

Abstract
Prenatal testosterone (T) treatment recapitulates the reproductive and metabolic phenotypes of polycystic ovary syndrome in female sheep. At the neuroendocrine level, prenatal T treatment results in disrupted steroid feedback on gonadotropin release, increased pituitary sensitivity to GnRH, and subsequent LH hypersecretion. Because prenatal T-treated sheep manifest functional hyperandrogenism and hyperinsulinemia, gonadal steroids and/or insulin may play a role in programming and/or maintaining these neuroendocrine defects. Here, we investigated the effects of prenatal and postnatal treatments with an androgen antagonist (flutamide [F]) or an insulin sensitizer (rosiglitazone [R]) on GnRH-stimulated LH secretion in prenatal T-treated sheep. As expected, prenatal T treatment increased the pituitary responsiveness to GnRH leading to LH hypersecretion. Neither prenatal interventions nor postnatal F treatment normalized the GnRH-stimulated LH secretion. Conversely, postnatal R treatment completely normalized the GnRH-stimulated LH secretion. At the tissue level, gestational T increased pituitary LHβ, androgen receptor, and insulin receptor-β, whereas it reduced estrogen receptor (ER)α protein levels. Although postnatal F normalized pituitary androgen receptor and insulin receptor-β, it failed to prevent an increase in LHβ expression. Contrarily, postnatal R treatment restored ERα and partially normalized LHβ pituitary levels. Immunohistochemical findings confirmed changes in pituitary ERα expression to be specific to gonadotropes. In conclusion, these findings indicate that increased pituitary responsiveness to GnRH in prenatal T-treated sheep is likely a function of reduced peripheral insulin sensitivity. Moreover, results suggest that restoration of ERα levels in the pituitary may be one mechanism by which R prevents GnRH-stimulated LH hypersecretion in this sheep model of polycystic ovary syndrome-like phenotype.
Polycystic ovary syndrome (PCOS), a major cause of infertility and one of the most common endocrine disorders in women of reproductive age (1–3), is characterized by manifestations that include oligo-anovulation, hyperandrogenism, polycystic ovarian morphology, and insulin resistance (4). At the reproductive neuroendocrine level, women with PCOS exhibit reduced sensitivity to the inhibitory effects of sex steroids on gonadotropin secretion (5, 6) and increased pituitary responsiveness to GnRH (7, 8), with both contributing to LH hypersecretion. Although epidemiological data and animal studies indicate that prenatal androgen excess may contribute to the development of PCOS traits during adult life (9–12), the pathophysiological mechanisms underlying these neuroendocrine defects remain unclear.
In recent years, several animal models have evolved to investigate the impact of perinatal exposure to steroid excess on the offspring's health (13). In female sheep, prenatal testosterone (T) treatment disrupts the developmental trajectory of the fetus, culminating in adult reproductive and metabolic perturbations that recapitulate those seen in women with PCOS (14). Because the use of sheep as an animal model allows detailed and repetitive hormonal profiling, our previous studies have indicated that the progressive reproductive deterioration seen in prenatal T-treated females may stem, at least in part, from tonic activation of the reproductive neuroendocrine axis (15–17). Female sheep exposed to T excess in utero manifest disrupted steroid negative feedback (18, 19), increased pituitary responsiveness to GnRH (20), and subsequent LH hypersecretion. Although studies have documented the involvement of hypothalamic neural pathways controlling GnRH secretion (21–24), the mechanisms underlying LH hypersecretion at the anterior pituitary level in prenatal T-treated sheep remain poorly understood.
When considering the potential mediators of LH excess in this sheep model, it is important to recognize that gestational T treatment increases not only maternal T levels but also fetal concentrations of T and estradiol (25). Furthermore, gestational T treatment leads to maternal hyperinsulinemia and disrupts insulin signaling in fetal metabolic tissues (26) during critical periods of development that encompass pituitary gonadotroph differentiation (27). Because gonadal steroids and insulin are important regulators of pituitary development (28–31), it is possible that endocrine imbalances during gestation contribute to the adult LH hypersecretion seen in prenatal T-treated sheep. Additionally, postnatal alterations in androgen (functional hyperandrogenism) and insulin homeostasis (peripheral insulin resistance and compensatory hyperinsulinemia) may contribute to the phenotypic expression of these neuroendocrine defects. Insulin augments the effects of GnRH on LH synthesis and secretion (32, 33), and selective deletion of the insulin receptor (IR) in the pituitary has been shown to prevent LH hypersecretion and to restore fertility in obese female mice (34). Moreover, studies in various species have shown that androgens play an important role on LH secretion. In the female rat, T is required to facilitate GnRH stimulation of LHβ mRNA (35). Furthermore, selective deletion of the androgen receptor (AR) in gonadotropes reduces the magnitude of the preovulatory LH surge in female mice (36).
To investigate the organizational contribution of T and insulin in programming LH excess in this sheep model of PCOS-like phenotype, we used pharmacological approaches to negate androgen action and to improve peripheral insulin sensitivity during gestation. We hypothesized that prenatal cotreatment with rosiglitazone (R), an insulin sensitizer, or flutamide (F), an androgen antagonist, prevents the development of LH hypersecretion in prenatal T-treated sheep. Moreover, because postnatal alterations in insulin and androgen signaling in the pituitary may contribute to LH excess, we hypothesized that postnatal treatment with either an insulin sensitizer or an androgen antagonist would normalize the pituitary responsiveness to GnRH in these females. This possibility is supported by our recent findings that postnatal treatment with R decreased the number of aberrant estrous cycles and prevented further reproductive deterioration in prenatal T-treated sheep (37).
Materials and Methods
All animal-related procedures were approved by the Institutional Animal Care and Use Committee of the University of Michigan and are consistent with the National Institutes of Health Guide for Use and Care of Animals.
Animals and experimental groups
Female sheep from 2 cohorts were used for this investigation. The first cohort included the following groups of female offspring: control (C) (n = 7); prenatal T treated (T, n = 6); prenatal cotreated with T and F (TF), an androgen antagonist (n = 7); prenatal cotreated with T and R (TR), an insulin sensitizer (n = 7); prenatal treatment with T plus postnatal treatment with F (T+F, n = 6); and prenatal treatment with T plus postnatal treatment with R (T+R, n = 8) (Figure 1, upper panel). Female offspring from study 1 were used to investigate the pituitary responsiveness to GnRH in vivo, and pituitary tissues were used for immunohistochemistry. Prenatal treatments spanned gestational day (GD)30–GD90 (term pregnancy, ∼147 d), and postnatal treatments began at weaning (∼8 wk of age) and continued until the time of euthanasia. Findings relative to preovulatory LH surge dynamics in this cohort of animals have been published previously (25). Additionally, metabolic parameters, including adiposity, adipocyte morphology, peripheral insulin sensitivity, and insulin signaling in metabolic tissues from this cohort, have also been reported (26, 38).
Figure 1.

Schematic showing the temporal sequence of experimental procedures. In the first study (upper panel), female sheep were treated prenatally (GD30–GD90) with vehicle (C, n = 7 female offspring), T (n = 6 female offspring), TF (n = 7 female offspring), or TR (n = 7 female offspring). Additionally, 2 groups of females prenatally treated with T were subjected to postnatal intervention with either F (T+F, n = 6 female offspring) or R (T+R, n = 8 female offspring). Postnatal treatments started at approximately 8 weeks of age and continued throughout the study. Pituitary sensitivity to exogenous GnRH was tested in vivo at approximately 15 months of age during the seasonal anestrous period. At approximately 24 months of age, ewes were euthanized and pituitary tissues were harvested, fixed in paraformaldehyde, and processed for immunohistochemistry (IHC). Female offspring (C, n = 8; T, n = 10; T+F, n = 9; T+R, n = 9) in study 2 (bottom panel) were subjected to similar experimental procedures, with the exception that prenatal T treatment spanned from GD60 to GD90 (as opposed to GD30–GD90 in study 1). At approximately 3 years of age, females were euthanized, and pituitary tissues were harvested, snap frozen, and processed for Western blot analysis (WB). No TF or TR groups were generated in study 2.
In study 2, pituitary tissues from C, T, T+F, and T+R animals were used for Western blot analyses. Females in this cohort (C, n = 9; T, n = 10; T+F, n = 9; T+R, n = 9) were generated following the exact same protocol as in study 1, with the only exception that prenatal T treatment was administered between GD60 and GD90 as opposed to GD30 and GD90 in study 1 (Figure 1, bottom panel). In previous studies, we found that prenatal T treatment between GD60 and GD90 also results in progressive reproductive deterioration, albeit at a slower rate than GD30- to GD90-treated females (15). Furthermore, we observed that peripheral insulin resistance is also a phenotypic trait common to the female offspring from both GD30–GD90 and GD60–GD90 treatment paradigms, indicating that the critical period for programming this metabolic alteration lies primarily between days 60 and 90 of gestation (39). Metabolic tissues from this cohort (study 2) have been previously used to investigate tissue sensitivity to insulin in vitro (26).
Husbandry and treatments
Information regarding prenatal T treatment, husbandry, and nutrition has been described in detail previously (40). Briefly, beginning approximately 3 weeks before breeding, adult ewes were group fed daily with 0.5 kg of shelled corn and 1.0–1.5 kg of alfalfa hay per animal. After mating to rams with proven fertility, all ewes were housed in pasture and group fed daily with 1.25 kg of alfalfa hay per animal.
In order to generate T females, pregnant Suffolk sheep were treated twice weekly with 100 mg of T propionate suspended in 2 mL of corn oil (∼1.2 mg/kg, im; Sigma-Aldrich Co) between GD30 and GD90 for study 1 and GD60 and GD90 for study 2. This dose of T has been shown to promote circulating concentrations of T in pregnant sheep and umbilical artery similar to those seen in intact adult males and 60-day-old male fetuses, respectively (17). C females received injections of vehicle (corn oil, im) during the same period. Female sheep in the TF group were cotreated with TF (15 mg/kg · d, sc; Sigma-Aldrich), whereas TR females were cotreated with TR (8 mg/d, orally, Avandia; GlaxoSmithKline). When twin births were involved, 1 offspring from each mother was randomly selected for use in the study.
After birth, lambs were supplemented with a commercial pelleted diet (Shur-Gain; Nutreco Canada, Inc) containing 3.6-Mcal/kg digestible energy and 18% crude protein. Female lambs were weaned at approximately 8 weeks of age and maintained outdoors at the University of Michigan Sheep Research Facility (Ann Arbor, MI; 42°18′ N) until the end of the studies. Postnatal interventions with F (15 mg/kg · d, orally; Sigma-Aldrich) or R (0.11 mg/kg · d, orally, Avandia; GlaxoSmithKline) started at weaning (∼8 wk of age) and continued throughout the study (Figure 1, upper panel). The dose of F used in this study has been demonstrated to block the effects of both exogenous and endogenous androgens on phenotypic virilization in males and prenatal T-treated female sheep (41). The R dose is within the range used to treat women with PCOS (42, 43) and has been shown to restore insulin sensitivity in prenatal T-treated sheep (37).
Pituitary responsiveness to GnRH and tissue harvest
To investigate the impact of prenatal T and pharmacological interventions on GnRH-stimulated LH secretion, a pituitary responsiveness test was performed at approximately 15 months of age during the anestrous season, when GnRH episodic release is markedly suppressed (study 1). Because we previously observed that FSH secretion in response to GnRH stimulation is not affected by prenatal T treatment in sheep (20), we have focused particularly on LH release in this study. Pituitary testing involved the administration of 4 GnRH bolus injections (2 ng/kg, iv; Sigma) 1.5 hours apart and collection of jugular blood samples at 15-minute intervals beginning 45 minutes before the first GnRH injection and ending 75 minutes after the last injection (total sampling, 6.5 h). The GnRH dose used has been shown to produce LH responses of similar magnitude as those seen in prepubertal females (44). Blood samples (3 mL) were collected into heparinized tubes and plasma was stored at −20°C until assessment of LH concentration.
At approximately 22 months of age, estrous cycle was synchronized with 2 injections of prostaglandin F2α (20 mg, im, Lutalyse; Pfizer Animal Health) administered 11 days apart, and ovariectomy was performed 24 hours after the second injection (natural follicular phase). At approximately 24 months of age (adult), all females were treated for 7 days with a controlled internal drug release progesterone implant (InterAG). Sixteen hours after removal of progesterone implants, females were treated with 4 3-cm-long estradiol implants to simulate ovarian steroid levels during the normal estrous cycle (45). After a 48-hour fasting period to assess metabolic parameters (26) and 24 hours after insertion of estradiol implants (artificial follicular phase), ewes were euthanized by administration of barbiturate overdose (10–15 mL, iv, Fatal Plus; Vortech Pharmaceuticals), and pituitary glands were fixed overnight in 4% paraformaldehyde for immunohistochemistry.
In order to investigate the protein level of several key regulators of gonadotropin secretion by Western blot analysis, fresh-frozen pituitary tissues from a different cohort of animals were used (study 2). Pituitary tissues were collected at approximately 3 years of age (adult) from ovariectomized, estradiol-replaced females (artificial follicular phase; as described above for study 1). All animals in both studies were fasted for 48 hours before euthanasia. Figure 1 illustrates the temporal sequence of experimental procedures conducted in these studies.
LH RIA
Plasma concentrations of LH were measured in duplicate using a validated RIA (46). The mean (±SEM) sensitivity of the assays was 0.85 ± 0.07 ng/mL (n = 10 assays). Mean intraassay coefficients of variation based on 4 reference pools was 6.5%. The mean interassay coefficient of variation for the same reference pools was 7.8%.
Western blot analysis
Fresh-frozen pituitary tissues collected from females in study 2 (half pituitary divided by a midsagittal cut) were homogenized in radioimmunoprecipitation assay buffer (Pierce RIPA buffer; Thermo Scientific) containing protease inhibitors (Complete Mini; Roche Diagnostics) and phosphatase inhibitors (Phos STOP; Roche Diagnostics). Tissue homogenates were centrifuged at 10 000g for 15 minutes at 4°C, and the whole-cell protein extract was used for the analysis. Equal amounts of protein (40–50 μg) were resolved on SDS-PAGE and transferred into a nitrocellulose membrane (Bio-Rad). Membranes were incubated in blocking buffer (5% nonfat milk diluted in Tris-buffered saline) for 60 minutes and incubated overnight (4°C) with primary antibodies (Table 1). Levels of phosphorylated and total forms of proteins as well as the corresponding loading Cs were determined in the same membrane after stripping and reblotting. Samples from all experimental groups were distributed through 4 SDS-PAGE gels (9 samples per gel) that were run under the same conditions. Protein bands were visualized using enhanced chemiluminescence (Pierce ECL Western Blotting Substrate; Thermo Scientific), and band density was determined using the ImageJ software (National Institutes of Health). The specificity of the antibodies was confirmed by visualization of protein bands of the correct size.
Table 1.
List of Antibodies Used in the Present Study
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used | RRID |
|---|---|---|---|---|---|---|
| Ovine LHβ | oβLH | NIDDK | Rabbit; polyclonal | WB 1:50K; IHC 1:1K | AB 2629449 | |
| Ovine GnRH-R | FSQCVTHCS FPQWWHQA FYN | Ovine GnRH-R | Dr Donal Skinner (University of Wyoming) | Rabbit, polyclonal | WB 1:1K | AB 2629448 |
| AR | AR (N-20) | Santa Cruz Biotechnology, SC-816 | Rabbit; polyclonal | WB 1:1K | AB 1563391 | |
| ERα | ERα (clone 1D5) | Thermo Scientific, MS-354 | Mouse; monoclonal | WB 1:500 | AB 61341 | |
| ERα | ERα (clone 1D5) | Dako, M7047 | Mouse; monoclonal | IHC 1:100 | AB 2101946 | |
| IRβ | IRβ (Ab-6; clone CT-3) | Thermo Scientific, MS-636 | Mouse; monoclonal | WB 1:200 | AB 142261 | |
| PTEN | PTEN (D4.3) XP | Cell Signaling, 9188 | Rabbit; monoclonal | WB 1:1K | AB 2253290 | |
| GAPDH | GAPDH (14C10) | Cell Signaling, 3683 | Rabbit; monoclonal | WB 1:1K | AB 1642205 | |
| p-AKT | p-AKT (Thr308) (D25E6) | Cell Signaling, 13038 | Rabbit; monoclonal | WB 1:1K | AB 2629447 | |
| AKT | AKT 9pan) (C67E7) | Cell Signaling, 4691 | Rabbit; monoclonal | WB 1:1K | AB 915783 | |
| p-mTOR | p-mTOR (S2448) | Cell Signaling, 2971 | Rabbit; monoclonal | WB 1:1K | AB 330970 | |
| mTOR | mTOR | Cell Signaling, 2972 | Rabbit; monoclonal | WB 1:1K | AB 330978 | |
| p-ERK | p-p44/42 MAPK (T202/Y204) (D13.14.4E) | Cell Signaling, 4370 | Rabbit; monoclonal | WB 1:1K | AB 2315112 | |
| ERK | p44/42 MAPK (Erk1/2) (137F5) | Cell Signaling, 4695 | Rabbit; monoclonal | WB 1:1K | AB 390779 |
WB, Western blotting; IHC, immunohistochemistry.
Immunohistochemistry and image analysis
Paraformaldehyde-fixed pituitary tissues collected in study 1 were serially dehydrated with graded ethanol, embedded in paraffin blocks, and cut in 5-μm sections using a microtome (Leica RM 2155; Leica Microsystems, Inc). The sections were mounted on Fisher Superfrost Plus slides (Fisher Scientific) and placed on a slide warmer (37°C) overnight. Four comparable sections/animal from the midsagittal plane were processed for double-label detection of LHβ and estrogen receptor (ER)α by immunofluorescence. To minimize variation, all the slides were processed in 2 batches, with all treatment groups distributed evenly among each batch. Slides were deparaffinized in 2 xylene washes followed by ethanol rehydration. Next, slides were washed in Tris-buffered saline and submitted to heat-induced epitope retrieval by incubation in 10mM sodium citrate buffer (pH 6.0) at 95°C–98°C for 25 minutes. Sections were blocked with 5% normal goat serum and incubated overnight at 4°C with mouse monoclonal anti-ERα (1:100; Dako). Next, slides were incubated with biotinylated goat antimouse IgG (1:400, 1 h at room temperature; Jackson ImmunoResearch) followed by streptavidin-conjugated horseradish peroxidase (1:250, 1 h at room temperature, Vectastain Elite ABC; Vector Labs). Alexa Fluor 555-conjugated streptavidin (1:300, 30 min at room temperature; Invitrogen) was used to label ERα immunoreactivity. Detection of LHβ was performed by incubation with rabbit antiovine LHβ (1:1500; obtained from Dr A. F. Parlow, National Hormone and Pituitary Program, National Institute of Diabetes and Kidney Diseases [NIDDK]) overnight at 4°C, followed by incubation with Alexa Fluor 488-conjugated donkey antirabbit IgG (1:300, 1 h at room temperature; Invitrogen). After completion of immunostaining, sections were cover slipped using mounting media containing 4′,6-diamidino-2-phenylindole (DAPI) (Prolong Gold Antifade with DAPI; Invitrogen).
The controls for the dual-label immunofluorescence procedure included omission of primary antibodies (anti-ERα and anti-LHβ) and preabsorption of the anti-LHβ antiserum with blocking peptide (5 μg/mL, ovine LHβ; NIDDK). For ERα antiserum, blocking peptide was not commercially available. These procedures eliminated the fluorescent signal for each corresponding antigen.
Sections were analyzed using a confocal laser-scanning microscope (Leica SP5X inverted; Leica Microsystems), and images were acquired with a ×40 PL APO objective at a resolution of 1024 × 1024 pixels. The pinhole aperture was set to 70.89 μm in order to obtain an optical section thickness of 1 μm. Excitation was done with 405-, 488-, and 555-nm laser lines, and acquisition bandwidths were set to 420–480, 505–550, and 570–650 nm, respectively.
All quantification analyses were performed by an observer blind to the assignment of tissue sections within experimental groups. Each pituitary section was divided into 3 main regions depending on the proximity to the median eminence (dorsal, mid, and ventral). The number of cells immunoreactive to LHβ, ERα, or both (colocalization) was determined in 0.5-mm2 fields (∼300 LHβ-immunoreactive cells/field) randomly located in the 3 different regions of the pituitary in 3 sections from each animal (total of 9 images/animal) using the ImageJ software (National Institutes of Health). This procedure resulted in 3 values for each region, and the mean value per region was computed for each animal. Care was taken to avoid the pars and zona tuberalis that contain a high density of gonadotropes or areas that contained large portal vessels (and therefore no cells) as previously reported (47).
Statistical analysis
The JMP software (SAS Institute, Inc) was used for statistical analyses. For assessment of the pituitary responsiveness to GnRH stimulation, differences in mean LH pulse peak and amplitude among treatment groups were analyzed using ANOVA with Tukey's honest significant difference (HSD) post hoc test. Pulse amplitude was defined as the mean amplitude (peak minus the preceding nadir) arising from the 4 GnRH injections for each animal.
For protein expression analysis (study 2), comparison of pituitary protein level was carried out after normalization of each protein (band density) with its corresponding loading C (Glyceraldehyde 3-phosphate dehydrogenase [GAPDH] band density). For phosphorylation level, expression of phosphorylated protein was normalized with its corresponding total protein. Comparisons between groups were performed using ANOVA with Tukey's HSD post hoc test.
For the immunohistochemistry data, the percentage of LHβ-immunoreactive cells that expressed ERα was normalized using the arcsine square root transformation method as described previously (48). The numbers of LHβ- and ERα-immunoreactive cells as well as the normalized percentage of LHβ cells that expressed ERα were compared among treatment groups using ANOVA with Tukey's HSD post hoc test. Because no differences were observed between the different pituitary regions (dorsal, middle, and ventral) for all the variables analyzed, data from these regions for each animal were pooled together for analyses.
Results
Pituitary responsiveness to GnRH
Patterns of circulating concentrations of LH during the pituitary sensitivity test from 2 representative females from each group are depicted in Figure 2, A, B, and D. For all animals, each GnRH injection induced a detectable LH pulse. Confirming our previous data (20), prenatal T-treated females presented greater (P < .05) mean LH pulse peak and pulse amplitude compared with Cs (Figure 2C). Barring a partial improvement in the LH pulse peak with prenatal R treatment, neither prenatal intervention (TF or TR) restored LH pulse parameters to C levels (Figure 2C). Conversely, postnatal R treatment (T+R) completely restored both the GnRH-stimulated LH pulse peak and pulse amplitude to C levels (Figure 2E). Postnatal treatment with F (T+F) failed to prevent an increase in the LH pulse peak and amplitude in females prenatally exposed to T excess (Figure 2E).
Figure 2.

Effects of gestational T excess and pharmacological intervention with an androgen antagonist (F) or an insulin sensitizer (R) on the GnRH-stimulated LH secretion in female sheep. A, B, and D, Two representative LH profiles of females treated with prenatal vehicle (C, n = 7), prenatal T (T, n = 6), prenatal T and prenatal F (TF, n = 7), prenatal T and prenatal R (TR, n = 7), prenatal T plus postnatal F (T+F, n = 6), or prenatal T plus postnatal R (T+R, n = 8). Arrowheads indicate time of GnRH injections. Bar graphs depict mean (±SEM) LH pulse peak and pulse amplitude of prenatal intervention (C) and postnatal intervention groups (E). Please note that, to facilitate comparison between groups, mean LH pulse peak and amplitude for C and T groups are repeated in C and E. Means with different superscripts are significantly (P < .05) different.
Protein expression of LHβ and key regulators of LH synthesis and secretion
Considering the positive effects of postnatal treatment with an insulin sensitizer on the GnRH-stimulated LH secretion, we took advantage of pituitary tissue available from a different cohort of animals (study 2) to investigate the potential mechanisms by which R improves pituitary function. In agreement with observations from study 1, prenatal T treatment (GD60–GD90) significantly increased (P < .05) the LHβ protein level in the anterior pituitary compared with Cs (Figure 3A). Levels of GnRH receptor (GnRH-R) followed a similar pattern, although no statistical differences were observed (Figure 3B). Prenatal T treatment also significantly (P < .05) increased AR and decreased ERα expression in the pituitary (Figure 3, C and D). Moreover, prenatal T-treated females presented greater (P < .05) pituitary levels of IRβ (Figure 3E), whereas no changes were observed in the expression of Phosphatase and tensin homolog (PTEN) (Figure 3F), a negative regulator of the Phosphoinositide 3-kinase/Protein kinase B (Akt) pathway closely associated with insulin resistance (48, 49). Although postnatal F treatment partially normalized IRβ (Figure 3E) and completely restored AR expression to C levels (Figure 3C), it failed to prevent an increase (P < .05) in LHβ expression in the pituitary (Figure 3A). Contrarily, postnatal treatment with R completely restored ERα to C level (Figure 3D) and partially prevented an increase in LHβ expression in the pituitary from females prenatally exposed to T excess (Figure 3A).
Figure 3.

Effects of gestational T excess and postnatal intervention with an androgen antagonist (F) or an insulin sensitizer (R) on the protein expression of LHβ and key regulators of LH synthesis/secretion in the pituitary from female sheep. Representative Western blottings and mean (±SEM) protein level ratio of LHβ to GAPDH (A), GnRH-R to GAPDH (B), AR to GAPDH (C), ERα to GAPDH (D), IRβ to GAPDH (E), and PTEN to GAPDH (F) in the anterior pituitary from females treated with prenatal vehicle (C, n = 8), prenatal T (T, n = 10), prenatal T plus postnatal F (T+F, n = 9), or prenatal T plus postnatal R (T+R, n = 9). Means with different superscripts are significantly (P < .05) different.
Insulin signaling in the pituitary
Because the pituitary expression of IRβ was elevated in T females and postnatal treatment with an insulin sensitizer normalized the GnRH-stimulated LH secretion, we investigated the activation (phosphorylation) of 3 important signaling pathways activated by IRβ, namely the Phosphoinositide 3-kinase/Akt, the mechanistic target of rapamycin (mTOR) and the MAPK/ERK pathways. Although a treatment effect was observed for the ratio of phospho (p)-mTOR/mTOR in the pituitary (ANOVA, P = .04), none of the group comparisons reached statistical significance in the post hoc analysis (Tukey's HSD test) (Figure 4B). No differences were observed in the phosphorylation of AKT (Figure 4A) or ERK (Figure 4C) among treatment groups.
Figure 4.

Effects of gestational T excess and postnatal intervention with an androgen antagonist (F) or an insulin sensitizer (R) on insulin signaling in the pituitary from female sheep. Representative Western blottings and mean (±SEM) protein level ratio of p-AKT to AKT, AKT to GAPDH (A), p-mTOR to mTOR, mTOR to GAPDH (B), p-ERK to ERK, and ERK to GAPDH (C) in the anterior pituitary from females treated with prenatal vehicle (C, n = 8), prenatal T (T, n = 10), prenatal T plus postnatal F (T+F, n = 9), or prenatal T plus postnatal R (T+R, n = 9). A treatment effect (P = .04) was observed for the ratio of p-mTOR to mTOR; however, none of the group comparisons reached statistical significance in the post hoc analysis (Tukey's HSD test).
LHβ and ERα immunoreactivity in the pituitary
Because the anterior pituitary is comprised of multiple cell types, we performed double-label immunofluorescence using pituitary tissue from study 1 (GD30–GD90) to investigate whether the normalization of ERα expression by postnatal R treatment is specific to gonadotropes (LHβ-immunoreactive cells). None of the experimental treatments altered the number of LHβ-immunoreactive cells in the pituitary (Figure 5A). Prenatal T treatment significantly (P < .05) reduced both the number of ERα-immunoreactive cells (Figure 5B) and the percentage of LHβ-immunoreactive cells that coexpressed ERα in the anterior pituitary (Figure 5C). Neither prenatal intervention (TF or TR) significantly prevented the reduction in ERα cell number (Figure 5B) or LHβ/ERα colocalization in the pituitary (Figure 5B). Although postnatal treatment with F also failed to normalize the number of ERα-immunoreactive cells (Figure 5B) and LHβ/ERα colocalization in the pituitary (Figure 5C), postnatal R treatment tended (P = .078) to restore the number of ERα-immunoreactive cells to C levels (Figure 5B). More importantly, postnatal treatment with the insulin sensitizer R completely restored the percentage of LHβ-immunoreactive cells that coexpressed ERα to C levels (Figure 5C).
Figure 5.
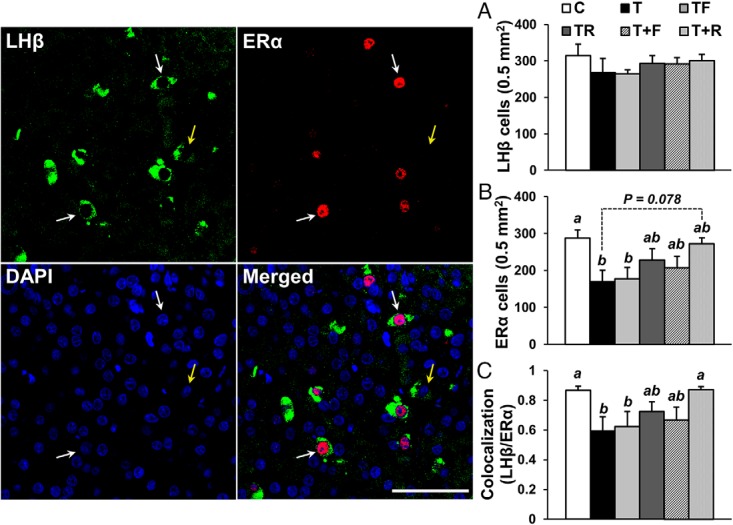
Effects of gestational T excess and pharmacological intervention with an androgen antagonist (F) or an insulin sensitizer (R) on LHβ and ERα immunoreactivity in the pituitary from female sheep. Confocal microscope images (1-μm optical sections) depicting LHβ (green), ERα (red), and DAPI (blue) staining in the anterior pituitary from females treated with prenatal vehicle (C, n = 7), prenatal T (T, n = 6), prenatal T and prenatal F (TF, n = 7), prenatal T and prenatal R (TR, n = 7), prenatal T plus postnatal F (T+F, n = 6), or prenatal T plus postnatal R (T+R, n = 8). White arrows indicate LHβ-immunoreactive cells that colocalize with ERα, and yellow arrow illustrates an LHβ-immunoreactive cell that does not colocalize with ERα. Bar graphs demonstrate the mean (±SEM) number of LHβ-immunoreactive cells (A), number of ERα-immunoreactive cells (B), and percentage of LHβ-immunoreactive cells that colocalize with ERα (C). Scale bar, 50 μm. Means with different superscripts are significantly (P < .05) different.
Discussion
Using an animal model of PCOS-like phenotype, our results demonstrate that postnatal treatment with the insulin sensitizer R prevents the GnRH-stimulated LH hypersecretion in female sheep prenatally exposed to T excess, thus suggesting that this neuroendocrine alteration is mainly secondary to perturbations in insulin-glucose homeostasis. Furthermore, our observations indicate that restoration of ERα levels in the anterior pituitary is likely a mechanism by which R normalizes pituitary responsiveness to GnRH in these females. The finding that prenatal intervention with either R or the androgen antagonist F failed to prevent the LH hypersecretion induced by gestational T excess suggests this pituitary defect may be programmed via the estrogenic effects of T (facilitated by the conversion of T to estradiol). Alternatively, both androgen and insulin signaling pathways act in synergy during fetal development to program LH hypersecretion in prenatal T-treated sheep. These possibilities and the potential relevance of the present results to women with PCOS and other metabolic disorders are discussed below.
Prenatal programming of pituitary LH excess
To identify the potential mediators involved in the programming of PCOS-like traits in prenatal T-treated sheep, we have extensively characterized the main endocrine and metabolic imbalances occurring during gestation in both the mother and the fetus. These studies showed that gestational T treatment increases the maternal concentrations of T, androstenedione, and insulin, as well as the fetal levels of T and estradiol (25). Moreover, gestational T treatment disrupts insulin signaling in metabolic tissues of the developing fetus (26). Because gonadal steroids and insulin are important regulators of pituitary development and function (28–31), changes in prenatal androgen, estrogen, and/or insulin actions might contribute to the development of adult LH hypersecretion in prenatal T-treated sheep.
Findings from this study, which revealed that prenatal intervention with either an androgen antagonist or an insulin sensitizer failed to prevent the GnRH-stimulated LH hypersecretion in adult females, suggest that increased estrogenic actions may contribute to the programming of this neuroendocrine defect. Increased estradiol levels found in fetuses of gestational T-treated sheep support this premise (25). Further support also comes from studies in transgenic mouse models that demonstrate that estrogens, acting primarily through ERα, influence the development and function of the anterior pituitary (51, 52). For example, selective deletion of ERα in the anterior pituitary markedly increased the mRNA levels of the 3 gonadotropin subunits (α-glycoprotein subunit, LHβ, and FSHβ) and resulted in complete infertility in mice (51). Direct effects of estrogen at the pituitary level have also been reported in the female sheep (53). Therefore, it is possible that increased levels of estradiol in gestational T-treated fetuses (25) promote organizational changes in the anterior pituitary that culminate in LH hypersecretion in adult female sheep. Future studies evaluating the effects of prenatal cotreatment with an estrogen antagonist are necessary to confirm this possibility.
Postnatal role of insulin in the manifestation of pituitary LH excess
Postnatal treatment with R completely normalized the pituitary sensitivity to GnRH in prenatal T-treated sheep, thus suggesting that postnatal perturbations in insulin-glucose homeostasis contribute, at least in part, to the phenotypic expression of this neuroendocrine defect. The findings of normalization of LH secretion after R treatment are in line with the proposed “2-hit” hypothesis used to explain the adult onset of some diseases. This hypothesis suggests that a genetic susceptibility combined with an insult occurring during prenatal life (“first-hit”) leads to reorganization of the various organ systems, which alone may be insufficient to alter the adult phenotype. However, endocrine imbalances and/or adverse stressors/exposures during postnatal life may act as a “second hit,” which can unmask or amplify the underlying defects culminating in disease states (54–58). Therefore, the hyperinsulinemic status evidenced in prenatal T-treated sheep likely acts as a second hit to promote GnRH-stimulated LH hypersecretion in adult females. In previous studies, we have demonstrated that prenatal T treatment leads to peripheral insulin resistance and compensatory hyperinsulinemia in prenatal T-treated sheep (37, 39), which likely result from reduced insulin sensitivity in the liver and skeletal muscle (26). Importantly, consistent with a role of hyperinsulinemia as a second hit in the manifestation of LH excess, postnatal treatment with R improves peripheral insulin sensitivity, normalizes the circulating concentrations of insulin, and prevents premature reproductive deterioration in prenatal T-treated sheep (37). Although perturbations in insulin-glucose homeostasis can impact different levels of the reproductive axis, the present results indicate that the effects of R improving reproductive function in prenatal T-treated sheep are mediated, at least in part, at the anterior pituitary level (Figure 6).
Figure 6.

Schematic drawing of the proposed events leading to pituitary LH hypersecretion in prenatal T-treated sheep. Adult sheep prenatally exposed to T excess manifest reduced insulin sensitivity in the liver and skeletal muscle resulting in peripheral insulin resistance and compensatory hyperinsulinemia. In turn, increased circulating concentrations of insulin stimulate the anterior pituitary (AP), which remains insulin sensitive, to synthesize and secrete large amounts of LH. Although the exact mechanisms by which insulin promotes LH hypersecretion in prenatal T-treated sheep remain unclear, a reduction in ERα levels in the AP appears to be involved.
In support of a role for insulin in the manifestation of LH excess, in vitro studies using primary cultures of rat anterior pituitary cells have shown that insulin enhances not only basal LH release, but it also augments the effects of GnRH on LH secretion (33, 59). A later study using LβT2 gonadotrope cells, a cell line derived from pituitary tumors in transgenic mice, demonstrated that insulin stimulates LH secretion largely by modulating LHβ gene expression (32). Similar to our findings in prenatal T-treated sheep, diet-induced obese female mice are hyperinsulinemic and exhibit a marked increase in LH release after GnRH stimulation (34). Notably, selective deletion of IRβ in the anterior pituitary prevented the LH hypersecretion and restored fertility in these obese females, indicating that direct insulin actions in the pituitary are required for the manifestation of LH excess in obese mice. In the murine model, however, changes in the pituitary responsiveness to GnRH are explained largely by changes in the expression of GnRH-R in the anterior pituitary (34). Using a well-validated antibody raised against the ovine GnRH-R (60, 61), we did not observe significant changes in the protein levels of GnRH-R between prenatal T-treated and C females. However, because changes in GnRH-R levels (although not significant) follow a similar pattern to those observed for LHβ, the involvement of GnRH-R in the LH hypersecretion in prenatal T-treated sheep cannot be discarded.
Pituitary changes associated with LH excess
Previously, we have reported that prenatal T-treated sheep exhibit a marked reduction in the sensitivity of the neuroendocrine system to the inhibitory effects of estradiol on LH secretion (18). Although this regulatory mechanism has been classically proposed to occur primarily at the hypothalamic level, observations in women (62), rhesus monkey (63), mice (51), and sheep (64) demonstrate a direct pituitary effect. For instance, studies in hypothalamo-pituitary disconnected sheep reported a reduction in the amplitude of the GnRH-stimulated LH pulses after estrogen administration (64, 65). Therefore, the present finding that prenatal T treatment reduces the pituitary levels of ERα in adult sheep suggests that the reduced responsiveness to the estradiol inhibitory feedback on LH secretion is likely mediated to a certain extent at the pituitary level. In addition, results from the present study indicate that restoration of ERα levels in the pituitary may be one mechanism by which R prevents LH hypersecretion in this sheep model. Importantly, immunohistochemical results confirming changes in ERα expression specifically in gonadotropes (LHβ-immunoreactive cells) substantiate a role for ERα in this process.
The findings that 1) postnatal treatment with R normalized the pituitary responsiveness to GnRH without preventing the increase in pituitary AR expression and 2) postnatal treatment with the androgen antagonist F restored AR expression to C levels but failed to normalize the GnRH-stimulated LH secretion suggest that increased postnatal androgen action in the pituitary is likely not a mechanism underlying this neuroendocrine perturbation in prenatal T-treated sheep. Similarly, conditional knockout of the AR in gonadotropes did not impact basal LH secretion, age at puberty, and fertility in female mice (36).
In addition to the adult hyperinsulinemic status reported previously in these females (37, 39), the present studies demonstrate that prenatal exposure to T excess increases the levels of IRβ in the anterior pituitary of adult sheep. Thus, increased insulin actions in the pituitary may stem not only from increased circulating concentrations of insulin but also due to an upregulation of IRβ in the pituitary. Although postnatal R treatment did not normalize IRβ expression in the pituitary, we have previously reported that it improves insulin sensitivity in metabolic tissues and reduces circulating concentrations of insulin (26, 37). The low and similar circulating concentrations of insulin across groups at the time of euthanasia (data not shown), a consequence of the 48-hour fast before tissue collection, likely prevented our ability to detect changes in activation (phosphorylation) of the main intracellular signaling pathways stimulated by insulin (baring a trend for greater phosphorylation of mTOR in T compared with C females). In agreement, basal activation of the insulin signaling pathways in metabolic tissues from this cohort of animals was similar among groups, despite striking changes after in vitro insulin stimulation (26). Therefore, future studies in which tissue is collected without previous fasting will be necessary to confirm whether prenatal T treatment increases the activation of the insulin signaling pathways in the anterior pituitary of adult sheep. Nevertheless, the fact that PTEN levels did not change among groups further suggests that the pituitary remains insulin sensitive in prenatal T-treated sheep despite the increased peripheral insulin resistance and hyperinsulinemia (26, 37).
Translational relevance
Most women with PCOS manifest LH excess, a neuroendocrine hallmark of this syndrome (66). Notably, the diurnal changes in the circulating concentrations of LH and insulin in PCOS women follow a similar time course (67), thus suggesting a positive association between them. Furthermore, similar to what is observed in prenatal T-treated sheep, treatment with R not only improves peripheral insulin sensitivity, but it also decreases the concentrations of LH in PCOS women (68, 69). Other insulin-sensitizing agents, such as troglitazone (70) and metformin (71, 72), have also been shown to reduce LH secretion in women with PCOS. Based on the finding that treatment with insulin sensitizer reduced the amplitude of LH pulses after exogenous GnRH stimulation in approximately 55% of women with PCOS, one study suggested that the effects of insulin are mediated, at least in part, at the pituitary level (73). In contrast, another study found no effect of 12-hour insulin infusion on GnRH-stimulated LH secretion in women with PCOS (74), suggesting that LH hypersecretion in PCOS may be mediated by factors other than insulin. Although it is possible that chronic hyperinsulinemia is required for the manifestation of pituitary LH hypersecretion in PCOS, one cannot rule out the possibility that diminished LH secretion after treatment with insulin-sensitizing agents may be secondary to ovulatory outcomes and luteal progesterone production.
Although we did not observe beneficial effects of F on GnRH-stimulated LH secretion in prenatal T-treated sheep, F treatment has been shown to restore the sensitivity of the neuroendocrine axis to the inhibitory effects of gonadal steroids on LH secretion in women with PCOS (5). These effects appear to be mediated largely at the hypothalamic level, since changes in LH secretion after F treatment are associated with a reduction in LH pulse frequency in PCOS subjects (5). Our earlier finding that postnatal F treatment prevents the advancement of puberty induced by prenatal T treatment in female sheep (75) suggests that F may restore the sensitivity to the inhibitory effects of estradiol on LH secretion. Thus, in conjunction with previous findings, the present observation that postnatal F treatment failed to normalize the GnRH-stimulated LH secretion indicates that the effects of F on neuroendocrine function in prenatal T-treated sheep are likely mediated primarily at the brain level.
For obvious reasons, tissue-specific alterations that may be associated with LH hypersecretion in women with PCOS are difficult to be determined. Although extrapolation of findings in animal models to human pathology should be done carefully, our results in prenatal T-treated sheep, an animal model that recapitulates the neuroendocrine phenotype of PCOS (14, 76), suggest that changes in insulin and estrogen actions in the pituitary may contribute to the LH hypersecretion in PCOS.
Acknowledgments
We thank Mr Douglas Doop and Gary McCalla for their valuable assistance in breeding, lambing, and careful animal care; Dr Almudena Veiga-Lopez, Dr Bachir Abi Salloum, Mr Evan Beckett, and Mrs Carol Herkimer for the help provided with administration of treatments and radioimmunoassays; and the Undergraduate Research Opportunity Program (University of Michigan) students, Nohal Mekkaoui and Jessica Peck, for their assistance with immunohistochemistry procedures and image analysis.
This work was supported by the National Institutes of Health Grant P01 HD44232 (to V.P.) and by a Lalor Foundation postdoctoral fellowship (R.C.C.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Akt
- Protein kinase B
- AR
- androgen receptor
- DAPI
- 4′,6-diamidino-2-phenylindole
- ER
- estrogen receptor
- F
- flutamide
- GAPDH
- Glyceraldehyde 3-phosphate dehydrogenase
- GD
- gestational day
- GnRH-R
- GnRH receptor
- HSD
- honest significant difference
- IR
- insulin receptor
- mTOR
- mechanistic target of rapamycin
- NIDDK
- National Institute of Diabetes and Kidney Diseases
- p
- phospho
- PCOS
- polycystic ovary syndrome
- PTEN
- Phosphatase and tensin homolog
- R
- rosiglitazone
- T
- testosterone.
References
- 1. Homburg R. Polycystic ovary syndrome - from gynaecological curiosity to multisystem endocrinopathy. Hum Reprod. 1996;11(1):29–39. [DOI] [PubMed] [Google Scholar]
- 2. Kousta E, White DM, Cela E, McCarthy MI, Franks S. The prevalence of polycystic ovaries in women with infertility. Hum Reprod. 1999;14(11):2720–2723. [DOI] [PubMed] [Google Scholar]
- 3. Balen A, Michelmore K. What is polycystic ovary syndrome? Are national views important? Hum Reprod. 2002;17(9):2219–2227. [DOI] [PubMed] [Google Scholar]
- 4. Dumesic DA, Oberfield SE, Stener-Victorin E, Marshall JC, Laven JS, Legro RS. Scientific statement on the diagnostic criteria, epidemiology, pathophysiology, and molecular genetics of polycystic ovary syndrome. Endocr Rev. 2015;36(5):487–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eagleson CA, Gingrich MB, Pastor CL, et al. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone 1. J Clin Endocrinol Metab. 2000;85(11):4047–4052. [DOI] [PubMed] [Google Scholar]
- 6. Christman GM, Randolph JF, Kelch RP, Marshall JC. Reduction of gonadotropin-releasing hormone pulse frequency is associated with subsequent selective follicle-stimulating hormone secretion in women with polycystic ovarian Disease. J Clin Endocrinol Metab. 1991;72(6):1278–1285. [DOI] [PubMed] [Google Scholar]
- 7. Rebar R, Judd HL, Yen SS, Rakoff J, Vandenberg G, Naftolin F. Characterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest. 1976;57(5):1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patel K, Coffler MS, Dahan MH, Malcom PJ, Deutsch R, Chang RJ. Relationship of GnRH-stimulated LH release to episodic LH secretion and baseline endocrine-metabolic measures in women with polycystic ovary syndrome. Clin Endocrinol. 2004;60:67–74. [DOI] [PubMed] [Google Scholar]
- 9. Dumesic DA, Abbott DH, Padmanabhan V. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord. 2007;8(2):127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davies MJ, Norman RJ. Programming and reproductive functioning. Trends Endocrinol Metab. 2002;13(9):386–392. [DOI] [PubMed] [Google Scholar]
- 11. Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79(5):1328–1333. [DOI] [PubMed] [Google Scholar]
- 12. Abbott DH, Dumesic DA, Franks S. Developmental origin of polycystic ovary syndrome - a hypothesis. J Endocrinol. 2002;174(1):1–5. [DOI] [PubMed] [Google Scholar]
- 13. Padmanabhan V, Veiga-Lopez A. Animal models of the polycystic ovary syndrome phenotype. Steroids. 2013;78(8):734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Padmanabhan V, Veiga-Lopez A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol. 2013;373(1–2):8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Birch RA, Padmanabhan V, Foster DL, Unsworth WP, Robinson JE. Prenatal programming of reproductive neuroendocrine function: fetal androgen exposure produces progressive disruption of reproductive cycles in sheep. Endocrinology. 2003;144(4):1426–1434. [DOI] [PubMed] [Google Scholar]
- 16. Veiga-Lopez A, Ye W, Phillips DJ, Herkimer C, Knight PG, Padmanabhan V. Developmental programming: deficits in reproductive hormone dynamics and ovulatory outcomes in prenatal, testosterone-treated sheep. Biol Reprod. 2008;78(4):636–647. [DOI] [PubMed] [Google Scholar]
- 17. Robinson JE, Birch RA, Foster DL, Padmanabhan V. Prenatal exposure of the ovine fetus to androgens sexually differentiates the steroid feedback mechanisms that control gonadotropin releasing hormone secretion and disrupts ovarian cycles. Arch Sex Behav. 2002;31(1):35–41. [DOI] [PubMed] [Google Scholar]
- 18. Sarma HN, Manikkam M, Herkimer C, et al. Fetal programming: excess prenatal testosterone reduces postnatal luteinizing hormone, but not follicle-stimulating hormone responsiveness, to estradiol negative feedback in the female. Endocrinology. 2005;146(10):4281–4291. [DOI] [PubMed] [Google Scholar]
- 19. Savabieasfahani M, Lee JS, Herkimer C, Sharma TP, Foster DL, Padmanabhan V. Fetal programming: testosterone exposure of the female sheep during midgestation disrupts the dynamics of its adult gonadotropin secretion during the periovulatory period. Biol Reprod. 2005;72(1):221–229. [DOI] [PubMed] [Google Scholar]
- 20. Manikkam M, Thompson RC, Herkimer C, et al. Developmental programming: impact of prenatal testosterone excess on pre-and postnatal gonadotropin regulation in sheep. Biol Reprod. 2008;78(4):648–660. [DOI] [PubMed] [Google Scholar]
- 21. Cheng G, Coolen LM, Padmanabhan V, Goodman RL, Lehman MN. The kisspeptin/neurokinin B/dynorphin (KNDy) cell population of the arcuate nucleus: sex differences and effects of prenatal testosterone in sheep. Endocrinology. 2010;151(1):301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sheppard KM, Padmanabhan V, Coolen LM, Lehman MN. Prenatal programming by testosterone of hypothalamic metabolic control neurones in the ewe. J Neuroendocrinol. 2011;23(5):401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cernea M, Padmanabhan V, Goodman RL, Coolen LM, Lehman MN. Prenatal testosterone treatment leads to changes in the morphology of KNDy neurons, their inputs, and projections to GnRH cells in female sheep. Endocrinology. 2015;156(9):3277–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jansen HT, Hershey J, Mytinger A, Foster DL, Padmanabhan V. Developmental programming: reproductive endocrinopathies in the adult female sheep after prenatal testosterone treatment are reflected in altered ontogeny of GnRH afferents. Endocrinology. 2011;152(11):4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abi Salloum B, Veiga-Lopez A, Abbott DH, Burant CF, Padmanabhan V. Developmental programming: exposure to testosterone excess disrupts steroidal and metabolic environment in pregnant sheep. Endocrinology. 2015;156(6):2323–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu C, Cardoso RC, Puttabyatappa M, Padmanabhan V. Developmental programming: prenatal testosterone excess and insulin signaling disruptions in female sheep. Biol Reprod. 2016;94(5):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brooks A, Hagan D, Sheng C, McNeilly A, Sweeney T. Prenatal gonadotrophins in the sheep. Anim Reprod Sci. 1996;42:471–481. [Google Scholar]
- 28. Dubois P. [Ontogenesis of gonadotropic cells]. Ann Endocrinol (Paris). 1989;47–53. [PubMed] [Google Scholar]
- 29. Mollard P, Hodson DJ, Lafont C, Rizzoti K, Drouin J. A tridimensional view of pituitary development and function. Trends Endocrinol Metab. 2012;23(6):261–269. [DOI] [PubMed] [Google Scholar]
- 30. Wu YJ, Chen DW, Liu JL, Zhang JH, Luo HS, Cui S. Estradiol promotes pituitary cell proliferation and gonadotroph differentiation at different doses and with different mechanisms in chick embryo. Steroids. 2009;74(4–5):441–448. [DOI] [PubMed] [Google Scholar]
- 31. Burks DJ, Font de Mora J, Schubert M, et al. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature. 2000;407(6802):377–382. [DOI] [PubMed] [Google Scholar]
- 32. Buggs C, Weinberg F, Kim E, Wolfe A, Radovick S, Wondisford F. Insulin augments GnRH-stimulated LHβ gene expression by Egr-1. Mol Cell Endocrinol. 2006;249(1–2):99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Soldani R, Cagnacci A, Yen SS. Insulin, insulin-like growth factor I (IGF-I) and IGF-II enhance basal and gonadotrophin-releasing hormone-stimulated luteinizing hormone release from rat anterior pituitary cells in vitro. Eur J Endocrinol. 1994;131(6):641–645. [DOI] [PubMed] [Google Scholar]
- 34. Brothers KJ, Wu S, DiVall SA, et al. Rescue of obesity-induced infertility in female mice due to a pituitary-specific knockout of the insulin receptor. Cell Metab. 2010;12(3):295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yasin M, Dalkin AC, Haisenleder DJ, Marshall JC. Testosterone is required for gonadotropin-releasing hormone stimulation of luteinizing hormone-β messenger ribonucleic acid expression in female rats. Endocrinology. 1996;137(4):1265–1271. [DOI] [PubMed] [Google Scholar]
- 36. Wu S, Chen Y, Fajobi T, et al. Conditional knockout of the androgen receptor in gonadotropes reveals crucial roles for androgen in gonadotropin synthesis and surge in female mice. Mol Endocrinol. 2014;28(10):1670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Veiga-Lopez A, Lee JS, Padmanabhan V. Developmental programming: insulin sensitizer treatment improves reproductive function in prenatal testosterone-treated female sheep. Endocrinology. 2010;151(8):4007–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cardoso RC, Veiga-Lopez A, Moeller J, et al. Developmental programming: impact of gestational steroid and metabolic milieus on adiposity and insulin sensitivity in prenatal testosterone-treated female sheep. Endocrinology. 2015;157(2):522–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Padmanabhan V, Veiga-Lopez A, Abbott DH, Recabarren SE, Herkimer C. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology. 2010;151(2):595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Manikkam M, Crespi EJ, Doop DD, et al. Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology. 2004;145(2):790–798. [DOI] [PubMed] [Google Scholar]
- 41. Jackson LM, Timmer KM, Foster DL. Sexual differentiation of the external genitalia and the timing of puberty in the presence of an antiandrogen in sheep. Endocrinology. 2008;149(8):4200–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Legro RS, Zaino RJ, Demers LM, et al. The effects of metformin and rosiglitazone, alone and in combination, on the ovary and endometrium in polycystic ovary syndrome. Am J Obstet Gynecol 2007;196:402.e10–e11. [DOI] [PubMed] [Google Scholar]
- 43. Roy KK, Baruah J, Sharma A, et al. A prospective randomized trial comparing the clinical and endocrinological outcome with rosiglitazone versus laparoscopic ovarian drilling in patients with polycystic ovarian disease resistant to ovulation induction with clomiphene citrate. Arch Gynecol Obstet. 2010;281(5):939–944. [DOI] [PubMed] [Google Scholar]
- 44. Wood RI, Ebling FJ, I'Anson H, Bucholtz DC, Yellon SM, Foster DL. Prenatal androgens time neuroendocrine sexual maturation. Endocrinology. 1991;128(5):2457–2468. [DOI] [PubMed] [Google Scholar]
- 45. Evans NP, Dahl GE, Mauger DT, Padmanabhan V, Thrun LA, Karsch FJ. Does estradiol induce the preovulatory gonadotropin-releasing hormone (GnRH) surge in the ewe by inducing a progressive change in the mode of operation of the GnRH neurosecretory system. Endocrinology. 1995;136(12):5511–5519. [DOI] [PubMed] [Google Scholar]
- 46. Niswender GD, Reichert LE, Jr, Midgley AR, Jr, Nalbandov AV. Radioimmunoassay for bovine and ovine luteinizing hormone. Endocrinology. 1969;84(5):1166–1173. [DOI] [PubMed] [Google Scholar]
- 47. Robinson JE, Hastie PM, Shah A, Smith A, Evans NP. Developmental programming: prenatal androgen exposure alters the gonadotroph population of the ovine pituitary gland. J Neuroendocrinol. 2012;24(3):434–442. [DOI] [PubMed] [Google Scholar]
- 48. Cardoso RC, Alves BR, Sharpton SM, Williams GL, Amstalden M. Nutritional programming of accelerated puberty in heifers: involvement of pro-opiomelanocortin neurones in the arcuate nucleus. J Neuroendocrinol. 2015;27(8):647–657. [DOI] [PubMed] [Google Scholar]
- 49. Ozes ON, Akca H, Mayo LD, et al. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci USA. 2001;98(8):4640–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wijesekara N, Konrad D, Eweida M, et al. Muscle-specific Pten deletion protects against insulin resistance and diabetes. Mol Cell Biol. 2005;25(3):1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Scully KM, Gleiberman AS, Lindzey J, Lubahn DB, Korach KS, Rosenfeld MG. Role of estrogen receptor-α in the anterior pituitary gland. Mol Endocrinol. 1997;11(6):674–681. [DOI] [PubMed] [Google Scholar]
- 52. Keri RA, Wolfe MW, Saunders TL, et al. The proximal promoter of the bovine luteinizing hormone β-subunit gene confers gonadotrope-specific expression and regulation by gonadotropin-releasing hormone, testosterone, and 17 β-estradiol in transgenic mice. Mol Endocrinol. 1994;8(12):1807–1816. [DOI] [PubMed] [Google Scholar]
- 53. Clarke I. Multifarious effects of estrogen on the pituitary gonadotrope with special emphasis on studies in the ovine species. Arch Physiol Biochem. 2002;110(1–2):62–73. [DOI] [PubMed] [Google Scholar]
- 54. Tang WY, Newbold R, Mardilovich K, et al. Persistent hypomethylation in the promoter of nucleosomal binding protein 1 (Nsbp1) correlates with overexpression of Nsbp1 in mouse uteri neonatally exposed to diethylstilbestrol or genistein. Endocrinology. 2008;149(12):5922–5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bayer TA, Falkai P, Maier W. Genetic and non-genetic vulnerability factors in schizophrenia: the basis of the “two hit hypothesis.” J Psychiatr Res. 1999;33(6):543–548. [DOI] [PubMed] [Google Scholar]
- 56. Puttabyatappa M, Cardoso RC, Padmanabhan V. Effect of maternal PCOS and PCOS-like phenotype on the offspring's health. Mol Cell Endocrinol. 2016;435:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bell MR, Hart BG, Gore AC. Two-hit exposure to polychlorinated biphenyls at gestational and juvenile life stages: 2. Sex-specific neuromolecular effects in the brain. Mol Cell Endocrinol. 2016;420:125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Padmanabhan V, Cardoso RC, Puttabyatappa M. Developmental programming, a pathway to disease. Endocrinology. 2016;157(4):1328–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Adashi EY, Hsueh AJ, Yen SS. Insulin enhancement of luteinizing hormone and follicle-stimulating hormone release by cultured pituitary cells. Endocrinology. 1981;108(4):1441–1449. [DOI] [PubMed] [Google Scholar]
- 60. Bliss SP, Navratil AM, Breed M, Skinner DC, Clay CM, Roberson MS. Signaling complexes associated with the type I gonadotropin-releasing hormone (GnRH) receptor: colocalization of extracellularly regulated kinase 2 and GnRH receptor within membrane rafts. Mol Endocrinol. 2007;21(2):538–549. [DOI] [PubMed] [Google Scholar]
- 61. Albertson AJ, Navratil A, Mignot M, Dufourny L, Cherrington B, Skinner DC. Immunoreactive GnRH type I receptors in the mouse and sheep brain. J Chem Neuroanat. 2008;35(4):326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shaw ND, Histed SN, Srouji SS, Yang J, Lee H, Hall JE. Estrogen negative feedback on gonadotropin secretion: evidence for a direct pituitary effect in women. J Clin Endocrinol Metab. 2010;95(4):1955–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nakai Y, Plant TM, Hess DL, Keogh EJ, Knobil E. On the sites of the negative and positive feedback actions of estradiol in the control of gonadotropin secretion in the rhesus monkey. Endocrinology. 1978;102(4):1008–1014. [DOI] [PubMed] [Google Scholar]
- 64. Clarke IJ, Cummins JT. Direct pituitary effects of estrogen and progesterone on gonadotropin secretion in the ovariectomized ewe. Neuroendocrinology. 1984;39(3):267–274. [DOI] [PubMed] [Google Scholar]
- 65. Clarke IJ, Cummins JT, Crowder ME, Nett TM. Long-term negative feedback effects of oestrogen and progesterone on the pituitary gland of the long-term ovariectomized ewe. J Endocrinol. 1989;120(2):207–214. [DOI] [PubMed] [Google Scholar]
- 66. Berga SL, Guzick DS, Winters SJ. Increased luteinizing hormone and α-subunit secretion in women with hyperandrogenic anovulation. J Clin Endocrinol Metab. 1993;77(4):895–901. [DOI] [PubMed] [Google Scholar]
- 67. Yen SS, Laughlin GA, Morales AJ. Interface between extra- and intraovarian factors in polycystic ovarian syndrome. Ann NY Acad Sci. 1993;687:98–111. [DOI] [PubMed] [Google Scholar]
- 68. Belli SH, Graffigna MN, Oneto A, Otero P, Schurman L, Levalle OA. Effect of rosiglitazone on insulin resistance, growth factors, and reproductive disturbances in women with polycystic ovary syndrome. Fertil Steril. 2004;81(3):624–629. [DOI] [PubMed] [Google Scholar]
- 69. Ghazeeri G, Kutteh WH, Bryer-Ash M, Haas D, Ke RW. Effect of rosiglitazone on spontaneous and clomiphene citrate-induced ovulation in women with polycystic ovary syndrome. Fertil Steril. 2003;79(3):562–566. [DOI] [PubMed] [Google Scholar]
- 70. Dunaif A, Scott D, Finegood D, Quintana B, Whitcomb R. The insulin-sensitizing agent troglitazone improves metabolic and reproductive abnormalities in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1996;81(9):3299–3306. [DOI] [PubMed] [Google Scholar]
- 71. Nestler JE, Jakubowicz DJ. Decreases in ovarian cytochrome P450c17 α activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. N Engl J Med. 1996;335(9):617–623. [DOI] [PubMed] [Google Scholar]
- 72. Velázquez E, Acosta A, Mendoza SG. Menstrual cyclicity after metformin therapy in polycystic ovary syndrome. Obstet Gynecol. 1997;90(3):392–395. [DOI] [PubMed] [Google Scholar]
- 73. Ulloa-Aguirre A, Portocarrero L, Zariñán T, et al. Effects of metformin on inappropriate LH release in women with polycystic ovarian syndrome and insulin resistance. Reprod Biomed Online. 2006;12(6):669–683. [DOI] [PubMed] [Google Scholar]
- 74. Patel K, Coffler MS, Dahan MH, et al. Increased luteinizing hormone secretion in women with polycystic ovary syndrome is unaltered by prolonged insulin infusion. J Clin Endocrinol Metab. 2003;88(11):5456–5461. [DOI] [PubMed] [Google Scholar]
- 75. Padmanabhan V, Veiga-Lopez A, Herkimer C, et al. Developmental programming: prenatal and postnatal androgen antagonist and insulin sensitizer interventions prevent advancement of puberty and improve LH surge dynamics in prenatal testosterone-treated sheep. Endocrinology. 2015;156(7):2678–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cardoso RC, Puttabyatappa M, Padmanabhan V. Steroidogenic versus metabolic programming of reproductive neuroendocrine, ovarian and metabolic dysfunctions. Neuroendocrinology. 2015;102(3):226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]