

Abstract
We present a novel approach to detect potential cis-acting regulatory loci that combines the functional potential, an empirical DNase-seq based estimate of the allele-specificity of DNase-I hypersensitivity sites, with kernel-based variance component association analyses against expression phenotypes. To test our method we used public ENCODE whole genome DNase-I sequencing data, from a single sample, to estimate the functional potentials of the subset of 10,552 noncoding heterozygous single-nucleotide polymorphisms (SNPs) that were also present in the Genetic Analysis Workshop 19 (GAW19) family-based data set. We then built two covariance kernels, one nonweighted and one weighted by the functional potentials, and conducted kernel-based variance component association analyses against the 20,527 transcript expression phenotypes in the GAW19 family-based data set. We found signals of potential cis-regulatory effects, that surpassed the Bonferroni significance threshold, for ten transcripts. Stepwise removal of the cis-located SNPs from the weighted kernel lead to the disappearance of the association signal from our top transcript hit. We found compelling evidence of allele-specific cis-regulation for four transcripts using both kernels, and our results agree with previous research that suggests the involvement of specific cis-located variants in the regulation of their neighboring gene.
Background
Variation found in noncoding regions of the genome is much more abundant and, perhaps, even more relevant than coding variation for certain human traits, but its biological meaning is hard to assess [1]. It has been noticed that between 34 and 88 % of the disease-associated variants detected by genome-wide association studies (GWAS) appear to cluster in noncoding regions of the genome, specifically in DNase-I hypersensitivity sites (DHSs) [2], and that some of the DHSs exhibit allele-specificity [2–4]. Chromatin remodeling processes, for example those associated with the transcription machinery, create openings in the chromatin, which can be detected as DHSs, that allow transcription factors to interact with the underlying DNA. Hence DHSs tend to correlate with known cis-acting regulatory elements, such as promoters and transcription factor binding sites [5].
We have been investigating a systematic approach that uses DHSs to determine if noncoding single-nucleotide variation changes the local allele-specific chromatin accessibility, something that would indicate a potential regulatory role for a variant [6]. We have also developed a variance component based burden test to determine the contribution of localized relationship kernels to the trait variance [7, 8]. Here, we test if by combining both lines of research we could detect potential cis-acting regulatory loci. Our approach differs from previous works [4, 9] in that (a) we evaluate the association of each expression phenotype against a single covariance kernel, in a 1 degree of freedom test, and (b) we use an allele-specific chromatin accessibility measure to filter and weight the variants.
Methods
Data set
We used single-nucleotide polymorphism (SNP) dosages from 959 genotyped individuals, transcript expression levels from 647 of those individuals, and the genealogies (1389 individuals in 20 families) that were provided as part of the Genetic Analysis Workshop 19 (GAW19) family-based data set [10]. In addition, we used publicly available data from a CEU-CEPH (Northern Europeans from Utah–Centre d’Etude du Polymorphisme Humain) female’s peripheral blood mononucleated cells, NA12878, and its derived lymphoblastoid cell line, GM12878. The specific data used were: whole genome sequencing (WGS) genotypes for NA12878, from Illumina’s Platinum Genomes [11], and mapped short-sequencing reads (reads) from all five replicates of the DHSs sequencing (DNase-seq) of GM12878, from ENCODE [12], were used in this study. Physical coordinates and annotations for genes, transcripts, and marker loci refer to release 19 of the human genome (hg19) from the University of California, Santa Cruz (UCSC).
Reference panel of heterozygous single-nucleotide polymorphism loci
We compiled a reference panel of heterozygous SNP sites from the genotype calls from the high-coverage/high-quality WGS of NA12878. This independent genotypes source allowed us to analyze heterozygous loci where, because of either low coverage or complete allele-specific accessibility, only 1 allele is represented in the DNase-seq reads.
Chromatin accessibility measurement
We defined our chromatin accessibility measure to be equal to the DNase-seq read depth of each allele at a heterozygous locus. Based on our previous experience [6] the DNase-seq reads from all five GM12878 replicates were pooled to increase the total sequencing coverage at the DHSs. Samtools [13] mpileup was then used to obtain genotype calls only for loci in the known NA12878 heterozygous reference panel, and allele-specific read depths were obtained from the count of forward and reverse mapped reference and alternative allele annotations stored in the DP4 tag of the generated variant call format (VCF) file.
Functional potential
A departure from the expectation of an equal chromatin accessibility measurement of the two alleles at a locus within a DHS is what we refer to as the locus functional potential (FP). We implemented the FP statistic as a likelihood ratio–based test that contrasts the observed allele read depths with their expected depth at known heterozygous loci within DHSs [6]. A significant bias toward 1 allele in the chromatin accessibility measure of a locus can indicate a putative allele-specific chromatin remodeling event that compromised the footprint left by a DHS. We estimated the FP for all known NA12878 heterozygous loci that were present in the DNase-seq of GM12878.
Trait and covariates
To test our approach we used the real expression phenotypes from approximately 20,000 transcripts provided in the GAW19 family data set [10]. In addition, we simulated 10,000 heritable quantitative phenotypes not associated with any of the SNP loci in the data set, using Sequential Oligogenic Linkage Analysis Routines (SOLAR) [14], to evaluate the performance of our test under a null hypothesis.
We also used the sex, age, their interactions, and the smoking status at the first visit as covariates in all models. The first two principal components (PC1, PC2) (estimated as described in Peralta et al. [7] and Almeida et al. [8]), were added to account for any unknown population substructure that might be present.
Covariance kernels
GAW19 SNP dosages were collected for all heterozygous loci from NA12878 with a FP estimate. Non informative loci were removed. A standardized dosages matrix, Z, was built from them, and the covariance matrix of the dosages, R, was obtained from
The covariance matrix was then scaled so that all diagonal elements were equal to 1, and the resulting matrix, K, was our nonweighted covariance kernel.
We also built a covariance kernel in which each locus contribution was weighted by its FP estimate. Because our FP statistic is a likelihood ratio test, we used the relative − loglikelihood from a locus against the sum of all loci − loglikelihoods as the locus weight, and thus all weights add up to 1. The covariance kernel, K, was constructed as before, with 1 exception. The covariance matrix of the dosages was obtained from
where D w is a diagonal matrix of weights.
Variance component model
We used the variance component model previously described in Peralta et al. [7] and Almeida et al. [8], in conjunction with the nonweighted and FP-weighted covariance kernels derived from the SNP dosages described above, to estimate the proportion of the phenotypic variance, h 2geff, explained by allele-specific genetic variants found within DHSs in an unrelated CEU-CEPH individual. The h 2geff variance component, and its significance, was estimated for each real and simulated expression phenotype using SOLAR, a flexible genetic variance component analysis program with a focus on general pedigrees [14].
Results
Our reference panel of heterozygous loci contained the 2,423,308 heterozygous SNPs that had been found in the WGS of NA12878. Only heterozygous loci are informative for allele-specific chromatin accessibility in a genome. Although heterozygous SNP sites can be directly inferred from DNase-seq data, it is not ideal, in part because of its very low coverage.
We were able to measure the allele-specific chromatin accessibility and estimate the FP for 48,236 (1.99 %) of those heterozygous SNPs but only 10,618 (22 %) of them were present in the GAW19 dosages. Of the 10,618 heterozygous-in-NA12878 SNPs with a FP estimation that were present in GAW19, 66 (0.62 %) were monomorphic in the GAW19 dosages and were therefore discarded from further analysis. The remaining 10552 SNPs with FP estimates were used for the construction of our weighted and nonweighted covariance kernels.
We conducted our variance component analysis of 10,000 simulated phenotypes using the weighted covariance kernel only and found no inflation or deflation of the p values of the estimated effects (Fig. 1), indicating that our test performed as expected when evaluated under the null hypothesis. Figure 2 shows the frequency distribution of the weights.
Fig. 1.
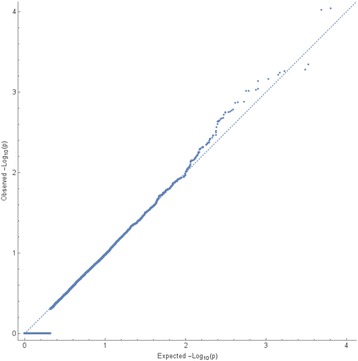
Quantile–quantile (Q-Q) plot of the p values obtained under a null hypothesis test. Analysis of 10,000 simulated phenotypes not associated with any of the GAW19 SNP loci. The obtained the p values follow the expected uniform distribution
Fig. 2.
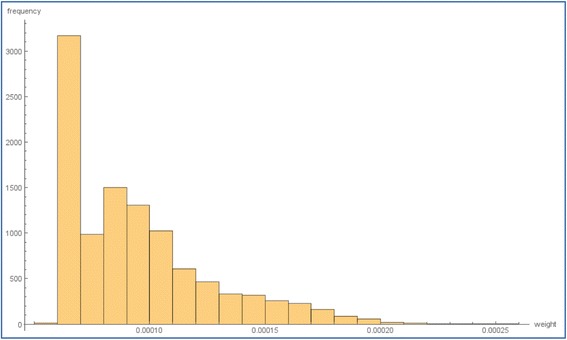
Frequency distribution of the weights used for variants in the weighted covariance kernel. Each weight represents the relative proportion of the functional potential − loglikelihood estimation of each variant in the kernel. The large proportion of variants in the first bin have a very small weight, and correspond to variants with a low confidence of having an allele-specific chromatin accessibility effect
We then analyzed the 20,527 transcript expression phenotypes in the GAW19 family data set using both the weighted and the nonweighted covariance kernels. After a genome-wide Bonferroni correction (−log10[α] = 5.6) we found significant evidence of potential cis-regulatory effects for ten transcripts (Table 1). Eight of the transcripts were detected by both covariance kernels but two of them, GI_4506738-S and GI_15451941-S, were only found to be significant when the weighted covariance kernel was used. In most of the cases, the use of the nonweighted covariance kernel tended to slightly decrease the proportion of the transcript expression variance explained by the kernel, which was on average very high in both cases (h 2geff,non − weighted = 0.6540, h 2geff, weighted = 0.7046). While most of the trait heritability was explained by the covariance kernel, a substantial amount (between 14 and 28 %) still remained. Table 2 lists these ten transcripts along with their annotations and closest SNPs in the covariance kernels. Table 3 shows how the signal from our top result, GI_42544126-I, decreases when SNPs within the transcript region are progressively removed from the kernel.
Table 1.
Transcripts for whom their variation in expression levels can be explained by a covariance kernel composed by SNP with FP estimates, at genome-wide significance
| Transcript | Covariance kernel | |||||||
|---|---|---|---|---|---|---|---|---|
| Non-weighted | Weighted | |||||||
| h2r | h2r_p | geff | geff_p | h2r | h2r_p | Geff | geff_p | |
| GI_42544126-I | 0.0000 | 0.5000 | 0.7074 | 4.03E-15 | 0.0000 | 0.5000 | 0.7145 | 4.55E-18 |
| GI_23097237-S | 0.0000 | 0.5000 | 0.7848 | 1.15E-14 | 0.0000 | 0.5000 | 0.7493 | 7.46E-12 |
| GI_10863968-S | 0.0000 | 0.5000 | 0.6109 | 5.68E-10 | 0.0000 | 0.5000 | 0.6122 | 9.05E-11 |
| Hs.283934-S | 0.0746 | 0.3138 | 0.8382 | 9.77E-10 | 0.1497 | 0.1443 | 0.7657 | 4.87E-09 |
| GI_12056480-A | 0.2357 | 0.0457 | 0.7069 | 1.69E-08 | 0.2792 | 0.0194 | 0.6628 | 5.43E-08 |
| GI_20986517-S | 0.0000 | 0.5000 | 0.7671 | 5.58E-08 | 0.0000 | 0.5000 | 0.7726 | 3.08E-08 |
| Hs.58104-S | 0.2230 | 0.0753 | 0.6886 | 6.89E-07 | 0.2705 | 0.0333 | 0.6415 | 8.47E-07 |
| GI_41393558-I | 0.0000 | 0.5000 | 0.5331 | 1.92E-06 | 0.0000 | 0.5000 | 0.5371 | 1.73E-06 |
| GI_4506738-S | NA | 0.0000 | 0.5000 | 0.4758 | 6.66E-07 | |||
| GI_15451941-S | NA | 0.2611 | 0.0441 | 0.6090 | 1.33E-06 | |||
geff, Gene-specific effect estimate (h 2 geff)
geff_p, significance of the gene-specific effect estimate
h2r, trait heritability estimate (h 2)
h2r_p, significance of the trait heritability estimate
Table 2.
Annotated transcript and SNP table
| Transcript | Gene | Chromosome | Start | Length | SNP | DBSnp rs | SNP annotation |
|---|---|---|---|---|---|---|---|
| GI_42544126-I | SF1 | chr11 | 64532075 | 14241 | 11_64511322 | rs2073798 | RASGRP2 intron |
| 11_64519345 | rs686171 | PYGM intron | |||||
| 11_64546106 | rs3741398 | SF1 2 kb upstream, nc transcript variant, 5’ UTR | |||||
| 11_64546257 | rs1633462 | SF1 2 kb upstream, nc transcript variant, 5’ UTR | |||||
| 11_64573589 | rs669976 | MEN1 intron | |||||
| 11_64576598 | rs67808744 | MEN1 intron | |||||
| 11_64577620 | rs7949944 | MEN1 5’ UTR, 2 kb upstream | |||||
| GI_23097237-S | CHST13 | chr3 | 126243130 | 19004 | 3_126218788 | rs6774768 | UROC1 intron |
| 3_126228953 | rs1873388 | UROC1 intron | |||||
| 3_126242964 | rs1388096 | CHST13 2 kb upstream | |||||
| 3_126245956 | rs4592980 | CHST13 intron/3’UTR | |||||
| 3_126246370 | rs1994642 | CHST13 intron/3’UTR | |||||
| 3_126247795 | rs11717719 | CHST13 intron | |||||
| 3_126247848 | rs11718493 | CHST13 intron | |||||
| GI_10863968-S | POLD4 | chr11 | 67119018 | 2034 | 11_67196237 | rs1476792 | |
| Hs.283934-S | TSPAN16 | chr19 | 11406815 | 30857 | 19_11340057 | rs17001244 | |
| 19_11358700 | rs4804579 | ||||||
| 19_11374675 | rs416231 | ||||||
| 19_11380295 | rs4804159 | ||||||
| 19_11406952 | rs374409 | ||||||
| GI_12056480-A | UTS2 | chr1 | 7907271 | 6280 | 1_7710810 | rs58905635 | CAMTA1 intron |
| 1_7725855 | rs4908471 | CAMTA1 intron | |||||
| 1_7749807 | rs3124797 | CAMTA1 intron | |||||
| GI_20986517-S | MAPK8IP1 | chr11 | 45907046 | 20970 | 11_45838926 | rs11038668 | |
| 11_45840939 | rs7112505 | ||||||
| 11_45891418 | rs7123390 | CRY2 intron | |||||
| Hs.58104-S | FAM101B | chr17 | 289771 | 8960 | 17_185027 | rs12951437 | |
| 17_198698 | rs11869174 | ||||||
| 17_206962 | rs11657163 | ||||||
| GI_41393558-I | KIF1B | chr1 | 10270763 | 97892 | 1_10270386 | rs3828081 | KIF1B 2 kb upstream |
| 1_10307453 | rs4240911 | KIF1B intron | |||||
| 1_10438687 | rs1536262 | KIF1B 3’UTR | |||||
| GI_4506738-S | RPS6KB2 | chr11 | 67195934 | 6945 | 11_67196237 | rs1476792 | RPS6KB2 intron |
| 11_67204342 | rs12787021 | PTPRCAP intron | |||||
| 11_67213956 | rs2109123 | ||||||
| 11_67253564 | rs7110021 | ||||||
| 11_67258805 | rs751567 | ||||||
| 11_67264679 | rs2276120 | ||||||
| GI_15451941-S | UBA52 | chr19 | 18682613 | 5657 | 19_18499151 | rs1059022 | |
| 19_18499238 | rs1804826 | ||||||
| 19_18715154 | rs72995445 | CRLF1 intron | |||||
| 19_18859680 | rs11085244 |
Gene symbols and coordinates for the ten transcripts that were detected as being potentially cis-regulated by SNPs in our covariance kernel. The closest SNPs to each gene are listed
Table 3.
Decrease in the association signal when cis-located SNPs are removed from the kernel
| Transcript | Gene | SNPs removed from the kernel | Covariance kernel | ||
|---|---|---|---|---|---|
| Weighted | |||||
| h2r | geff | geff_p | |||
| none | 0.0000 | 0.7145 | 4.55E-18 | ||
| GI_42544126-I | SF1 | 2 in SF1 | 0.0000 | 0.6809 | 1.32E-12 |
| all in transcript region | 0.1349 | 0.1349 | 2.00E-05 | ||
Discussion
The objective of this study was to investigate the prioritization of SNPs based on their potential as functional, cis-acting, regulatory elements. To that end we used a combined approach that integrates functional information, in the form of allele-specific chromatin accessibility measurements at DHSs, gene expression phenotypes, and a variance component model that estimates the proportion of a trait’s variance as a result of a localized relationship kernel.
We constructed nonweighted and weighted covariance kernels, using the 10,552 SNPs with an available FP estimate, and obtained the proportion of variance in the levels of transcript expression that could be explained by them in the family data set. We identified a clear signal for eight transcripts when using the nonweighted kernel, and for two additional transcripts when using the weighted kernel (see Table 1). In contrast, we found no signals when we performed our analysis using the set of 10,000 simulated phenotypes; an indication that our test statistic was not artificially inflated when evaluated under the null hypothesis (see Fig. 1).
Some of our results are difficult to interpret because of the distance between the transcript location and the closest SNPs to it in our kernels. For transcripts GI_12056480-A and GI_15451941-S our results might indicate the presence of long-acting cis-elements, but could also be the result of, for example, linkage disequilibrium with SNPs in closer proximity to the transcript.
However, close examination of the annotations of the significant transcripts in our results shows suggestive evidence of potential cis-acting variants. Particularly for the GI_23097237-S, GI_42544126-I, GI_4506738-S, and GI_41393558-I transcripts, corresponding to the CHST13, SF1, RP56KB2, and KIF1B genes, respectively. The SNPs with FP estimates that we incorporated in our covariance kernel near these genes are all located either within the gene or within the promoter region of the gene (see Table 2). The progressive removal of SNPs within and near the SF1 gene led to the degradation of the signal from the GI_42544126-I transcript (see Table 3), clearly suggesting a cis-acting effect of the variants in the transcript expression. Furthermore, previous research provides additional compelling evidence for the implication of rs11718493 in the allele-specific methylation of CpGs and the regulation of CHST13 [15, 16], a carbohydrate sulfotransferase that is present in the Golgi membrane [17], and rs1536262 has been reported to be a likely candidate for the regulation of KIF1B expression [18].
Conclusions
Our kernel-based variance component test was able to prioritize noncoding variation from whole-genome sequencing data based on their potential to regulate gene expression. An allele-specific chromatin accessibility measure was used as both a biologically meaningful filter for the selection of the variants and the weight of each variant in the covariance kernel. We observed compelling evidence to support the idea that four genes might be cis-regulated by the SNPs we identified in them.
Acknowledgements
Type 2 Diabetes Genetic Exploration by Next-generation sequencing in Ethnic Samples (T2D-GENES) is supported by National Institutes of Health grants: U01 DK085524, U01 DK085501, U01 DK085526, U01 DK085584, and U01 DK085545. The San Antonio Family Heart Study (SAFHS) is supported by P01 HL045222; the San Antonio Family Diabetes Study (SAFDS) is supported by R01 DK047482; the San Antonio Family Gallbladder Study (SAFGS) is supported by R01 DK053889. SOLAR is supported by National Institute of Mental Health grant MH059490, and the supercomputing facilities used for this work at the AT&T Genetics Computing Center were supported in part by a gift from the SBC Foundation.
Declarations
This article has been published as part of BMC Proceedings Volume 10 Supplement 7, 2016: Genetic Analysis Workshop 19: Sequence, Blood Pressure and Expression Data. Summary articles. The full contents of the supplement are available online at http://bmcproc.biomedcentral.com/articles/supplements/volume-10-supplement-7. Publication of the proceedings of Genetic Analysis Workshop 19 was supported by National Institutes of Health grant R01 GM031575.
Authors’ contributions
JB, LA, EM, and JMP conceived the overall study. JB, MA, and JMP developed the statistical analyses. JMP performed the analyses and drafted the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Juan Manuel Peralta, Email: juan.peralta@utrgv.edu.
Marcio Almeida, Email: marcio.almeida@utb.edu.
Lawrence J. Abraham, Email: lawrie.abraham@uwa.edu.au
Eric Moses, Email: eric.moses@uwa.edu.au.
John Blangero, Email: blangero@uthscsa.edu.
References
- 1.Ward LD, Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol. 2012;30(11):1095–106. doi: 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, et al. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol Syst Biol. 2011;7:522. doi: 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDaniell R, Lee BK, Song L, Liu Z, Boyle AP, Erdos MR, Scott LJ, Morken MA, Kucera KS, Battenhouse A, et al. Heritable individual-specific and allele-specific chromatin signatures in humans. Science. 2010;328(5975):235–9. doi: 10.1126/science.1184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell O, Tiwari VK, Thomä NH, Schübeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12(8):554–64. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- 6.Peralta JM, Blangero J, Abraham LJ, Moses E. A genome-wide assay for regulatory functional potential of sequence variants (Abstract/Program #1699F). Presented at the 63rd Annual Meeting of The American Society of Human Genetics. Boston, MA; 2013.
- 7.Peralta JM, Almeida M, Kent JW, Blangero J. A variance component-based gene burden test. BMC Proc. 2014;8(Suppl 1):S49. doi: 10.1186/1753-6561-8-S1-S49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almeida M, Peralta JM, Farook V, Puppala S, Kent JW, Jr, Duggirala R, Blangero J. Pedigree-based random effect tests to screen gene pathways. BMC Proc. 2014;8(Suppl 1):S100. doi: 10.1186/1753-6561-8-S1-S100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ge B, Pokholok DK, Kwan T, Grundberg E, Morcos L, Verlaan DJ, Le J, Koka V, Lam KC, Gagné V, et al. Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat Genet. 2009;41(11):1216–22. doi: 10.1038/ng.473. [DOI] [PubMed] [Google Scholar]
- 10.Blangero J, Teslovich TM, Sim X, Almeida MA, Jun G, Dyer TD, Johnson M, Peralta JM, Manning AK, Wood AR, et al. Omics squared: human genomic, transcriptomic, and phenotypic data for Genetic Analysis Workshop 19. BMC Proc. 2015;9 Suppl 8:S2. doi: 10.1186/s12919-016-0008-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Illumina. http://www.illumina.com/platinumgenomes/. Accessed 12 Sept 2016.
- 12.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet Hum Genet. 1998;62(5):1198–211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schalkwyk LC, et al. Allelic skewing of DNA methylation is widespread across the genome. Am J Hum Genet Hum Genet. 2010;86(2):196–212. doi: 10.1016/j.ajhg.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milani L, Lundmark A, Nordlund J, Kiialainen A, Flaegstad T, Jonmundsson G, Kanerva J, Schmiegelow K, Gunderson KL, Lönnerholm G, et al. Allele-specific gene expression patterns in primary leukemic cells reveal regulation of gene expression by CpG site methylation. Genome Res. 2009;19(1):1–11. doi: 10.1101/gr.083931.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, MD. MIM Number: 610124: 05/18/2006. http://omim.org/. Accessed 12 Sept 2016.
- 18.Ma H, Wang L-E, Zhensheng L, Sturgis EM, Wei Q. Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research. Orlando: AACR; 2011. Polymorphisms of PLCE1 and KIF1B and risk of squamous cell carcinoma of the head and neck. [Google Scholar]