

Abstract
Corticotropin-releasing factor (CRF) and the CRF-related peptides, urocortin (Ucn)-1, Ucn2, and Ucn3 signal through receptors CRFR1 and CRFR2 to restore homeostasis in response to stress. The Ucns exert potent cardioprotective effects and may have clinical utility in heart failure. To explore the activity of this system in the heart, we measured the levels of myocardial gene expression of the CRF/Ucn family of ligands/receptors and investigated genetic variation and alternative splicing of CRFR1 in 110 heart failure patients and 108 heart donors. Using quantitative real-time PCR, we detected CRFR1, CRFR2, CRF, Ucn1, Ucn2, and Ucn3 in all samples. CRFR2α was the most abundant receptor and Ucn3 the most abundant ligand, both in patients and donors. Compared with donors, cardiac expression of CRFR1, CRF, and Ucn3 was higher (P < .001) and CRFR2α lower (P = .012) in patients. In patients and donors, genetic variation within CRFR1, represented by the chromosome 17q21.31 inversion polymorphism, was associated with markedly higher CRFR1 expression (P < .001), making CRFR1 and CRFR2α expression almost equivalent in some patients. A novel, truncated splice variant of CRFR1, designated CRFR1j, was identified and shown to exert a dominant-negative effect on CRFR1 signaling in vitro. The novel variant was expressed in a greater proportion of patients (60%) than donors (3%, P < .001). In summary, cardiac expression of CRFR1, CRF, and Ucn3 genes is elevated in heart failure and may contribute to the activation of the CRF/Ucn system in these patients. A common variant within the CRFR1 gene and a novel CRFR1 splice variant may modulate CRFR1 expression and signaling.
The corticotropin releasing factor (CRF) family of ligands and receptors are key regulators of stress and metabolism and exert a wide range of systemic and local effects through endocrine, paracrine, and autocrine signaling (1). The CRF-related peptides, urocortin (Ucn)-1, Ucn2, and Ucn3 have emerged as an important group of cardiac regulators that have considerable therapeutic potential in the treatment of heart failure and ischemic heart disease (2). The Ucns exert multiple beneficial effects on the heart and vasculature including increased cardiac contractility and cardiac output; improved hemodynamic, neurohormonal, and renal function; and protection from ischemia/reperfusion injury (3). In patients with heart failure, plasma levels of Ucn1 are elevated and correlate with markers of disease severity and cardiac dysfunction (4). Antagonism of endogenous Ucns in experimental heart failure promotes adverse hemodynamic, neurohormone, and renal effects (5), suggesting that the Ucns form part of the beneficial compensatory response to this condition and may have prognostic utility in heart failure patients.
The actions of CRF and the Ucns are mediated via two families of class B1 G protein-coupled receptors, CRF receptor (CRFR)-1 and CRFR2, which exist in numerous alternatively spliced forms and exhibit distinct patterns of expression in the central nervous system and periphery that reflect their diverse physiological functions (1). CRF has high affinity for CRFR1 but not CRFR2 (6); Ucn1 has high affinity for both receptors (6); Ucn2 and Ucn3 bind exclusively to CRFR2 (7, 8). The CRFR2 receptor is abundantly expressed within the left ventricle (LV) and intramyocardial vasculature of the heart (predominantly CRFR2α in humans, CRFR2β in rodents) at levels that are approximately equivalent to other well-characterized cardiovascular receptors such as the angiotensin II type 1 receptor (9, 10). The genes encoding the Ucns are also expressed at high levels, particularly Ucn1 and Ucn3 (10–12), and Ucn1-like immunoreactivity in the LV is elevated in heart failure patients (13, 14). In contrast, the expression of CRFR1 and CRF is thought to be weak or absent in the human heart (10). However, a comprehensive analysis of the myocardial gene expression profile of all CRF family members in heart failure patients and controls has not been performed to date.
To contribute to our understanding of how the CRF family of ligands and receptors may influence heart function and pathology, we examined the relationship between levels of gene expression of the CRF family members in myocardial tissue from heart failure patients and from heart donors with no previously diagnosed heart disease and investigated genetic variation and alternative splicing as potential regulatory mechanisms of this system in the heart.
Materials and Methods
Collection of human heart tissue samples
Heart tissue from the midsection of the LV free wall of the myocardium of organ donors (n = 108) and heart failure patients (n = 110) was collected by the Cleveland Clinic Kaufman Center for Heart Failure human heart tissue bank between 1993 and 2006, as previously described (15). All donor hearts were originally donated for transplantation. Reasons for rejection for cardiac transplantation included histocompatibility mismatch, structural damage to the heart, subclinical cardiac disease, or other elements of the medical history that made the donor undesirable. The decision that the heart could not be used for transplantation was made by members of a clinical organ procurement team. Explanted and donor hearts donated for research were rapidly transported to the laboratory in cold cardioplegic solution, separated by heart chamber, frozen in liquid nitrogen, and stored at −80°C. We have repeatedly verified the isolation of high-quality RNA from these specimens. The study was approved by the Cleveland Clinic Internal Review Board (Internal Review Board protocol 2378) and adhered to the principles outlined in the Declaration of Helsinki. All procedures followed were in accordance with institutional guidelines and all families and/or patients provided informed consent.
Quantitative real-time PCR
Total RNA was extracted from frozen tissue as previously described (16). RNA concentration and purity were determined using a Nanodrop spectrophotometer (Nanodrop Technologies). The mean RNA concentration and 260:280 ratio (±SD) in the heart donors was 768 ± 282 ng/L and 2.06 ± 0.03, and in heart failure patients was 902 ± 426 ng/L and 2.04 ± 0.03. Although the donor tissues were stored longer than the heart failure tissues prior to the RNA extraction (donor samples 6.1 ± 3.2 y, heart failure patients 1.9 ± 1.0 y, P < .001), the assessment of the integrity of the RNA by gel electrophoresis confirmed intact nondegraded RNA in all samples.
Quantitative real-time PCR was performed in duplicate for CRFR1, CRFR2α, CRFR2β, CRF, Ucn1, Ucn2, and Ucn3 on a Lightcycler 480 real-time PCR system (Roche Diagnostics). All primer sets except for CRF were designed across exon/exon boundaries, and all assays included minus reverse transcriptase controls for greater than 10% of the samples to ensure there was no amplification of double-stranded DNA transcripts. For all assays, the samples were quantified using a standard curve consisting of five serial dilution points of purified DNA template in triplicate and a no-template control (absolute quantitation). Primer sequences and reaction efficiencies are provided in Supplemental Table 1 and reaction conditions are provided in the Supplemental Methods.
Genotyping chromosome 17q21.31 inversion polymorphism
Genomic DNA was extracted from frozen tissue as previously described (16). Heart donors and heart failure patients were genotyped for a 900-kb inversion polymorphism on chromosome 17q21.31 (chr17q21.31) by amplifying a 238-bp insertion within the 17q21.31 locus that is representative of the H1 haplotype, as published previously (17). This polymorphism segregates with much of the genetic variation within CRFR1 and was used as a marker for genetic variation at this locus. Amplimers were sequenced on a 3100-Avant genetic analyzer (Applied Biosystems) to confirm specific amplification. Genotypes were validated by regenotyping 38% of the samples selected at random, which gave 100% concordance with original genotypes. Hardy-Weinberg statistics were calculated using the online calculator www.oege.org/software/hwe-mr-calc.shtml (18).
Semiquantitative RT-PCR
Nested primer sets were used to identify alternatively spliced transcripts of CRFR1 as previously described (19). Alternatively spliced forms of CRFR1 were characterized by sequencing amplimers of different sizes. Nested primer sets specific for the previously unreported, novel CRFR1 splice variant identified in this study were designed using Primer3 (20). The novel variant was recorded as present in samples in which the predicted 121-bp band was detected in both replicates. For samples in which amplification was detected in one replicate (17%), the samples were retested in triplicate and the novel variant recorded as present if amplification was detected in at least two replicates. Primer sequences are provided in Supplemental Table 2, and reaction conditions for nested PCR assays are provided in the Supplemental Methods.
Plasmid constructs
An expression plasmid encoding the novel CRFR1 splice variant, CRFR1j, was subcloned into the pcDNA3.1+ (Invitrogen) expression vector using nested PCR primers that incorporated EcoR1 or Not1 restriction sites (for primer sequences and reaction conditions, see the Supplemental Methods) and transformed into One Shot TOP10 chemically competent Escherichia coli (Invitrogen) according to the manufacturer's instructions. Vector DNA was purified with plasmid minikits (QIAGEN) and sequenced on a 3100-Avant genetic analyzer (Applied Biosystems) to confirm the presence of the expected cloning insert. Expression plasmids for human CRFR1α were generated as previously described (21).
Transient transfections and luciferase reporter assays
Human embryonic kidney (HEK)-293T cells were cultured in DMEM supplemented with 10% fetal bovine serum (Sigma) and 2 mM glutamine (Invitrogen). Cells (1.8 × 106) were seeded in 10-cm tissue culture dishes in 10 mL of medium, incubated overnight, washed in Optimem (Invitrogen) without serum, and transfected for 6 hours with a mix of Lipofectamine 2000 (Invitrogen), CRE-luciferase reporter plasmid, β-galactosidase plasmid, and varying amounts of the expression plasmids encoding the novel CRFR1 splice variant, CRFR1 or empty vector diluted in Optimem (Invitrogen), as indicated. Cells were then incubated with complete media (containing serum) overnight, trypsinized, and seeded at a density of 1 × 105 cells/well in a poly-D-lysine-coated 48-well plate in 300 μL complete media. After 24 hours the cells were washed in DMEM+0.1% BSA and treated for 4 hours with varying concentrations of the CRFR1 agonist, sauvagine (0–100 nM) or vehicle, in triplicate, in 300 μL serum-free media. The cells were harvested in lysis buffer (1% Triton X-100; 25 mM glyclglycine, pH 7.8; 15 mM MgSO4; 4 mM EGTA; and 1 mM dithiothreitol). Luciferase reporter activity was measured using D-luciferin luciferase substrate on a Modulus Microplate Reader (Turner Biosystems) and normalized to β-galactosidase (measured using o-nitrophenyl-β-galactopyranoside) to control for transfection efficiency. Reported data correspond to the luciferase activity of each plasmid (or combination of plasmids) relative to the no-treatment control.
Cell surface expression assays
COSM6 cells, cultured in DMEM supplemented with 3% fetal bovine serum (Sigma), were transiently transfected as described above, with a mix of Lipofectamine 2000 (Invitrogen) and varying amounts of the expression plasmids encoding myc-tagged CRFR1, the novel CRFR1 splice variant, soluble mouse CRFR2α (22), or empty vector diluted in Optimem (Invitrogen) as indicated. After 24 hours, the cells were seeded across a poly-D-lysine-coated 24-well plate (1 × 106 cells/well, in triplicate), incubated overnight in 500 μL complete media, washed in Hepes dissociation buffer (HDB)+0.1% BSA, and incubated for 2 hours with a mouse antimyc antibody (Sigma; diluted 1:200) in 500 μL HDB+0.1% BSA. The cells were washed, incubated for 1 hour with a horseradish peroxidase-conjugated antimouse antibody (Sigma) diluted 1:1000 in 250 μL HDB+0.1% BSA, washed again, and incubated with 3′, 5, 5′-tetramethyl benzidine substrate until a blue color change was observed. The reaction was stopped with 0.18 M sulfuric acid and absorbance at 450 nm measured on a Modulus microplate reader. Reported data correspond to the level of myc-tag detected on the cell surface for each plasmid (or combination of plasmids) relative to the vector control.
Statistical analysis
Clinical characteristics and sample storage time were compared between the heart donors and patients using χ2 and ANOVA analyses. Gene expression data displayed consistently skewed distributions and were log transformed before analysis and geometric means and 95% confidence intervals have been reported. All gene expression analyses were adjusted for potential confounding factors (age, gender, ethnicity, and sample storage time). Gene expression levels of CRFR1, CRFR2α, CRF, Ucn1, Ucn2, and Ucn3 were compared between the heart donors and patients using a linear regression. The relationship between the expression levels of each gene was analyzed using Pearson correlation. Correlation matrices were plotted with R software (http://www.R-project.org) and the Corrplot package version 0.30 (23, 24). The association between the chromosome 17q21.31 inversion polymorphism and levels of CRFR1 and CRFR2α gene expression was analyzed, assuming an additive genetic model with linear regression. Associations between the presence of the predicted novel CRFR1 splice variant and clinical characteristics were performed using χ2 and ANOVA tests. Functional assays were analyzed with linear regression. All statistical analyses were performed with SPSS Statistics version 21.0 (SPSS Inc), and a value of P < .05 was taken to indicate statistical significance.
Results
Heart donor and heart failure patient characteristics
The clinical characteristics of the heart failure patients are shown in Table 1. The clinical characteristics of the heart donors were described previously (16). Compared with heart donors, heart failure patients were, on average, 7.4 years older (P < .001), and a greater proportion were male (P < .001) and of non-European ancestry (P = .003).
Table 1.
Baseline Characteristics of Heart Failure Patients and Heart Donors
| Baseline Characteristics | n | Heart Failure Patients | n | Donors |
|---|---|---|---|---|
| Age, ya | 110 | 55.3 ± 12.2 | 107 | 48.0 ± 12.5 |
| Gender (male/female) | 110 | 89/21 (81% male) | 107 | 55/52 (51% male) |
| Ethnicity | 110 | 82% European | 106 | 96% European |
| 15% African American | ||||
| 3% Hispanic | 4% African American | |||
| 38/2 (81%/4%) | ||||
| Cigarette smoking (past/present) | 47 | 27.0 ± 5.1 | ||
| Body mass index, kg/m2a | 64 | 63.9 ± 8.7 | ||
| Diastolic blood pressure, mm Hga | 78 | 97.2 ± 12.4 | ||
| Systolic blood pressure, mm Hga | 78 | 3.7 (3.0–4.5) | ||
| Total cholesterol, mmol/Lb | 87 | 1.9 (1.5–2.5) | ||
| LDL, mmol/Lb | 87 | 1.1 ± 0.4 | ||
| HDL, mmol/La | 87 | 1.2 (0.9–1.6) | ||
| Triglycerides, mmol/Lb | 87 | 5.7 (4.9–6.8) | ||
| Glucose, mmol/Lb | 94 | 174 (95–351) | ||
| Brain natriuretic peptide, pmol/Lb | 82 | 15.7 ± 5.5 | ||
| LVEF, %a | 94 | 106 (88–132) | 61 | 52% ± 15.3% |
| Creatinine, μmol/Lb | 91 | 6.1 (2.6–10.1) | ||
| Duration of heart failure, yb | 70 | 16 (17.4%) | ||
| Family history of heart disease | 92 | 47 (47.0%) | ||
| Previously diagnosed hypertension | 100 | 24 (24.0%) | ||
| Type 2 diabetes | 100 | 48 (48.0%) | ||
| Myocardial infarction | 100 | 58 (52.7%) | ||
| ACE inhibitor treatment | 110 | 61 (55.5%) | ||
| β-Blocker treatment | 110 | 69 (69.0%) | ||
| Loop diuretic treatment | 100 | 52 (52.0%) | ||
| Spironolactone treatment | 100 |
Abbreviations: ACE, angiotensin-converting enzyme; HDL, high-density lipoprotein; ; LDL, low-density lipoprotein; LVEF, LV ejection fraction.
Mean and SD.
Median and interquartile range.
Myocardial gene expression of the CRF family of ligands and receptors
Expression of CRFR1, CRFR2α, CRF, Ucn1, Ucn2, and Ucn3 was detected within the LV of the myocardium of heart donors and heart failure patients (Figure 1). In heart donors, CRFR2α was the most abundantly expressed receptor. On average, the expression levels of CRFR1 and CRFR2β were markedly lower than CRFR2α, with CRFR2β reaching detectable levels in only 39% of the samples. The most abundantly expressed ligands were Ucn3 and Ucn1. Expression levels of several family members were correlated (Figure 2A). The strongest relationships were between CRFR2α and Ucn2 and CRF and Ucn1.
Figure 1.
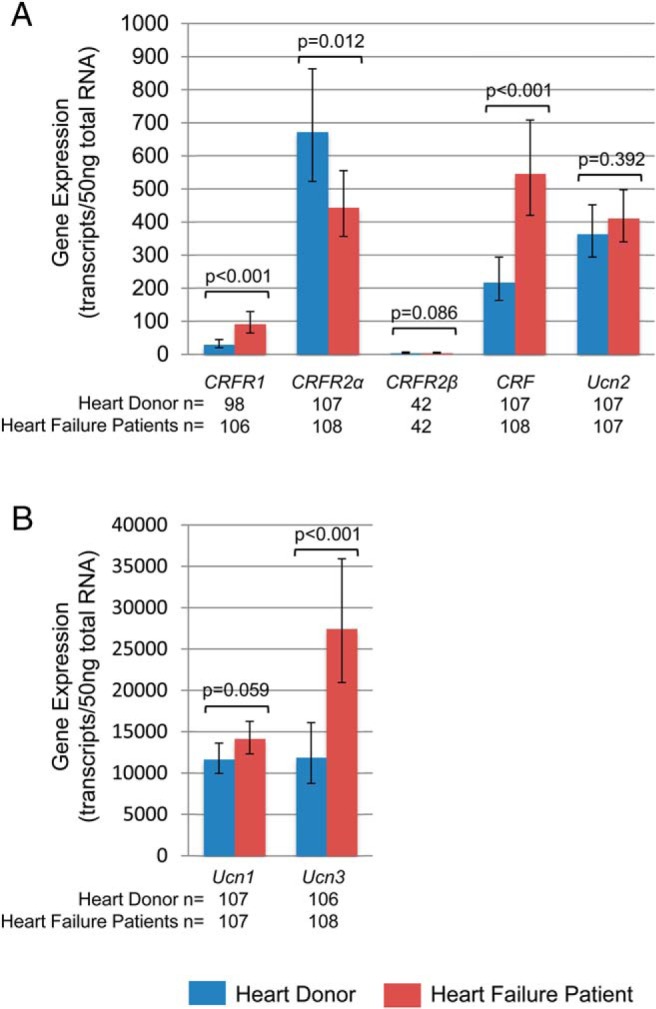
Myocardial gene expression of the CRF family of ligands and receptors in heart donors (blue) and heart failure patients (red). Expression levels of the CRF receptors, CRF and Ucn2 (A) and Ucn1 and Ucn3 (B) in the LV of the human heart adjusted for age, gender, ethnicity, and sample storage time are shown. In patients, the expression levels of CRFR1, CRF, and Ucn3 were higher and CRFR2α were lower compared with donors. Data represent geometric means and 95% confidence intervals.
Figure 2.
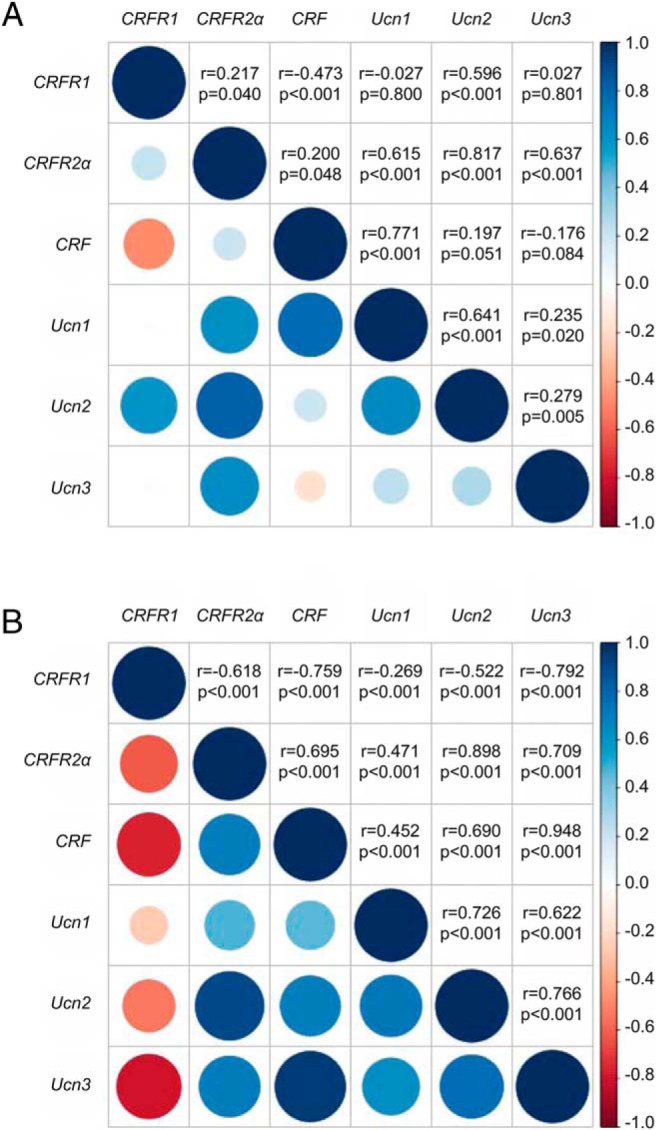
Correlation matrix of gene expression levels of the CRF family of ligands and receptors in the LV of the human heart in heart donors (A) and heart failure patients (B). Gene expression levels were adjusted for age, gender, ethnicity, and sample storage time. The upper half of each figure displays the Pearson correlation coefficients and P value for each association. The lower half of each figure gives a pictorial representation of these values: the Pearson correlation coefficient is represented by the color of each circle (positive correlations are blue, negative correlations are red), whereas the P value is indicated by the size of the circle (larger circle sizes indicate highly statistically significant relationships).
In the heart failure patients, the expression levels of CRFR1, CRF, and Ucn3 were significantly higher, and the levels of CRFR2α were significantly lower, compared with the heart donors (Figure 1). Although CRFR2α remained the most abundant receptor subtype, the ratio of CRFR2α to CRFR1 decreased from greater than 20:1 in the heart donors to approximately 4:1 in the heart failure patients. The expression levels of CRF and the Ucn genes were even more strongly correlated in the patients than in the donors (Figure 2B).
Chr17q21.31 haplotype and CRFR1 gene expression
Much of the genetic variation within the CRFR1 gene segregates with a large approximately 900-kb inversion polymorphism at chromosome 17q21.31, which occurs as two distinct haplotypes, H1 and H2. The H1/H2 haplotype frequency in the heart donors was 52% H1H1, 47% H1H2, and 1% H2H2 and in the heart failure patients was 75% H1H1, 23% H1H2, and 2% H2H2. The haplotype frequency was in Hardy-Weinberg equilibrium in patients (P = .956) but not in donors (P = .004).
In both the heart donors and heart failure patients, individuals carrying one or more copies of the rarer H2 allele had higher levels of CRFR1 compared with other individuals (heart donors: 3.0-fold, P < .001; heart failure patients: 3.8-fold, P < .001, Figure 3A). These individuals also tended to have lower levels of CRFR2α, although this was significant only in heart donors (heart donors: 1.5-fold, P = .016; heart failure patients: 1.1-fold, P = .677, Figure 3B). Consequently, the ratio of CRFR2α to CRFR1 was markedly reduced in individuals with the H2 allele, with CRFR2α and CRFR1 expression nearly equivalent in the heart failure patients carrying the H2 allele (heart donors: H1H1 vs H2 carriers, 30:1 vs 6:1, P < .001; heart failure patients: H1H1 vs H2 carriers, 6:1 vs 1.6:1, P < .001, Figure 3C).
Figure 3.
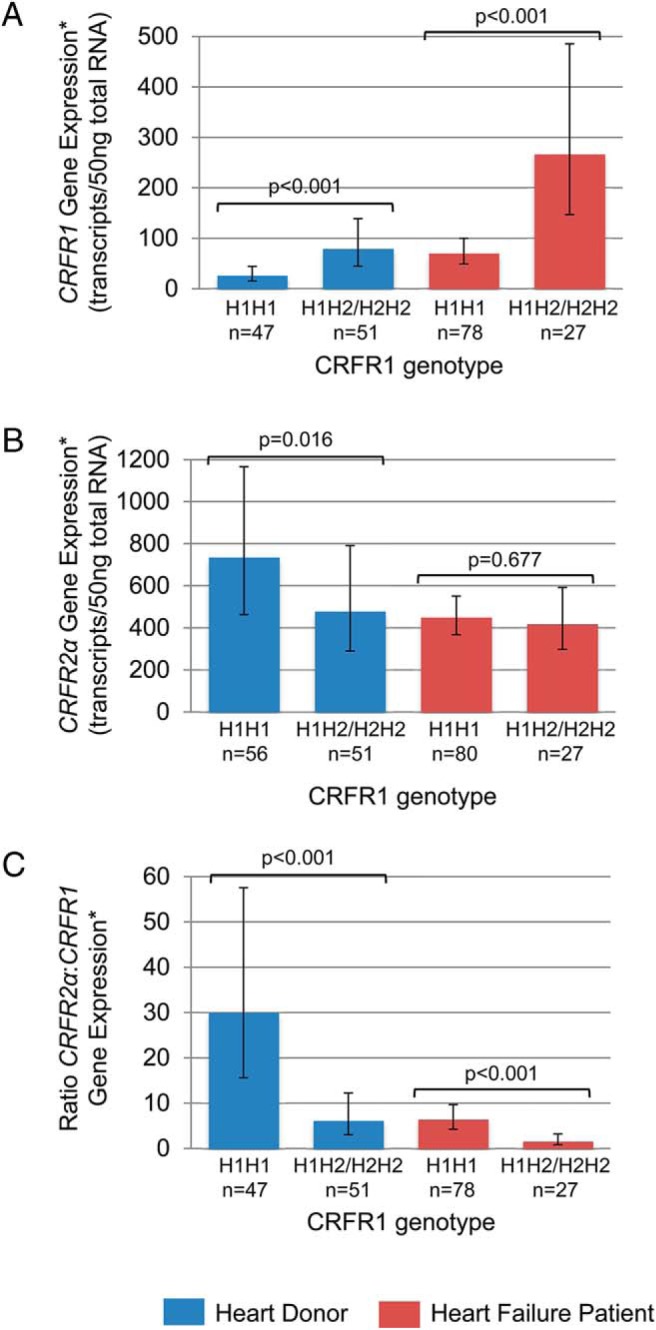
Associations between a large inversion polymorphism on chromosome 17q21.31 (which spans the CRFR1 gene) and CRFR1 and CRFR2α expression in heart donors (blue) and heart failure patients (red). A, Levels of CRFR1 expression were higher in donors and patients with the H2 haplotype. B, Levels of CRFR2α expression were lower in donors with the H2 haplotype. C, Consequently, the ratio of CRFR2α:CRFR1 expression was markedly lower in donors and patients with the H2 haplotype, making CRFR1 expression almost equivalent to CRFR2α expression in patients with the H2 haplotype. Gene expression data were adjusted for age, gender, ethnicity, and sample storage time. Data represent geometric means and 95% confidence intervals.
Characterizing a novel CRFR1 splice variant, CRFR1j
A previously unreported splice variant of CRFR1, designated Homo sapiens CRH receptor variant 1j, was discovered in a subset of heart donors and heart failure patients, in which exons 4 and 6 of the CRFR1 gene were spliced out (Figure 4, A and B). The deletion causes a frameshift and a premature stop signal in exon 9 of the CRFR1 gene. Analysis of the Ambion FirstChoice total RNA survey panel showed that CRFR1j is expressed in brain, thyroid, thymus, esophagus, trachea, adipose, small intestine, colon, bladder, prostate, testes, ovary, cervix, and placenta and, to a lesser extent, lung, heart, skeletal muscle, and kidney (Figure 4C). CRFR1j gene expression was not detected in the liver or spleen. The full-length functional CRFR1 isoform (CRFR1α) was present in all tissues expressing CRFR1j, except for the esophagus and skeletal muscle (Figure 4C). The CRFR1j mRNA transcript was detected in a greater proportion of the heart failure patients than the heart donors (62% of patients vs 3% of donors, P < .001). In the heart failure patients, the presence of CRFR1j mRNA was associated with a higher myocardial expression of CRFR1α (3.0-fold, P < .001). Patients in whom CRFR1j could be detected were more likely to carry the chr17q21.31 H2 allele (34% vs 11%, P = .011).
Figure 4.

Sequence and structural representation of the novel CRFR1 splice variant j. A, DNA and protein sequence of CRFR1j, illustrating the predicted protein sequence that results from the loss of exons 4 and 6, which leads to a shift in reading frame and a new stop codon in exon 9 (underlined sequence). Alternate exons are indicated by black and red text. B, Schematic illustration of the structure of the CRFR1j and the full-length, functional CRFR1 isoform (CRFR1α), showing that only the first three exons of CRFR1j are predicted to be identical to the functional receptor. The stop codon in exon 9 of CRFR1j results in a truncated protein compared with CRFR1α. Black boxes indicate exons of the CRFR1 gene sequence. Gray boxes indicate exons translated in the correct reading frame to generate functional CRFR1α, and white boxes indicate exons predicted to be translated in an alternative reading frame. Arrows indicate the location of nested primers used for semiquantitative PCR of novel CRFR1j splice variant. The reverse primer for the second PCR spans the unique CRFR1j splice junction between exons 3 and 5. CT-CD, carboxy-terminal cytoplasmic domain; NT-ECD1, amino-terminal extracellular domain 1; 7 TM, seven-transmembrane domains. C, RT-PCR indicating the presence or absence of CRFR1α and CRFR1j mRNA in range normal human tissues (expected band size for CRFR1α is 69 bp and for CRFR1j is 121 bp).
Functional assays demonstrated that HEK293T cells transfected with CRFR1α and treated with increasing doses of sauvagine (a CRFR1 agonist) showed expected proportional increases in the activation of the cAMP response element in a luciferase reporter assay (Figure 5A). In contrast, cells transfected with the CRFR1j construct failed to show a response, indicating that CRFR1j was unable to activate the cAMP signaling pathway (Figure 5B). Coexpression of CRFR1α with increasing doses of CRFR1j proportionally reduced the activation of the cAMP response element by CRFR1α, suggesting that CRFR1j may exert a dominant-negative effect on CRFR1α (P < .001 for 0.1 and 1 nmol/L doses of sauvagine, Figure 6A). Cell surface assays indicated that CRFR1j may inhibit the expression of CRFR1α at the cell surface (P = .053, Figure 6B). Coexpression of a control gene, soluble mouse CRFR2α, with CRFR1α showed that the dominant-negative effect on the cell surface expression was specific to CRFR1j, rather than a consequence of the nonspecific competitive transcription/translation of another mRNA (Figure 6C).
Figure 5.
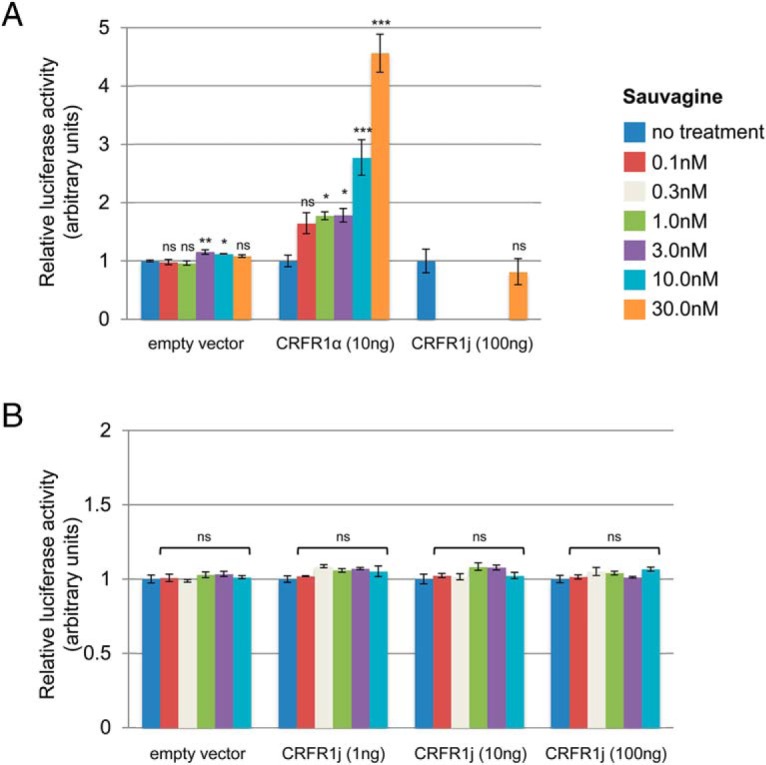
The novel CRFR1j splice variant is unable to activate the intracellular cAMP signaling pathway. A, Activation of a cAMP-response element luciferase reporter in HEK293T cells transfected with CRFR1α (full length, functional isoform) or CRFR1j, after treatment with the CRFR1 agonist, sauvagine (0–30 nM). Cells transfected with CRFR1α showed increasing activity with increasing sauvagine concentration. In contrast, cells transfected with 10 times the amount of CRFR1j showed no activity at the highest dose of sauvagine. B, Activation of a cAMP-response element luciferase reporter by sauvagine (0–10 nM) in HEK293T cells transfected with varying amounts of the novel CRFR1j splice variant (1–100 ng), indicating a complete lack of activation at all concentrations. Experiments were performed in triplicate. Data represent means and SEs. Statistics compare sauvagine treatment with the no-treatment group for each transfection. *, P < .05, **, P < .01, ***, P < .001 (ns, P > .05).
Figure 6.
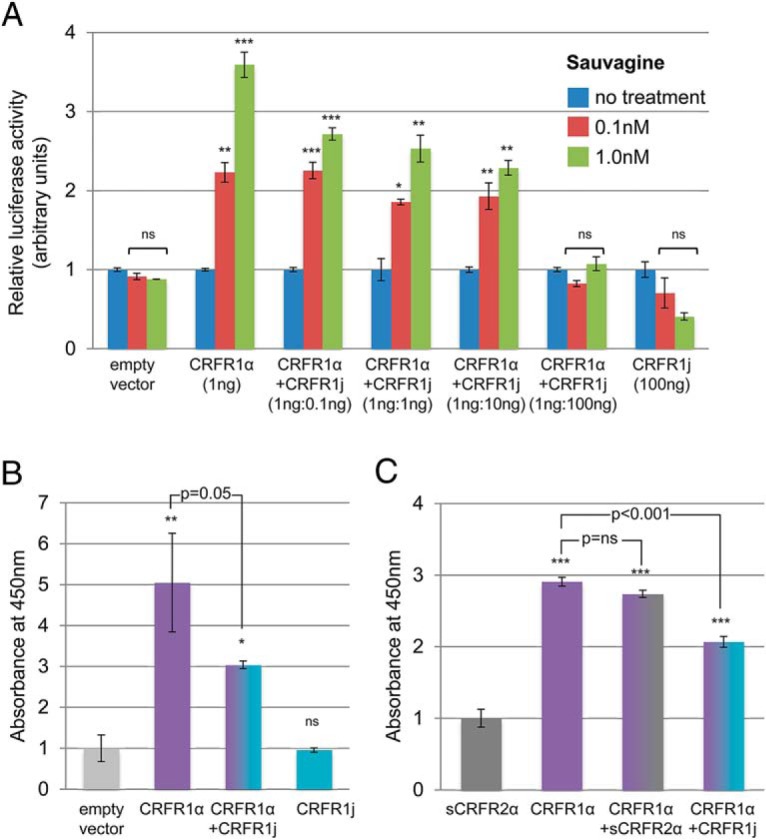
Dominant-negative activity of the novel CRFR1 splice variant j on the full-length CRFR1α receptor. A, Activation of a cAMP-response element luciferase reporter by sauvagine (0.1 nM, 1 nM) in HEK293T cells transfected with a fixed amount of CRFR1α and increasing amounts of CRFR1j. Luciferase activity decreased with increasing CRFR1j at both concentrations of sauvagine (P < .001 for both analyses), indicating a potential dominant-negative effect of CRFR1j on CRFR1 signaling. B, Cell surface expression of CRFR1α in COSM6 cells transfected with myc-tagged CRFR1α alone or in combination with CRFR1j, showing reduced cell surface expression in cotransfected cells. Experiments were performed in triplicate. Data represent means and SEs. Statistics compare sauvagine treatment with the no-treatment group for each transfection, unless otherwise indicated. *, P < .05, **, P < .01, ***, P < .001 (ns, P > .05).
Discussion
Our findings demonstrate that all members of the CRF family of ligands and receptors are expressed within the LV of the human myocardium, including CRFR1 and CRF. In heart failure, the down-regulation of CRFR2α and the up-regulation of CRFR1, CRF, and Ucn3 suggest that both CRFR1 and CRFR2 signaling are activated in this condition. Furthermore, our data suggest that CRFR1 expression may be altered in association with genetic variation at the CRFR1 gene locus and that CRFR1 signaling may be modulated by a novel, truncated splice variant of CRFR1, designated CRFR1j. This novel variant was detected in a wide range of human tissues and has a dominant-negative effect on CRFR1 signaling in vitro. CRFR1j was rarely detected in donor heart tissue but was detected in greater than 60% of LV tissues from heart failure patients, in whom it was associated with a higher expression of CRFR1. This suggests that the expression of the novel variant may be induced when CRFR1 expression is high.
Previous studies have shown that CRFR2α and the Ucns are abundantly expressed in the LV of the normal heart (10, 12). Our study confirms these findings and demonstrates a robust expression of CRFR1 and CRF in the LV in normal and diseased states. This suggests that the distribution of CRFRs in humans is considerably more heterogeneous than in rodents and that peripheral tissues, including the heart, express both receptor subtypes (1). However, it should be noted that most heart donors were on life support as a result of head trauma or cerebral vascular accident, and thus, the myocardial expression profile of genes and splice variants reported in this study may have been affected by traumatic injury, acute drug treatments, and underlying subclinical cardiac disease prior to the donation of tissue. Similarly, the myocardial expression profile in heart failure patients could have been affected by chronic drug treatments for heart failure. However, despite this, our findings are supported by observations in human heart failure and gene expression data from several animal models of cardiac disease. Plasma levels of Ucn1 are elevated in human heart failure (4), and two studies have reported increased Ucn1-like immunoreactivity in cardiac tissue from heart failure patients compared with controls (13, 14). Furthermore, myocardial expression of CRFR2 was reduced and Ucn was increased in a rat model of LV hypertrophy (DOCA-salt) (14, 25), and Ucn expression was increased in cardiac myocytes in response to ischemia in vitro (26, 27) and in isolated rat hearts (28). These findings are consistent with our findings of a lower expression of CRFR2 and a higher expression of the Ucns in heart failure patients and suggest minimal confounding from drug treatments or traumatic brain injury.
Genetic variants within the genes encoding the CRF family of ligands and receptors have been associated with a range of predominantly anxiety- and stress-related conditions including depression (29), panic disorder (30), and suicide (31). Our data suggest that a genetic variation at the CRFR1 gene locus is strongly associated with levels of CRFR1 gene expression. CRFR1 is located at chromosome 17q21.31, one of the most structurally complex and evolutionarily dynamic regions of the human genome (32). The region occurs as two distinct haplotypes, H1 and H2, which span approximately 1.5 Mb and includes an approximately 900-kb inversion that encompasses several genes including CRFR1 (33). Much of the genetic variation within CRFR1 segregates with this polymorphism. Among European populations the H1 haplotype has been associated with an increased risk of a group of neurodegenerative diseases including Alzheimer's disease, which are characterized by accumulation of fibrillar aggregates of microtubule-associated protein-τ (MAPT) in the brain cortex (34). The MAPT gene is located within the 17q21.31 inversion region, near CRFR1. MAPT is expressed at elevated levels within the cerebral cortex of H1 carriers, and this most likely represents the functional mechanism underlying the association between the inversion polymorphism and increased risk of neurodegenerative disease (34).
Our data provide a second example of altered expression of a gene located within this region in association with the inversion haplotype, with individuals with the H2 haplotype having higher levels of CRFR1 gene expression compared with H1 homozygotes, regardless of disease status. If individuals with the H2 haplotype are subsequently shown to have an increased protein expression of CRFR1 in cells, this relationship may help explain one of the mechanisms by which genetic variation at the CRFR1 locus contributes to an increased risk of anxiety and stress-related conditions. Thus, further studies are needed to determine whether genetic variation at the CRFR1 locus is associated with CRFR1 activity in the brain and heart and has the potential to influence disease susceptibility or progression in heart failure.
In contrast with other classes of G protein-coupled receptors, the exon-intron organization of class B1 G protein-couple receptors, of which CRFR1 and CRFR2 are members, permits extensive alternative splicing enabling expression of numerous splice variants that differ in their ligand specificity, binding, and G protein coupling (1). Although the physiological relevance of many of the CRFR1 and CRFR2 splice variants is uncertain, their complex pattern of expression suggests they may not be functionally redundant (19, 35–38). We have identified a novel, truncated form of CRFR1, designated CRFR1j, that was observed to down-regulate CRFR1 signaling by reducing the expression of the full-length CRFR1 receptor at the cell surface. This splice variant lacks exons that encode part of the receptor's ligand-binding and transmembrane domains, and the predicted protein sequence shares homology with only the first three exons of the full-length CRFR1 receptor. CRFR1j expression was predominantly detected in heart failure patients and was associated with a higher CRFR1 expression, leading us to speculate that it may be induced in disease or when CRFR1 expression is high, as a compensatory mechanism. However, several questions regarding the physiological and clinical relevance of CRFR1j in the heart remain. Our in vitro experiments in transfected cultured cells do not provide information on the basal levels or subcellular localization of endogenous CRFR1j in relevant tissues, nor do they show whether endogenous levels of this splice variant are sufficient to influence cell surface expression and signaling of endogenous CRFR1. Furthermore, because CRFR2 is the dominant CRF family receptor in the heart, it is unknown whether the modulation of cardiac CRFR1 signaling has the potential to influence the development and progression of disease, or clinical outcome, in heart failure.
The limitations of this study include potential confounding of gene expression by traumatic brain injury (heart donors), variation in tissue processing time (heart donors), acute and chronic drug treatments (heart donors and heart failure patients), and differences in age, gender, and ethnicity between donors and heart failure patients. In addition, our data are descriptive in nature, and more research is needed to understand the clinical relevance of our findings in the development and progression of heart failure. It is unclear whether the putative changes in the expression of genes and alternative splicing observed in the current study would be sufficient to promote the development of heart failure or occur as a consequence of it. However, in mice, deletion of CRFR2 did not influence basal LV function (39, 40), and several studies have shown the down-regulation of this receptor in response to elevated circulating Ucn. Consequently, we speculate that the putative changes in gene expression and alternative splicing seen in the current study do not drive the development of heart failure but may form part of the beneficial compensatory response to poor cardiac output in this condition.
In summary, we have demonstrated a robust expression of genes encoding all members of the CRF family of ligands and receptors within the LV of the human myocardium. Our data suggest that both CRFR1 and CRFR2 contribute to the coordinated compensatory response to heart failure and that the genetic variation at the CRFR1 locus and alternative splicing may modulate CRFR1 activity in the heart.
Acknowledgments
We gratefully acknowledge the donation of the human myocardium for research purposes.
This work was supported by the Heart Foundation of New Zealand, the Cleveland Clinic Foundation, and the National Institute of Diabetes and Digestive and Kidney Diseases (Award P01 DK26741–30). A.P.P. was supported by a New Zealand Foundation for Research, Science, and Technology Postdoctoral Fellowship and the Clayton Medical Research Foundation, Inc.
The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Disease or the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CRF
- corticotropin releasing factor
- CRFR
- CRF receptor
- HDB
- Hepes dissociation buffer
- HEK
- human embryonic kidney
- LV
- left ventricle
- MAPT
- microtubule-associated protein-τ
- Ucn
- urocortin.
References
- 1. Hillhouse EW, Grammatopoulos DK. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev. 2006;27:260–286. [DOI] [PubMed] [Google Scholar]
- 2. Boonprasert P, Lailerd N, Chattipakorn N. Urocortins in heart failure and ischemic heart disease. Int J Cardiol. 2008;127:307–312. [DOI] [PubMed] [Google Scholar]
- 3. Emeto TI, Moxon JV, Rush C, Woodward L, Golledge J. Relevance of urocortins to cardiovascular disease. J Mol Cell Cardiol. 2011;51:299–307. [DOI] [PubMed] [Google Scholar]
- 4. Wright SP, Doughty RN, Frampton CM, Gamble GD, Yandle TG, Richards AM. Plasma urocortin 1 in human heart failure. Circ Heart Fail. 2009;2:465–471. [DOI] [PubMed] [Google Scholar]
- 5. Rademaker MT, Charles CJ, Espiner EA, Frampton CM, Lainchbury JG, Richards AM. Endogenous urocortins reduce vascular tone and renin-aldosterone/endothelin activity in experimental heart failure. Eur Heart J. 2005;26:2046–2054. [DOI] [PubMed] [Google Scholar]
- 6. Vaughan J, Donaldson C, Bittencourt J, et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature. 1995;378:287–292. [DOI] [PubMed] [Google Scholar]
- 7. Lewis K, Li C, Perrin MH, et al. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc Natl Acad Sci USA. 2001;98:7570–7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reyes TM, Lewis K, Perrin MH, et al. Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc Natl Acad Sci USA. 2001;98:2843–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wiley KE, Davenport AP. CRF2 receptors are highly expressed in the human cardiovascular system and their cognate ligands urocortins 2 and 3 are potent vasodilators. Br J Pharmacol. 2004;143:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kimura Y, Takahashi K, Totsune K, et al. Expression of urocortin and corticotropin-releasing factor receptor subtypes in the human heart. J Clin Endocrinol Metab. 2002;87:340–346. [DOI] [PubMed] [Google Scholar]
- 11. Hsu SY, Hsueh AJ. Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nat Med. 2001;7:605–611. [DOI] [PubMed] [Google Scholar]
- 12. Takahashi K, Totsune K, Murakami O, et al. Expression of urocortin III/stresscopin in human heart and kidney. J Clin Endocrinol Metab. 2004;89:1897–1903. [DOI] [PubMed] [Google Scholar]
- 13. Ikeda K, Tojo K, Tokudome G, et al. Cardiac expression of urocortin (Ucn) in diseased heart; preliminary results on possible involvement of Ucn in pathophysiology of cardiac diseases. Mol Cell Biochem. 2003;252:25–32. [DOI] [PubMed] [Google Scholar]
- 14. Nishikimi T, Miyata A, Horio T, et al. Urocortin, a member of the corticotropin-releasing factor family, in normal and diseased heart. Am J Physiol Heart Circ Physiol. 2000;279:H3031—H3039. [DOI] [PubMed] [Google Scholar]
- 15. Yang J, Moravec CS, Sussman MA, et al. Decreased SLIM1 expression and increased gelsolin expression in failing human hearts measured by high-density oligonucleotide arrays. Circulation. 2000;102:3046–3052. [DOI] [PubMed] [Google Scholar]
- 16. Pilbrow AP, Folkersen L, Pearson JF, et al. The chromosome 9p21.3 coronary heart disease risk allele is associated with altered gene expression in normal heart and vascular tissues. PloS One. 2012;7:e39574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baker M, Litvan I, Houlden H, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–715. [DOI] [PubMed] [Google Scholar]
- 18. Rodriguez S, Gaunt TR, Day IN. Hardy-Weinberg equilibrium testing of biological ascertainment for Mendelian randomization studies. Am J Epidemiol. 2009;169:505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pisarchik A, Slominski AT. Alternative splicing of CRH-R1 receptors in human and mouse skin: identification of new variants and their differential expression. FASEB J. 2001;15:2754–2756. [DOI] [PubMed] [Google Scholar]
- 20. Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. [DOI] [PubMed] [Google Scholar]
- 21. Chen R, Lewis KA, Perrin MH, Vale WW. Expression cloning of a human corticotropin-releasing-factor receptor. Proc Natl Acad Sci USA. 1993;90:8967–8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen AM, Perrin MH, Digruccio MR, et al. A soluble mouse brain splice variant of type 2α corticotropin-releasing factor (CRF) receptor binds ligands and modulates their activity. Proc Natl Acad Sci USA. 2005;102:2620–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Friendly M. Corrgrams: exploratory displays for correlation matrices. Am Stat. 2002;56:316–324. [Google Scholar]
- 24. Murdoch DJ, Chow ED. A graphical display of large correlation matrices. Am Stat. 1996;50:178–180. [Google Scholar]
- 25. Makino S, Asaba K, Takao T, Hashimoto K. Type 2 corticotropin-releasing hormone receptor mRNA expression in the heart in hypertensive rats. Life Sci. 1998;62:515–523. [DOI] [PubMed] [Google Scholar]
- 26. Brar BK, Stephanou A, Okosi A, et al. CRH-like peptides protect cardiac myocytes from lethal ischaemic injury. Mol Cell Endocrinol. 1999;158:55–63. [DOI] [PubMed] [Google Scholar]
- 27. Okosi A, Brar BK, Chan M, et al. Expression and protective effects of urocortin in cardiac myocytes. Neuropeptides. 1998;32:167–171. [DOI] [PubMed] [Google Scholar]
- 28. Knight RA, Chen-Scarabelli C, Yuan Z, et al. Cardiac release of urocortin precedes the occurrence of irreversible myocardial damage in the rat heart exposed to ischemia/reperfusion injury. FEBS Lett. 2008;582:984–990. [DOI] [PubMed] [Google Scholar]
- 29. Polanczyk G, Caspi A, Williams B, et al. Protective effect of CRHR1 gene variants on the development of adult depression following childhood maltreatment: replication and extension. Arch Gen Psychiatry. 2009;66:978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smoller JW, Yamaki LH, Fagerness JA, et al. The corticotropin-releasing hormone gene and behavioral inhibition in children at risk for panic disorder. Biol Psychiatry. 2005;57:1485–1492. [DOI] [PubMed] [Google Scholar]
- 31. Wasserman D, Wasserman J, Rozanov V, Sokolowski M. Depression in suicidal males: genetic risk variants in the CRHR1 gene. Genes Brain Behav. 2009;8:72–79. [DOI] [PubMed] [Google Scholar]
- 32. Zody MC, Garber M, Adams DJ, et al. DNA sequence of human chromosome 17 and analysis of rearrangement in the human lineage. Nature. 2006;440:1045–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stefansson H, Helgason A, Thorleifsson G, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–137. [DOI] [PubMed] [Google Scholar]
- 34. Caffrey TM, Wade-Martins R. Functional MAPT haplotypes: bridging the gap between genotype and neuropathology. Neurobiol Dis. 2007;27:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grammatopoulos DK, Dai Y, Randeva HS, et al. A novel spliced variant of the type 1 corticotropin-releasing hormone receptor with a deletion in the seventh transmembrane domain present in the human pregnant term myometrium and fetal membranes. Mol Endocrinol. 1999;13:2189–2202. [DOI] [PubMed] [Google Scholar]
- 36. Karteris E, Markovic D, Chen J, Hillhouse EW, Grammatopoulos DK. Identification of a novel corticotropin-releasing hormone type 1β-like receptor variant lacking Exon 13 in human pregnant myometrium regulated by estradiol-17β and progesterone. Endocrinology. 2010;151:4959–4968. [DOI] [PubMed] [Google Scholar]
- 37. Pisarchik A, Slominski A. Corticotropin releasing factor receptor type 1: molecular cloning and investigation of alternative splicing in the hamster skin. J Invest Dermatol. 2002;118:1065–1072. [DOI] [PubMed] [Google Scholar]
- 38. Ross PC, Kostas CM, Ramabhadran TV. A variant of the human corticotropin-releasing factor (CRF) receptor: cloning, expression and pharmacology. Biochem Biophys Res Commun. 1994;205:1836–1842. [DOI] [PubMed] [Google Scholar]
- 39. Bale TL, Hoshijima M, Gu Y, et al. The cardiovascular physiologic actions of urocortin II: acute effects in murine heart failure. Proc Natl Acad Sci USA. 2004;101:3697–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coste SC, Kesterson RA, Heldwein KA, et al. Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat Genet. 2000;24:403–409. [DOI] [PubMed] [Google Scholar]