulation such as DNA methylation, histone modifications, and acetylation. An alteration in these processes leads to epigenetic modification. Therefore, in this review, we summarize the role of Hcy metabolism abnormalities in alcohol-induced toxicity with epigenetic adaptation and their influences on cerebrovascular pathology.
Keywords: DNA methylation, cystathionine-β-synthase, cystathionine-γ-lyase, cerebrovascular pathology, vascular dementia
Introduction
Alcohol intake is one of the leading threats to the health and safety of people in Western countries, affecting about 14 million people in the United States (Ron 2004). Alcohol is readily spread throughout the body in the blood stream and crosses biological membranes, which affect virtually all biological processes. Ethanol exposure causes profound damages to both the adult and developing brain. Drinking excessive amounts of alcohol (chronic) regularly for years is toxic to almost every tissue of the body. Many of the toxic effects of alcohol are due to disturbances of a wide variety of metabolic functions and organ damage. Long-term alcohol use increases the risk of various cerebrovascular disorders and different types of cancers (Agarwal and Seitz et al., 2001). Alcohol depresses the central nervous system (CNS), interfering with mood behavior, cognition and coordination.
Hyperhomocysteinemia (HHcy) is characterized by an elevated concentration of Hcy in the blood, greater than 15 μmol/L. There are different levels of HHcy: mild (15–30 μmol/L), moderate (31–100 μmol/L), or severe (>100 μmol/L). Excess production of Hcycan occur as a result of impaired metabolism due to a deficiency in cofactors (vitamin B6, B12, folate) or to genetic alteration in metabolic enzymes (methionine synthase; MS, methyltetrahydrofolate reductase; MTHFR, cystathionine-β-synthase; CBS, and cystathionine-γ-lyase; CSE). HHcy is commonly seen in about 5% to 12% of the general population and is recognized as a risk factor for cerebrovascular diseases (Okura et al., 2014; Feng et al., 2013; Clarke et al., 2000) diabetes, obesity, and hepatic steatosis (Dara and Kaplowitz et al., 2011; Kaplowitz et al., 2004). Various epidemiological and longitudinal studies suggested a causal link between Hcy and cognitive impairment (Nurk et al., 2005). This may be due to cerebrovascular as well as to direct neurotoxic mechanisms (Sachdev et al., 2005). Changes of Hcy overtime predicted the decline in memory scores in elderly subjects (Nurk et al., 2005) and several follow up studies demonstrated a positive association between Hcy at baseline and a worsening in some measures of cognitive function after several years (Ravaglia et al., 2005; McCaddon et al., 2001).
Chemistry of Homocysteine
Hcy is a naturally occurring homologue of the amino acid cysteine and differs by only an additional methylene (-CH2) group in normal physiological conditions. Hcy does not come from the diet; it is primarily synthesized from methionine (Met) through a multi-step process within the body. As previous reports suggest, excess accumulation of Hcy in the body causes cell damage and promotes vascular and microvascular disease, leading to cerebrovascular dysfunction (Dayal et al., 2004; Lominadze et al., 2006; Eren et al., 2014; Kamat et al., 2013; Muradashvili et al., 2014). This occurs because of an error in biochemical transformation in metabolic processes. It may also occur due to a lack of necessary and essential vitamins required for physiological processes within the body. The lack of these vitamin cofactors in transformation pathways can produce elevated homocysteine levels that place patients at risk for vascular disease. HHcy is associated with damage to the vascular system by a mechanism related to oxidative stress, resulting in a build-up of damaging free radicals (Tyagi et al., 2005; Kalani et al., 2014). Free radical oxygen species and other known oxidants may trigger many brain diseases including stroke and vascular dementia. These reactive chemical, oxygen, or nitrogen species generate free radicals, which can oxidize the neuronal lipid bilayer by oxidizing lipids and proteins in the vascular endothelial wall. Hcy itself can undergo auto-oxidation, thus causing the disruption of redox homeostasis and affecting the redox signaling pathways in vascular and neuronal cells (Zou et al., 2005;Perna et al., 2003; Weiss et al., 2003). Also, Hcy has been found to induce neurological dysfunction via oxidative stress (James et al., 2004; Ho et al., 2003). A large body of evidence suggests that chronic alcohol exposure is associated with impaired sulfur amino acid metabolism, and thus can alter redox stress in the brain (Worrall and Thiele et al., 2001). The proposed mechanisms by which HHcy and alcohol exposure induce injury in the brain are both underlined by vascular damage, cognitive impairment, and neurological complications that can result in a severe cerebrovascular event, such as stroke or dementia.
Metabolism of Homocysteine
The association between HHcy and cerebrovascular diseases has been suggested to be an independent risk factor for many pathological conditions, including cerebral stroke (Joseph et al., 2009). Hcy is produced as an intermediate product in Met metabolism, and its concentrations vary extensively depending on Met concentration. Hcy is metabolized by way of transsulfuration to cysteine by cystathionine-β-synthase (CBS) or re-methylated to Met through Methionine synthase (MS) or Betaine methyltransferase (BMT) (Selhub et al., 1999). Other metabolic products such as Vitamin B6, which acts as the cofactor of CBS, are essential for Hcy metabolism. In addition, folate and vitamin B12 are needed as co-substrates and cofactors for MS. It has been shown that Hcy is regulated through a series of pathways, which are dependent on B vitamins, particularly folate (Halsted et al., 2002). However, B12 deficiency, especially in elderly, is often associated with neurodegenerative disorders including vascular dementia (VAD), Parkinson’s disease (PD), multiple sclerosis, and Alzheimer’s disease (AD). Several studies have suggested that folate is very important for brain functions (Kennedy et al., 2016) and deprivation of folate induce robust increase in Hcy, oxidative stress and increased Amyloid beta (Aβ)-induced apoptosis, while folate supplementation prevented the generation of oxidative stress by Aβ (Ho et al., 2003). Treatment with the S-adenosyl hydrolase inhibitor (3-deaza adenosine), provides neuroprotection in normal, apolipoprotein E-deficient mice and in cultured neuronal cells deprived of folate and vitamin E and subjected to oxidative challenge (Tchantchou et al., 2004). In agreement with those studies, it has also been reported that intake of vitamin B12 and B6 improve brain function by lowering the levels of Hcy that exist in HHcy condition (Quadri et al., 2004).
Methylene tetrahydrofolate reductase (MTHFR) is an enzyme in Met metabolism, and it reduced activity of the MTHFR gene increases the mean plasma Hcy level, which is influenced by a single-nucleotide polymorphism in the MTHFR gene. A collective genetic variant of the MTHFR gene, also known as a key enzyme in Hcy metabolism, has been associated with an increase in Hcy levels (Delvin et al., 2004). Hcy is also involved in the metabolism of methyl groups (DNA-methylation) plays an important role in the regulation of gene expression. Therefore, it has been suggested that Hcy is an important epigenetic factor.
Homocysteine is methylated to methionine and then to S-adenosyl methionine (SAM), which is a substrate and primary methyl donor in methylation reactions. Since S-adenosyl homocysteine (SAH) is both a product and potent inhibitor of methylation reactions, the SAM/SAH ratio is considered a useful indicator for proper methylation (Tyagi et al., 2005). SAH is also the substrate for SAH hydrolase (SAHH), a reversible reaction that generates homocysteine but increases SAH when homocysteine concentrations are high. In trans-sulfuration reactions, homocysteine is a substrate for the CBS reaction (Ovechkin et al., 2006; Tyagi et al., 2007), which is facilitated by SAM to generate cystathionine and finally produces glutathione (GTH), the principal antioxidant in the body as well as in the brain (Figure. 1). Studies have revealed that drinking ethanol/alcohol is linked to elevated homocysteine levels in the plasma (Stickel et al., 2000) as well as in the liver. Reports have also suggested that consumption of ethanol along with a folate-deficient diet is associated with increased levels of homocysteine (Halsted et al., 2002). Thus, it may be concluded that increased ethanol intake and low folate diets may lead to HHcy.
Figure 1. Interaction of Alcohol and Folic acid pathways.
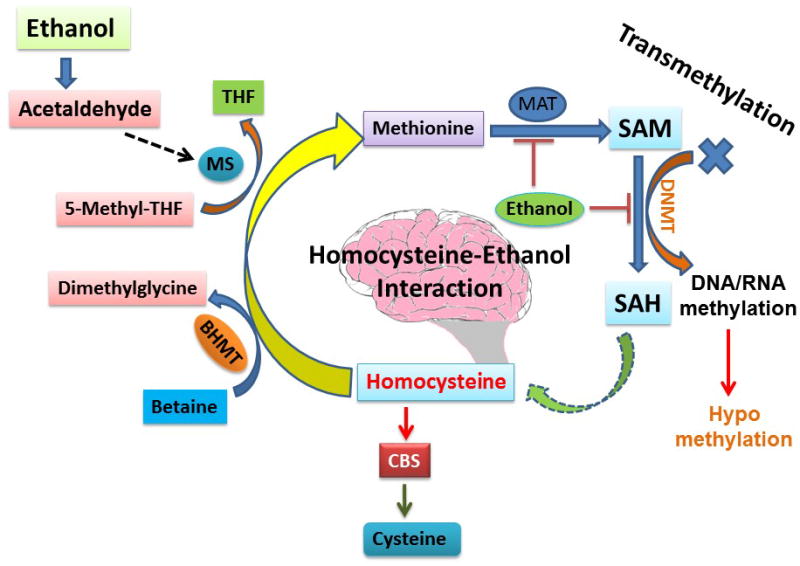
Acetaldehyde, the end product of ethanol, interacts with methionine synthase (MS), which is responsible for the conversion of Hcy to methionine. One of the possible reasons for HHcy during alcohol exposure may be due to inhibition of the enzyme MS by ethanol; then Hcy is not converted into methionine and thus leads to the condition of HHcy. On the other hand, other pathways such as transmethylation pathways may also be affected by the inhibition of MS and may cause abrupt disturbance of S-adenosyl methionine (SAM) and S-Adenosyl-L-homocysteine (SAH). SAM to SAH conversion is controlled by DNA methyltransferase (DNMT). If the pathway disturbed, the transmethylation process also gets disturbed. On the other hand, high Hcy also causes the low expression of CBS, an antioxidant enzyme in the brain which maintains the redox system. In this way, ethanol can cause HHcy and thus affect the transmethylation pathway, redox system, and impair brain function.
Homocysteine, Alcohol and metabolic disturbances in the brain
Genetic alteration and its consequences lead to decreased expression of Hcy-metabolizing enzymes such as CBS, MTHFR and methionine synthase (MS) that leads to HHcy (Faraci and Lentz et al., 2004). Changes in plasma Hcy concentrations reflect one aspect of the metabolic consequences of methyl group deficiencies or nutrient supplementations. Folic acid supplementation spares betaine as a methyl donor. Betaine is a significant determinant of plasma Hcy, particularly in cases of folate deficiency, methionine overload, or alcohol consumption. Betaine supplementation has a lowering effect on methionine load and high Hcy levels. Increasing choline or betaine levels can reduce hypo methylation and lower Hcy levels. Folic acid supplementation also lowers the risk factor for stroke by reducing total plasma homocysteine levels. Increased levels of Hcy can cause elevated blood pressure and are considered a risk factor for recurrent strokes. There is convincing evidences about the effectiveness of vitamin-B complex supplementation in lowering the stroke risk (Gustavo et al., 2011). In particular, Hcy levels are increased in the body when metabolism of cysteine or methionine is impaired. This often occurs due to dietary deficiencies in vitamin B6, vitamin B12 and folic acid. The chronic alcoholic tends to be undernourished and is usually deficient in B vitamins. However, it remains obscure whether alcohol-dependent patients benefit from homocysteine-lowering strategies through folate, vitamin B6 and B12 supplementation, particularly in those who have low folate status (Figure. 2).
Figure 2. Effect of Alcohol on Folic acid pathways.
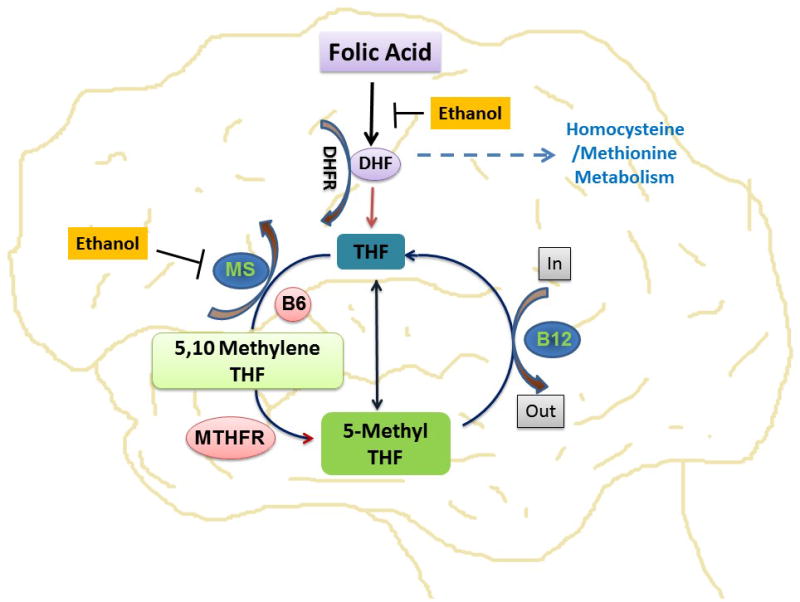
Representative diagram showing the interaction of ethanol (alcohol) on folic acid pathways. Folic acid is converted into tetrahydrofolate (THF) by the enzyme dihydrofolate reductase (DHFR). This THF is converted into methylene tetrahydrofolate (MTHF) by the cofactor vitamin B6. Again MTHF is transformed to THF by the key enzyme, MTHF reductase (MTHFR). In this diagram, the authors speculate that ethanol may inhibit the folic acid to DHF conversion and methionine synthase activity.
Homocysteine, Alcohol and Cerebrovascular dysfunction
Many studies suggest that heavy alcohol consumption increases the risk for cardiac disease and ischemic stroke (Hillbom et al., 1999a, b, c). It is even possible that heavy drinking and occasional drinking could trigger an ischemic stroke (Lai et al., 2012). Interestingly, moderate levels of alcohol consumption have a protective effect on coronary artery disease, cardiac disease, and hemorrhagic strokes, but alcohol consumption beyond moderate levels is risk factor for coronary artery disease, cardiac disease, hemorrhagic strokes, and chronic liver disease. An alcohol-induced stroke can occur after an acute intake of an intoxicating dose of alcohol causes a sudden marked increase of blood flow, which causes a dislodging of a thrombus present in the arterial wall. The circulatory effects of alcohol may not only dispatch emboli from the heart, but also artery-to-artery emboli from proximal arterial sources. In this context, the finding by Koehler et al., (2001) clearly suggests the strong association between total Hcy, folate intake and, alcohol consumption. Several studies suggest that brain infarction in stroke showed a positive correlation with heavy drinking of alcohol. Although chronic ethanol consumption affects plasma Hcy levels, which is associated with the risk of cerebrovascular disease (Hankey and Eikel boom et al., 1999), in consistencies with results deal with moderate alcohol consumption and increased levels of Hcy (Bleichet et al., 2001; Jacques et al., 2001), decreased levels of Hcy (Burger et al., 2004; de Bree et al., 2001), or unchanged (Ayaoriet et al., 2000) Hcy levels.
Epidemiological studies have revealed that vascular risk factors associated with diabetes are high Hcy levels and hypertension, which may play a role in the development of Alzheimer’s disease (AD). The elevated level of Hcy in the plasma is recognized as a risk factor for AD and predictive parameter for the decline in cognitive function. Ehrlich and Humpel et al., 2012 reported that homocysteine and ethanol treatments declined memory function and reduced the number of cholinergic neurons in rodents and induced blood-brain barrier leakage. Moreover, a combination of ethanol and cholesterol did not alter the memory and blood-brain barrier (BBB) leakage, but the combination of ethanol and homocysteine did promote BBB damage. These observations strongly suggest that homocysteine and alcohol in combination worsen conditions in the brain, such as cerebrovascular function (Bagheri et al., 2015). In addition, the damage accelerates the progression of brain pathologies, such as impaired cerebral blood flow, AD progression, and vascular dementia. Both AD and VD are the major forms of dementia affecting the elderly population, in which the levels of many metabolites are altered in cerebrospinal fluid (CSF) and serum (Lopamudra et al., 2013).
Alcohol and impaired brain function in association of NMDA and GABA receptor
Alcohol affects the brain’s neurons in several ways. It alters their membranes as well as their ion channels, enzymes, N-methyl-D-aspartate (NMDA) and gamma-aminobutyric acid (GABA) receptors. There are numerous reports showed ethanol action is directly associated with neurotransmitters like dopamine, serotonin, GABA, NMDA and its receptors (Fernando et al., 1997, George et al., 2007,2009,2010, Benjamin et al., 2016; Spencer et al., 2016; McClintick et al., 2016). Primary mechanism by which ethanol affects the brain is through regulation of neurotransmitters and ligands, voltage gated channels that are essential for neuronal excitability and synaptic function (Hoffman et al., 2003). By altering the function of these channels, ethanol alters neuronal properties and ultimately influences the function of the brain, and consequently behavior.
It has been found that Hcy induces neuronal cell damage by stimulating NMDA receptors as well as by producing free radicals. The neurotoxicity of Hcy via overstimulation of N-methyl-D-aspartate receptors may contribute to the pathogenesis of brain shrinkage and be linked to alcohol exposure. Short-term exposure for a few hours to ethanol produces only temporary functional changes. However, long-term ethanol exposure produces persistent neuro-adaptive changes that have serious long-term consequences to brain function. For example, chronic exposure to ethanol has been reported to increase the density of NMDA receptor binding and elevates the expression of specific NMDA receptor subunits in the brain (Hoffman et al., 2003). Similar changes in the expression of voltage-gated Ca2+ channels (VGCCs) have been reported following chronic alcohol exposure (Dolin et al., 1987). The alteration in NMDA receptors and VGCCs are thought to underlie the hyper-excitability and excitotoxicity associated with chronic alcohol use (Gulya et al., 1991; Whittington et al., 1995).
GABA receptors are probably the most common kind in the mammalian nervous system, and GABA is the primary inhibitory neurotransmitter in the brain (Tyagi et al., 2007a, b). Hcy, an excitatory amino acid, has a high binding affinity for GABA receptors, it can inhibit their activity and expression. It has been shown that alcohol mediates the rise in homocysteine, as well as the neurotransmitters GABA and glutamate and their respective receptors: GABA and NMDA (Davies, 2003). GABA reduces the activity of the signal-receiving neuron, whereas glutamate, the primary excitatory neurotransmitter, stimulates the activity of the signal receiving neuron. Alcohol has been shown to activate the GABA receptors and to inhibit the NMDA receptors (Figure. 3) and resulting in the impairment of brain functions
Figure 3. Interaction of Alcohol with receptors.
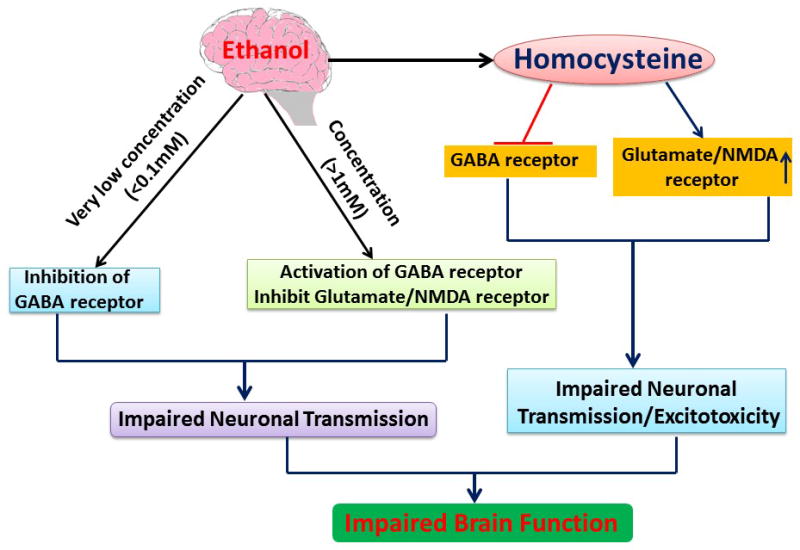
This flow chart summarizes the effects of ethanol on GABA and NMDA receptors. An interesting finding is that the concentration of ethanol determines the activation or inhibition of GABA and NMDA receptors. Alteration in GABA and NMDA receptors by ethanol affects the neuronal transmission and thus brain function. As per reports, ethanol increases the homocysteine concentration and binding affinity of Hcy with GABA and NMDA receptors also affect neuronal function.
Alcohol and Epigenetic changes (Acetylation/Deacetylation)
Long-term alcohol exposure causes widespread changes in brain gene expression in humans and animal models (George et al., 2010, Xiang et al., 2015, McClintick et al., 2016). Many of these contribute to cellular adaptations which leads to changes in DNA and histone methylation, histone acetylation, and microRNA expression that affect expression of multiple genes in various types of brain cells (i.e., neurons and glia), that contribute to brain pathology and brain plasticity associated with alcohol abuse. Histone proteins are the second major target of epigenetic changes. The work of Ghezzi et al., 2013 explored genes for the functional alcohol tolerance by using a novel genomic epigenetic approaches of two chemically distinct alcohols. In Drosophila melanogaster, ethanol and benzyl alcohol induce mutual cross-tolerance, indicating that they share a common mechanism for producing tolerance. Ethanol, its major metabolite acetate and HHcy have been shown to induce histone H3 acetylation (Shukla et al.,2015; Fang et al.,2016). Ethanol enhanced histone H3K9 acetylation in a dose- and time-dependent manner in primary cultures of hepatocytes, without affecting H3K14 acetylation, demonstrating the specificity of ethanol in regulating covalent modifications to histone proteins. Park and colleagues (2012) reported that chronic ethanol treatment did not change global H3K9 acetylation, but did result in an increase in H3K9 acetylation. Epigenetic regulation following ethanol administration showed an increase in H3K9 acetylation and a decrease in H3K27 trimethylation marks in the brains of rodents (D’Addario et al., 2013). Histone lysine methylation is an important modification that is associated with chromatin remodeling and gene regulation and has been implicated in drug-induced neuronal plasticity mechanisms, memory formation, and cognition (Gupta et al. 2010; Maze et al. 2010; Schaefer et al., 2009; Shinkai and Tachibana et al 2011). In brief, histone acetylation and deacetylation mechanisms, especially in the brain are an important neuronal regulatory mechanism that may be involved in the development and maintenance during alcohol exposure. Deacetylation i.e histone deacetylases (HDAC) involves removal of an acetyl group on lysine residues in the N-terminal tail and on the surface of the core of histone proteins. Histone deacetylation is associated with gene silencing and alcohol regulates metabolic activity which one ways regulates gene expression. Thus, it could be suggested that (histone deacetylases inhibitor therapy may provide an alternative option to treat alcohol exposure and its underlying mechanism and causes.
Alcohol and Epigenetic changes (DNA methylation)
Elevated Hcy levels have been previously linked to altered DNA methylation levels in various diseases such as cardiovascular disease (CVD) and cerebrovascular disease. Folate or vitamin B12 based methods of lowering Hcy have had limited effects in reducing CVD events. Vitamin based therapies have limitations and have failed to reverse epigenetic changes induced by HHcy (Kalani et al. 2014). DNA methylation is a covalent modification to DNA, which occurs on cytosine residues and involves the addition of a methyl group to the C5 position (5-mc) and is catalyzed by DNA methyltransferases (DNMTs) (Bestor et al., 2000). Methylation of DNA is thought to mediate transcriptional repression via two ways: directly hindering binding of DNA-binding proteins (Klose and Bird et al., 2006) and indirectly through binding of methyl-CpG (Goll and Bestor et al., 2005). The first identified and most abundant DNMT, DNMT1 also known as a “maintenance methyltransferase” can recognize hemi-methylated DNA and perform methylation of the complementary strand. The de novo methyltransferases, DNMT3a and DNMT3b, methylate previously un-methylated cytosine’s. They are essential during development and have been implicated in synaptic plasticity in the CNS and in learning and memory mechanisms (Feng et al., 2010; Levenson et al., 2006a,b; Miller and Sweatt et al., 2007 and 2008). The demethylation of DNA is a rapidly emerging study involving a complex inter play of interdependent pathways and mechanisms (Gavin et al., 2013). Activity-dependent DNA demethylation is a dynamic process crucial to neuronal function. The work of Robert A. Philibert et al., 2012 reported that chronic alcohol intake is associated with significant changes in CpG methylation, and in particular, increased hypermethylation of CpG islands there by it causes chronic alcohol tolerance. A genome-wide DNA methylome analysis suggests that widespread alcohol-induced alterations with significant hypermethylation of many regions of chromosomes particularly those associated with molecular pathways for metabolic processes, oxidative stress, and neuronal properties of stem cells, which it causes alcohol-induced terato-genesis in nervous system (Khalid et al., 2014). Since it has been continuously reported that chronic alcohol exposure leads to a rise in plasma homocysteine levels as well as increased methylation/demethylation, therapies targeting the epigenetic machinery as well as lowering circulating Hcy may have a more valuable effect in reducing the incidence of cerebrovascular events and complications (Figure. 3).
Hyperhomocysteinemia, Alcohol and Epigenetic regulation
Elevated homocysteine levels (HHcy) in the blood of alcoholic patients have been associated with hypermethylation at the promoter of the homocysteine-induced endoplasmic reticulum protein gene, corresponding to lower mRNA expression in blood cells (Bleich et al., 2006). Hyperhomocysteinemia levels have also been implicated in hypermethylation of the alpha-synuclein (SNCA) gene promoter, which may be responsible for altered mRNA and protein expression, and was linked to alcohol cravings (Bonsch et al., 2004, 2005). Chronic alcohol consumption could be associated with altered methylation patterns of various genes acting via SAM metabolism and altering homocysteine levels. However, hypomethylation of DNA has been reported. Alternation in promoter DNA methylation levels in alcohol-dependent patients have been reported of many genes, including nerve growth factor (NGF), homocysteine induced endoplasmic reticulum protein (HERP), opiod receptor (OPRMI), dopamine transporter (DAT). Increased DNA methylation of the SNCA gene in alcoholic patients may be eventually caused in disruption of dopaminergic neurotransmission. Elevated promoter methylation in HERP results in increased rate of seizure and neurologic damage (nieratschker et al., 2013). Approaches that can detect altered transcripts and corresponding changes in methylation levels in peripheral blood samples, such as lymphocytes and mononuclear cells can be used as potential tool for the development of biomarkers (Biermann et al., 2009; Hillemacher et al., 2009;Bleich and Hillemacher et al., 2009).
Future direction
The metabolic disturbances and epigenetic processes during alcohol exposure or the combination of alcohol intake and high homocysteine levels clearly facilitate the development of cerebrovascular pathology. Cerebrovascular dysfunction, such as cerebral stroke and vascular dementia, are extensively correlated with HHcy. However, more research on the metabolic and epigenetic processes of these mechanisms, which are promising but have limitations, need to be addressed. The major issue is that the same metabolic or epigenetic changes involved in alcohol mediated high homocysteine levels are also involved in hyperhomocysteinemia alone. It needs to be explored whether this is in fact implies the same mechanisms or if there are different mechanisms in each which create cerebrovascular pathology.
The metabolic disturbances and epigenetic processes during alcohol exposure or the combination of high homocysteine levels and chronic alcohol intake clearly facilitate the development of cerebrovascular pathology. Cerebrovascular dysfunction, such as cerebral stroke and vascular dementia, are extensively correlated with hyperhomocystenemia. However, more research into the metabolic and epigenetic processes of these mechanisms, which are promising but have some limitations, needs to be addressed. Also, continued exploration of how alcohol mediates high levels of homocysteine and/or alcohol intake plus HHcy induces cerebral stroke and related cerebrovascular pathologies is needed. The major issue is that the same metabolic or epigenetic changes involved in alcohol mediated high homocysteine levels are also involved in hyperhomocysteinemia alone. It needs to be explored whether this is in fact implies the same mechanisms or if there are different mechanisms in each which create cerebrovascular pathology.
Conclusion
In the present review, we discussed the metabolic and epigenetic changes during alcohol intake and their effects on brain function altered by plasma homocysteine levels. High Hcy levels lead to alteration of the activity of DNMTs in part acetylation/deacetylation that consequently causes hypo or hyper methylation of genes. This metabolic and epigenetic alteration further affects the genes that play an important role in pathophysiology of cerebrovascular disease, such as cerebral stroke and vascular dementia. However, the role of alcohol-induced plasma Hcy levels and how this mediates metabolic and epigenetic changes are poorly explored. The authors believe that alcohol induces HHcy and that mediates metabolic and epigenetic changes. The future exploration of these mechanisms appears to be promising. Extensive studies are needed to explore the metabolic and epigenetic changes during alcohol mediated HHcy and the therapeutic mechanisms that could improve cerebrovascular pathology (Figure. 4). Much epigenetic therapeutics have been developed for other diseases, and understanding the functional relationships between epigenetic processes and the transcriptome in the alcoholic brain may lead to new molecular targets for developing medications for solving the anomalies related to alcohol consumption and alcoholism.
Figure 4. Homocysteine, alcohol and its epigenetic mechanism.
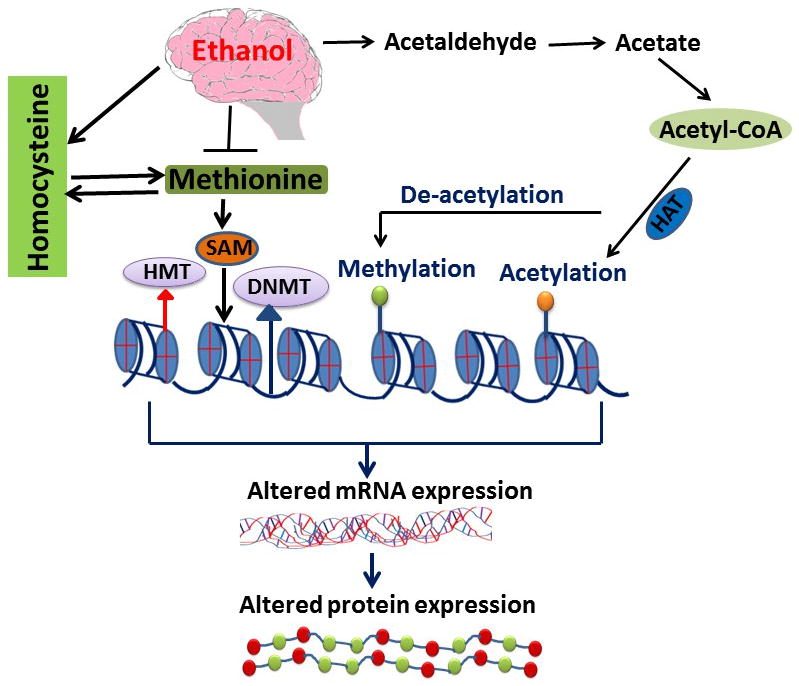
This diagram shows the role of ethanol by products such as acetaldehyde, acetate, and Acetyl-CoA that affect the acetylation and deacetylation of the gene. Deacetylation promotes the methylation of genes, which causes decreased expression of the genes. Alternatively as per the authors’ speculation, ethanol inhibits methionine formation by inhibiting methionine synthase (MS) gene expression. Increased ethanol consumption affects MS, which affects re-methylation process by inhibiting formation of methionine from homocysteine, excess homocysteine accumulation later on resulted in an altered epigenetic process.
Acknowledgments
Financial support from National Institute of health grant HL-107640 and AR-067667 are greatly acknowledged.
Abbreviations
- Hcy
Homocysteine
- tHcy
Total Homocysteine
- CBS
Cystathionine β-synthase
- BHMT
Betaine methyltransferase
- MTHFR
Methylene tetrahydrofolate reductase
- SAH
S-adenosyl homocysteine
- SAM
S-adenosyl methionine
- AD
Alzheimer’s disease
- VD
vascular dementia
- PD
Parkinson’s disease
- GABA
Gamma-aminobutyric acid
- DNMTs
DNA methyltransferases
- MS
Methionine synthase
- CSE
Cystathionine-γ-lyase
Footnotes
Conflict of interest: The authors have declared no conflict of interest.
References
- Ayaori M, Hisada T, Yoshida H, Shige H, Ito T, Nakajima K, Higashi K, Yonemura A, Ishikawa T, Ohsuzu F, Saionji K, Tamai S, Nakamura H. Effect of alcohol intake on the levels of plasma homocysteine in healthy males. J Nutr Sci Vitaminol. 2000;46:171–174. doi: 10.3177/jnsv.46.171. [DOI] [PubMed] [Google Scholar]
- Agarwal DP, Seitz HK. Alcohol in Health and Disease. Marcel Dekker; New York: 2001. [Google Scholar]
- Bagheri F, Goudarzi I, Lashkarbolouki T, Salmani ME. Melatonin prevents oxidative damage induced by maternal ethanol administration and reduces homocysteine in the cerebellum of rat pups. Behav Brain Res. 2015;287:215–25. doi: 10.1016/j.bbr.2015.03.022. [DOI] [PubMed] [Google Scholar]
- Benjamin F, Patricio AC, Gustavo MC, Luis GA. Potentiation of Gamma Aminobutyric Acid Receptors (GABAAR) by Ethanol: How Are Inhibitory Receptors Affected? Front Cell Neurosci. 2016;10:114. doi: 10.3389/fncel.2016.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor TH. The DNA methyltransferases of mammals. Hum mol genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Biermann T, Reulbach U, Lenz B, Frieling H, Muschler M, Hillemacher T, Kornhuber J, Bleich S. N-methyl-D-aspartate 2b receptor subtype (NR2B) promoter methylation in patients during alcohol withdrawal. J Neural Transm. 2009;116:615–622. doi: 10.1007/s00702-009-0212-2. [DOI] [PubMed] [Google Scholar]
- Bleich S, Bleich K, Kropp S, Bittermann HJ, Degner D, Sperling W, Ruther E, Kornhuber J. Moderate alcohol consumption in social drinkers raises plasma homocysteine levels: a contradiction to the ‘French Paradox’? Alcohol Alcohol. 2001;36:189–192. doi: 10.1093/alcalc/36.3.189. [DOI] [PubMed] [Google Scholar]
- Bleich S, Degner D, Sperling W, Bonsch D, Thurauf N, Kornhuber J. Homocysteine as a neurotoxin in chronic alcoholism. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:453–464. doi: 10.1016/j.pnpbp.2003.11.019. [DOI] [PubMed] [Google Scholar]
- Bleich S, Hillemacher T. Homocysteine, alcohol exposure and its molecular networks. Pharmacopsychiatry. 2009;42(Suppl 1):102–109. doi: 10.1055/s-0029-1214396. [DOI] [PubMed] [Google Scholar]
- Bleich S, Lenz B, Ziegenbein M, Beutler S, Frieling H, Kornhuber J, Bonsch D. Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcohol Clin Exp Res. 2006;30:587–591. doi: 10.1111/j.1530-0277.2006.00068.x. [DOI] [PubMed] [Google Scholar]
- Bonsch D, Greifenberg V, Bayerlein K, Biermann T, Reulbach U, Hillemacher T, Kornhuber J, Bleich S. Alpha-synuclein protein levels are increased in alcoholic patients and are linked to craving. Alcohol Clin Exp Res. 2005;29:763–765. doi: 10.1097/01.alc.0000164360.43907.24. [DOI] [PubMed] [Google Scholar]
- Bonsch D, Reulbach U, Bayerlein K, Hillemacher T, Kornhuber J, Bleich S. Elevated alpha synuclein mRNA levels are associated with craving in patients with alcohol exposure. Biol psychiat. 2004;56:984–986. doi: 10.1016/j.biopsych.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Bousiges O, Vasconcelos AP, Neidl R, Cosquer B, Herbeaux K, Panteleeva I, Loeffler JP, Cassel JC, Boutillier AL. Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus. Neuropsychopharmacology. 2010;35:2521–2537. doi: 10.1038/npp.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brattstrom L, Wilcken DE. Homocysteine and cardiovascular disease: cause or effect? Am J Clin Nutr. 2000;72:315–323. doi: 10.1093/ajcn/72.2.315. [DOI] [PubMed] [Google Scholar]
- Burger M, Mensink G, Bstrup A, Thierfelder W, Pietrzik K. Alcohol consumption and its relation to cardiovascular risk factors in Germany. Eur J Clin Nutr. 2004;58:605–614. doi: 10.1038/sj.ejcn.1601854. [DOI] [PubMed] [Google Scholar]
- Clarke R. Lowering blood homocysteine with folic acid-based supplements: meta-analysis of randomised trials. Indian Heart J. 2000;52:S59–64. [PubMed] [Google Scholar]
- Cravo ML, Gloria LM, Selhub J, Nadeau MR, Camilo ME, Resende MP, Cardoso JN, Leitao CN, Mira FC. Hyperhomocysteinemia in chronic alcohol exposure: correlation with folate, vitamin B-12, and vitamin B-6 status. Am J Clin Nutr. 1996;63:220–224. doi: 10.1093/ajcn/63.2.220. [DOI] [PubMed] [Google Scholar]
- D’Addario C, Caputi FF, Ekstrom TJ, Di Benedetto M, Maccarrone M, Romualdi P, Candeletti S. Ethanol induces epigenetic modulation of prodynorphin and pronociceptin gene expression in the rat amygdala complex. J mol Neurosci. 2013;49:312–319. doi: 10.1007/s12031-012-9829-y. [DOI] [PubMed] [Google Scholar]
- Dayal S, Arning E, Bottiglieri T, Boger RH, Sigmund CD, Faraci FM, Lentz SR. Cerebral vascular dysfunction mediated by superoxide in hyperhomocysteinemic mice. Stroke. 2004;35:1957–1962. doi: 10.1161/01.STR.0000131749.81508.18. [DOI] [PubMed] [Google Scholar]
- Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53(5):1752–63. doi: 10.1002/hep.24279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bree A, Verschuren WM, Blom HJ, Kromhout D. Lifestyle factors and plasma homocysteine concentrations in a general population sample. Am J Epidemiol. 2001;154:150–154. doi: 10.1093/aje/154.2.150. [DOI] [PubMed] [Google Scholar]
- Devlin AM, Arning E, Bottiglieri T, Faraci FM, Rozen R, Lentz SR. Effect of Mthfr genotype on diet-induced hyperhomocysteinemia and vascular function in mice. Blood. 2004;103:2624–2629. doi: 10.1182/blood-2003-09-3078. [DOI] [PubMed] [Google Scholar]
- Davies M. The role of GABAA receptors in mediating the effects of alcohol in the central nervous system. J Psychiatry Neurosci. 2003;28:263–274. [PMC free article] [PubMed] [Google Scholar]
- Dolin S, Little H, Hudspith M, Pagonis C, Littleton J. Increased dihydropyridine-sensitive calcium channels in rat brain may underlie ethanol physical dependence. Neuropharmacology. 1987;26:275–279. doi: 10.1016/0028-3908(87)90220-6. [DOI] [PubMed] [Google Scholar]
- Ehrlich D, Humpel C. Chronic vascular risk factors (cholesterol, homocysteine, ethanol) impair spatial memory, decline cholinergic neurons and induce blood-brain barrier leakage in rats in vivo. J Neurol Sci. 2012;322:92–95. doi: 10.1016/j.jns.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren E, Ellidag HY, Aydin O, Yilmaz N. Homocysteine, Paraoxonase-1 and Vascular Endothelial Dysfunction: Omnibus viis Romam Pervenitur. J Clin Diagn Res. 2014;8:CE01–04. doi: 10.7860/JCDR/2014/7827.4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci FM, Lentz SR. Hyperhomocysteinemia, oxidative stress, and cerebral vascular dysfunction. Stroke. 2004;35:345–347. doi: 10.1161/01.STR.0000115161.10646.67. [DOI] [PubMed] [Google Scholar]
- Feng C, Bai X, Xu Y, Hua T, Huang J, Liu XY. Hyperhomocysteinemia associates with small vessel disease more closely than large vessel disease. Int J Med Sci. 2013;10:408–12. doi: 10.7150/ijms.5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando V. Alcohol and Neurotransmitter Interactions Alcohol Health and Res World. 1997;21:44–148. [PMC free article] [PubMed] [Google Scholar]
- Gavin DP, Chase KA, Sharma RP. Active DNA demethylation in post-mitotic neurons: a reason for optimism. Neuropharmacology. 2013;75:233–245. doi: 10.1016/j.neuropharm.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AK, Anju TR, Paulose CS. Enhanced 5-HT(2A) receptors in brain stem and ALDH activity in brain stem and liver: 5-HT(2A) regulation on ALDH in primary hepatocytes cultures in vitro. Neurochem Res. 2009;34:1535–41. doi: 10.1007/s11064-009-9940-9. [DOI] [PubMed] [Google Scholar]
- George AK, Balarama Kaimal S, Paulose CS. Decreased dopamine D(2) receptor function in cerebral cortex and brain stem: their role in hepatic ALDH regulation in ethanol treated rats. Mol Cell Biochem 2007. 2007;304:181–8. doi: 10.1007/s11010-007-9498-2. [DOI] [PubMed] [Google Scholar]
- George AK, Paul J, Kaimal SB, Paulose CS. Decreased cerebral cortex and liver 5-HT2A receptor gene expression and enhanced ALDH activity in ethanol-treated rats and hepatocyte cultures. Neurol Res. 2010;325:10–18. doi: 10.1179/174313209X385554. [DOI] [PubMed] [Google Scholar]
- Giles WH, Kittner SJ, Croft JB, Wozniak MA, Wityk RJ, Stern BJ, Sloan MA, Price TR, McCarter RJ, Macko RF, Johnson CJ, Feeser BR, Earley CJ, Buchholz DW, Stolley PD. Distribution and correlates of elevated total homocyst(e)ine: the Stroke Prevention in Young Women Study. Ann Epidemiol. 1999;9:307–313. doi: 10.1016/s1047-2797(99)00006-x. [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Gulya K, Grant KA, Valverius P, Hoffman PL, Tabakoff B. Brain regional specificity and time-course of changes in the NMDA receptor-ionophore complex during ethanol withdrawal. Brain res. 1991;547:129–134. [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavo S. The Role of Vitamin B in Stroke Prevention. Emerging Therapy Critiques. 2011;42:838–842. [Google Scholar]
- a.Halsted CH, Villanueva JA, Devlin AM. Folate deficiency, methionine metabolism, and alcoholic liver disease. Alcohol. 2002;27:169–172. doi: 10.1016/s0741-8329(02)00225-2. [DOI] [PubMed] [Google Scholar]
- b.Halsted CH, Villanueva JA, Devlin AM, Chandler CJ. Metabolic interactions of alcohol and folate. J Nutr. 2002;132:2367S–2372S. doi: 10.1093/jn/132.8.2367S. [DOI] [PubMed] [Google Scholar]
- Hankey GJ, Eikelboom JW. Homocysteine and vascular disease. Lancet. 1999;354:407–413. doi: 10.1016/S0140-6736(98)11058-9. [DOI] [PubMed] [Google Scholar]
- a.Hillbom M. Oxidants, antioxidants, alcohol and stroke. Front Biosci. 1999;4:67–71. doi: 10.2741/A481. [DOI] [PubMed] [Google Scholar]
- b.Hillbom M, Juvela S, Numminen H. Alcohol intake and the risk of stroke. J Cardiovasc Risk. 1999;6:223–228. doi: 10.1177/204748739900600406. [DOI] [PubMed] [Google Scholar]
- c.Hillbom M, Numminen H, Juvela S. Recent heavy drinking of alcohol and embolic stroke. Stroke. 1999;30:2307–2312. doi: 10.1161/01.str.30.11.2307. [DOI] [PubMed] [Google Scholar]
- Hillemacher T, Weinland C, Heberlein A, Groschl M, Schanze A, Frieling H, Wilhelm J, Kornhuber J, Bleich S. Increased levels of adiponectin and resistin in alcohol dependence--possible link to craving. Drug Alcohol Depend. 2009;99:333–337. doi: 10.1016/j.drugalcdep.2008.07.019. [DOI] [PubMed] [Google Scholar]
- Hoffman PL. NMDA receptors in alcohol exposure. Int rev neurobiol. 2003;56:35–82. doi: 10.1016/s0074-7742(03)56002-0. [DOI] [PubMed] [Google Scholar]
- Hultberg B, Berglund M, Andersson A, Frank A. Elevated plasma homocysteine in alcoholics. Alcohol Clin Exp Res. 1993;17:687–689. doi: 10.1111/j.1530-0277.1993.tb00820.x. [DOI] [PubMed] [Google Scholar]
- Jacques PF, Bostom AG, Wilson PW, Rich S, Rosenberg IH, Selhub J. Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. Am J Clin Nutr. 2001;73:613–621. doi: 10.1093/ajcn/73.3.613. [DOI] [PubMed] [Google Scholar]
- James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr. 2004;80:1611–1617. doi: 10.1093/ajcn/80.6.1611. [DOI] [PubMed] [Google Scholar]
- Ji C, Deng Q, Kaplowitz N. Role of TNF-alpha in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology. 2004;40:442–51. doi: 10.1002/hep.20309. [DOI] [PubMed] [Google Scholar]
- Jiang R, Hu FB, Giovannucci EL, Rimm EB, Stampfer MJ, Spiegelman D, Rosner BA, Willett WC. Joint association of alcohol and folate intake with risk of major chronic disease in women. Am J Epidemiol. 2003;158:760–771. doi: 10.1093/aje/kwg221. [DOI] [PubMed] [Google Scholar]
- Joseph J, Handy DE, Loscalzo J. Quo vadis: whither homocysteine research? Cardiovasc Toxicol. 2009;9:53–63. doi: 10.1007/s12012-009-9042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalani A, Kamat PK, Givvimani S, Brown K, Metreveli N, Tyagi SC, Tyagi N. Nutri-epigenetics ameliorates blood-brain barrier damage and neurodegeneration in hyperhomocysteinemia: role of folic acid. J mol neurosci. 2014;52:202–215. doi: 10.1007/s12031-013-0122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat PK, Kalani A, Givvimani S, Sathnur PB, Tyagi SC, Tyagi N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience. 2013;252:302–319. doi: 10.1016/j.neuroscience.2013.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DO. B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review. Nutrients. 2016;8:68. doi: 10.3390/nu8020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends biochem sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Koehler KM, Baumgartner RN, Garry PJ, Allen RH, Stabler SP, Rimm EB. Association of folate intake and serum homocysteine in elderly persons according to vitamin supplementation and alcohol use. Am J clin nutr. 2001;73:628–637. doi: 10.1093/ajcn/73.3.628. [DOI] [PubMed] [Google Scholar]
- Lai CL, Liu MT, Yin SJ, Lee JT, Lu CC, Peng GS. Heavy binge drinking may increase risk of stroke in nonalcoholic hypertensives carrying variant ALDH2*2 gene allele. Acta Neurol Taiwan. 2012;21:39–43. [PubMed] [Google Scholar]
- a.Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. The J biol chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- b.Levenson JM, Sweatt JD. Epigenetic mechanisms: a common theme in vertebrate and invertebrate memory formation. Cell Mol Life Sci. 2006;63:1009–1016. doi: 10.1007/s00018-006-6026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lominadze D, Roberts AM, Tyagi N, Moshal KS, Tyagi SC. Homocysteine causes cerebrovascular leakage in mice. Am J Physiol Heart Circ Physiol. 2006;290:1206–1213. doi: 10.1152/ajpheart.00376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopamudra R, Vineet KK, Prajna B, Kausik B, Sandip P, Keya P, Debasis B, Sasanka C. Serum Homocysteine, Dehydroepiandrosterone Sulphate and Lipoprotein (a) in Alzheimer’s Disease and Vascular Dementia. Aging Dis. 4:57–64. [PMC free article] [PubMed] [Google Scholar]
- Lutz UC. Alterations in homocysteine metabolism among alcohol dependent patients--clinical, pathobiochemical and genetic aspects. Curr Drug Abuse Rev. 2008;1:47–55. doi: 10.2174/1874473710801010047. [DOI] [PubMed] [Google Scholar]
- Maze I, Covington HE, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaddon A, Hudson P, Davies G, Hughes A, Williams JH, Wilkinson C. Homocysteine and cognitive decline in healthy elderly. Dement Geriatr Cogn Disord. 2001;12:309–313. doi: 10.1159/000051275. [DOI] [PubMed] [Google Scholar]
- McClintick JN, McBride WJ, Bell RL, Ding ZM, Liu Y, Xuei X, Edenberg HJ. Gene Expression Changes in Glutamate and GABA-A Receptors, Neuropeptides, Ion Channels, and Cholesterol Synthesis in the Periaqueductal Gray Following Binge-Like Alcohol Drinking by Adolescent Alcohol-Preferring (P) Rats. Alcohol Clin Exp Res. 2016;40:955–68. doi: 10.1111/acer.13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Campbell SL, Sweatt JD. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem. 2008;89:599–603. doi: 10.1016/j.nlm.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi R, Metreveli N, Tyagi SC, Lominadze D. Ablation of MMP9 gene ameliorates paracellular permeability and fibrinogen-amyloid beta complex formation during hyperhomocysteinemia. J Cereb Blood Flow Metab. 2014;34:1472–1482. doi: 10.1038/jcbfm.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk E, Refsum H, Tell GS, Engedal K, Vollset SE, Ueland PM, Nygaard HA, Smith AD. Plasma total homocysteine and memory in the elderly: the Hordaland Homocysteine Study. Ann Neurol. 2005;58:847–57. doi: 10.1002/ana.20645. [DOI] [PubMed] [Google Scholar]
- Nieratschker V, Batra A, Fallgatter AJ. Genetics and epigenetics of alcohol dependence. J Mol Psychiarr. 2013;1:11. doi: 10.1186/2049-9256-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okura T, Miyoshi K, Irita J, Enomoto D, Nagao T, Kukida M, Tanino A, Kudo K, Pei Z, Higaki J. Hyperhomocysteinemia is one of the risk factors associated with cerebrovascular stiffness in hypertensive patients, especially elderly males. Sci Rep. 2014;4:56–63. doi: 10.1038/srep05663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovechkin AV, Tyagi N, Sen U, Lominadze D, Steed MM, Moshal KS, Tyagi SC. 3-Deaza adenosine mitigates arterial remodeling and hypertension in hyperhomocysteinemic mice. Am J Physiol Lung Cell Mol Physiol. 2006;291:905–911. doi: 10.1152/ajplung.00543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid Omar, Kim Jeffrey J, Kim Hyun-Sung, Hoang Michael, Tu Thanh G, Elie Omid, Lee Connie, Vu Catherine, Horvath Steve, Spigelman Igor, Kim Yong. Gene expression signatures affected by alcohol-induced DNA methylomic deregulation in human embryonic stem cells. Stem Cell Research. 2012 May;12(3):791–806. doi: 10.1016/j.scr.2014.03.009. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PH, Lim RW, Shukla SD. Gene-selective histone H3 acetylation in the absence of increase in global histone acetylation in liver of rats chronically fed alcohol. Alcohol Alcohol. 2012;47:233–239. doi: 10.1093/alcalc/ags004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna AF, Ingrosso D, De Santo NG. Homocysteine and oxidative stress. Amino Acids. 2003;25:409–417. doi: 10.1007/s00726-003-0026-8. [DOI] [PubMed] [Google Scholar]
- Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, Brunetti N, Porcellini E, Licastro F. Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr. 2005;82:636–643. doi: 10.1093/ajcn.82.3.636. [DOI] [PubMed] [Google Scholar]
- Ray L, Khemka VK, Behera P, Bandyopadhyay K, Pal S, Pal K, Basu D, Chakrabarti S. Serum Homocysteine, Dehydroepiandrosterone Sulphate and Lipoprotein (a) in Alzheimer’s Disease and Vascular Dementia. Aging Dis. 2013;4:57–64. [PMC free article] [PubMed] [Google Scholar]
- Ron D. Signaling Cascades Regulating NMDA Receptor Sensitivity to Ethanol. Neuroscientist. 2004;10:325–336. doi: 10.1177/1073858404263516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri P, Fragiacomo C, Pezzati R, Zanda E, Forloni G, Tettamanti M, Lucca U. Homocysteine, folate, and vitamin B-12 in mild cognitive impairment, Alzheimer disease, and vascular dementia. The Am J Clin Nutr. 2004;80:114–122. doi: 10.1093/ajcn/80.1.114. [DOI] [PubMed] [Google Scholar]
- Sachdev PS. Homocysteine and brain atrophy. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1152–1161. doi: 10.1016/j.pnpbp.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Schaefer A, Sampath SC, Intrator A, Min A, Gertler TS, Surmeier DJ, Tarakhovsky A, Greengard P. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron. 2009;64:678–691. doi: 10.1016/j.neuron.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selhub J. Homocysteine metabolism. Annu Rev Nutr. 1999;19:217–246. doi: 10.1146/annurev.nutr.19.1.217. [DOI] [PubMed] [Google Scholar]
- Selhub J, Jacques PF, Rosenberg IH, Rogers G, Bowman BA, Gunter EW, Wright JD, Johnson CL. Serum total homocysteine concentrations in the third National Health and Nutrition Examination Survey (1991–1994): population reference ranges and contribution of vitamin status to high serum concentrations. Ann Intern Med. 1999;131:331–339. doi: 10.7326/0003-4819-131-5-199909070-00003. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SD, Restrepo R, Fish P, Lim RW, Ibdah JA. Different mechanisms for histone acetylation by ethanol and its metabolite acetate in rat primary hepatocytes. J Pharmacol Exp Ther. 2015;354:18–23. doi: 10.1124/jpet.115.223867. [DOI] [PubMed] [Google Scholar]
- Spencer KB, Mulholland PJ, Chandler LJ. FMRP Mediates Chronic Ethanol-Induced Changes in NMDA, Kv4.2, and KChIP3 Expression in the Hippocampus. Alcohol Clin Exp Res. 2016;40:1251–61. doi: 10.1111/acer.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickel F, Choi SW, Kim Y, Bagley PJ, Seitz HK, Russell RM, Selhub J, Mason JB. Effect of Chronic Alcohol Consumption on Total Plasma Homocysteine Level in Rats. Alcohol Clin Exp Res. 2000;24:259–264. [PubMed] [Google Scholar]
- Tchantchou F, Graves M, Ortiz D, Rogers E, Shea TB. Dietary supplementation with 3-deaza adenosine, N-acetyl cysteine, and S-adenosyl methionine provide neuroprotection against multiple consequences of vitamin deficiency and oxidative challenge: relevance to age-related neurodegeneration. Neuromolecular Med. 2004;6:93–103. doi: 10.1385/NMM:6:2-3:093. [DOI] [PubMed] [Google Scholar]
- a.Tyagi N, Lominadze D, Gillespie W, Moshal KS, Sen U, Rosenberger DS, Steed M, Tyagi SC. Differential expression of gamma-aminobutyric acid receptor A (GABA(A)) and effects of homocysteine. Clin Chem Lab Med. 2007;45:1777–1784. doi: 10.1515/CCLM.2007.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- b.Tyagi N, Moshal KS, Tyagi SC, Lominadze D. Gamma-Aminbuturic acid A receptor mitigates homocysteine-induced endothelial cell permeability. Endothelium. 2007;14:315–323. doi: 10.1080/10623320701746164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC. Mechanisms of homocysteine-induced oxidative stress. Am J Physiol Heart Circ Physiol. 2005;289:H2649–2656. doi: 10.1152/ajpheart.00548.2005. [DOI] [PubMed] [Google Scholar]
- Weiss N, Heydrick SJ, Postea O, Keller C, Keaney JF, Jr, Loscalzo J. Influence of hyperhomocysteinemia on the cellular redox state–impact on homocysteine-induced endothelial dysfunction. Clin Chem Lab Med. 2003;41:1455–1461. doi: 10.1515/CCLM.2003.223. [DOI] [PubMed] [Google Scholar]
- Worrall S, Thiele GM. Protein modification in ethanol toxicity. Adverse Drug React Toxicol Rev. 2001;20:133–159. [PubMed] [Google Scholar]
- Whittington MA, Lambert JD, Little HJ. Increased NMDA receptor and calcium channel activity underlying ethanol withdrawal hyperexcitability. Alcohol Alcohol. 1995;30:105–114. [PubMed] [Google Scholar]
- Wilhelm J, Bayerlein K, Hillemacher T, Reulbach U, Frieling H, Kromolan B, Degner D, Kornhuber J, Bleich S. Short-term cognition deficits during early alcohol withdrawal are associated with elevated plasma homocysteine levels in patients with alcohol exposure. J Neural Transm. 2006;113:357–363. doi: 10.1007/s00702-005-0333-1. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Kim KY, Gelernter J, Park IH, Zhang H. Ethanol up regulates NMDA receptor subunit gene expression in human embryonic stem cell-derived cortical neurons. PLoS One. 2015;12:10. doi: 10.1371/journal.pone.0134907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou CG, Banerjee R. Homocysteine and redox signaling Antioxid. Redox Signal. 2005;7:547–559. doi: 10.1089/ars.2005.7.547. [DOI] [PubMed] [Google Scholar]