

Abstract
Small cell lung cancer (SCLC) is the most aggressive type of lung cancer, with almost 95% of patients succumbing to the disease. Although RBM5, a tumor suppressor gene, is downregulated in the majority of lung cancers, its role in SCLC is unknown. Using the GLC20 SCLC cell line, which has a homozygous deletion encompassing the RBM5 gene locus, we established stable RBM5 expressing sublines and investigated the effects of RBM5 re-expression. Transcriptome and target identification studies determined that RBM5 directly regulates the cell cycle and apoptosis in SCLC cells, as well as significantly downregulates other important transformation-associated pathways such as angiogenesis and cell adhesion. RNA sequencing of paired non-tumor and tumor SCLC patient specimens showed decreased RBM5 expression in the tumors, and expression alterations in the majority of the same pathways that were altered in the GLC20 cells and sublines. Functional studies confirmed RBM5 expression slows SCLC cell line growth, and increases sensitivity to the chemotherapy drug cisplatin. Overall, our work demonstrates the importance of RBM5 expression to the non-transformed state of lung cells and the consequences of its deletion to SCLC development and progression.
Keywords: Cancer research, Cell biology
1. Introduction
According to the American Cancer Society, more people die from primary cancers of the lung than from any other type of cancer. The most aggressive type of lung cancer occurs in the “small cells”, found in the main bronchi (Travis et al., 2004). Small cell lung cancer (SCLC) almost exclusively occurs in people with a history of tobacco smoking (Jackman and Johnson, 2005; Travis et al., 2004).
Lung cancer initiation and progression are attributed at the molecular level to many factors, but arguably the most interesting is the loss of heterozygosity in a few regions throughout the short arm of chromosome three. Notably, allelic loss within the 3p21.3 region is evidenced even in pre-neoplastic tissue from smokers (Wistuba et al., 2000). An overlapping homozygous deletion within 3p21.3, noted in lung and breast tumors, harbors a number of tumor suppressor genes (Lerman and Minna, 2000). RNA Binding Motif 5 (RBM5), a putative lung cancer tumor suppressor gene (Sutherland et al., 2010), resides near the end of the telomeric deletion breakpoint that was noted in three SCLC cell lines (Lerman and Minna, 2000). In the majority of lung cancers (including SCLC and non-small cell lung cancer (NSCLC)), expression of RBM5 is downregulated but present (Oh et al., 2002). The cause of this downregulation is unknown, but does not appear to result from gene mutation or promoter hypermethylation (Oh et al., 2008; Oh et al., 2007), suggesting allelic loss may be responsible. The almost universal downregulation of RBM5 in all types of lung cancer does suggest it plays an important role in lung cancer initiation and/or progression. RBM5 was, in fact, identified as one of nine downregulated genes within a 17 gene signature associated with metastasis in various human solid tumors, including lung (Ramaswamy et al., 2003).
Previous functional work relating to RBM5 in a variety of cancer cell lines identified it as a modulator of the cell cycle and apoptosis, partially via its influence on alternative splicing (Bechara et al., 2013; Oh et al., 2002). In regards to lung cancer specifically, some functional work regarding RBM5 has been performed using a lung adenocarcinoma cell line (A549), which showed that increased RBM5 expression correlated with (a) G1 cell cycle arrest (Network, 2014; Shao et al., 2012), and (b) increased apoptosis (Oh et al., 2006; Shao et al., 2012). No functional work, however, has been undertaken for RBM5 in SCLC. This project set out to determine the importance of RBM5 in SCLC, in order to better understand the consequences of its downregulation to the development and progression of this disease. Furthermore, since SCLC is the most aggressive type of lung cancer, with 95% of patients eventually succumbing to the disease (Govindan et al., 2006), it is clear that a better understanding of this disease, as well as more effective treatment options, are required.
GLC20 is a SCLC cell line derived from small cells within a lung tumor biopsy (Smit et al., 1992). The cells have two 3p21 homozygous deletions, one of which includes RBM5, making the cells an attractive model in which to study the functional consequences of RBM5 re-expression (Angeloni, 2007; Kok et al., 1994). We established two RBM5 expressing populations, with different levels of RBM5, and conducted transcriptome analyses to identify the pathways affected by altering the levels of RBM5. Target identification experiments were carried out to determine which of these pathways were directly affected by RBM5. To validate our findings, we (a) compared our in vitro transcriptomic results to transcriptomic data from two paired non-tumor/tumor patient specimens with a 50% downregulation of RBM5 expression, and (b) experimentally examined the effects of no versus low versus high RBM5 expression on cell proliferation and apoptosis. Our results suggest that RBM5 is a key SCLC suppressor and guardian of the non-transformed phenotype.
2. Results & discussion
2.1. Establishment of a GLC20 model for SCLC studies relating to RBM5
GLC20 cells are RBM5-null and were established from a tumor that was multidrug resistant (doxorubicin, cyclophosphamide and vincristine), a common characteristic of advanced SCLC. As depicted in Fig. 1A, the breakpoint for the GLC20 deletion is upstream of RBM5, and the entire RBM5 gene is deleted (Lerman and Minna, 2000). We verified the absence of RBM5 DNA, RNA and protein (Fig. 1B–E).
Fig. 1.

Characterization of wildtype GLC20 cells and RBM5 expressing sublines. (A) Cartoon of location of deletion breakpoints in various lung cell lines. (B) Genomic DNA PCR results from different cell lines. (C) RBM5 Southern Blot. (D) RT-PCR results from different cell lines. (E) Western Blot. (F) RBM5 expression in GLC20 stable transfectants by RT-PCR and Western Blot. (G) Cartoon of 5′ end of RBM5 gene, not drawn to scale, showing approximate locations of various probes. Box marked “W”: Western antibody LUCA-15 UK; box marked “S”: Southern probe; RT-PCR primers LU15(2) and LU15(3) (black thin open arrowheads); genomic PCR primers Gen1E2Fc and Gen2E3I2R (red thick arrows). See Figure S1 for full gel of B, D and F, and full blot of E and F.
To better understand the impact of RBM5 downregulation on SCLC, stable populations of RBM5 expressing cells were established. Two RBM5-expressing sublines were established, one clonal (designated GLC20.C4 or “C4”) and one from a pooled population of RBM5-expressing transfectants (designated GLC20.T2 or “T2”) (Fig. 1F). We also established an empty vector control subline (GLC20.pcDNA3 or “pcDNA3”).
To ensure that a GLC20 model would be useful for functional studies relating to RBM5, chiefly known as a modulator of apoptosis, we examined their cell death profile when exposed to various apoptogenic agents. The highly resistant nature of GLC20 cells to chemotherapy was demonstrated (Fig. 2), since cisplatin exposure, without etoposide, required a concentration of 80 μg/ml (226 μM) to be somewhat effective at promoting apoptosis by 24 h post exposure (Fig. 2A, lanes 1–3). GLC20 cells are, however, capable of undergoing apoptosis, since PARP cleavage was observed 24 h following co-administration of 10 μg/ml (33 μM) cisplatin and 10 or 20 μg/ml (17 or 34 μM) etoposide (Fig. 2A, lanes 4–9), and almost complete following 40 to 48 h incubation with these agents (Fig. 2B lanes 4–9). Thus, although these cells are highly drug resistant, they are capable of experiencing cisplatin-mediated apoptosis.
Fig. 2.

Apoptosis induction by various apoptogenic stimuli. GLC20 cells were treated with cisplatin with or without etoposide, for the various times indicated. Details in Materials & methods. See Figure S2 for full blots.
2.2. RNA-Seq shows that over 12% of the transcriptome is differentially expressed by RBM5 in GLC20 cells, SLC25A53 is the most altered gene, and pathways relating to cancer are the most likely impacted
To investigate if and how RBM5 expression influences GLC20 cells, deep sequencing of the transcriptome (RNA-Seq) of the parental GLC20 cells and three sublines was carried out. See Materials & methods for an explanation of the “control” used in sequencing analyses.
Firstly, we confirmed RBM5 expression levels within the transcriptomic data for T2 and C4 (Fig. 3A). Differential expression testing (refer to Materials & methods) identified that 12.5% of the transcriptome examined was significantly differentially expressed between control and T2, and 18.4% between control and C4 (Fig. 3B). Furthermore, over 50% of the genes that were differentially expressed in T2 were also differentially expressed in C4, suggesting that any effect in C4 is not likely the result of a clonal effect related to subclone establishment. Solute Carrier Family 25 Member 53 (SLC25A53) was the most significantly differentially expressed gene in both T2 and C4 compared to control (based on log2 (fold-change); 1112.1 FPKM (Fragments Per Kilobase of transcript per Million mapped reads) to 0.068 FPKM in control vs. C4, and 1127.3 FPKM to 0.073 FPKM in control vs. T2). This gene is part of a large family of transporters that control various cellular functions, although a particular role for SLC25A53 has yet to be identified (Palmieri, 2013). Other highly downregulated genes common to both T2 and C4 include CD9, Discs Large Homolog Associated Protein 1 (DLGAP1), and Feline Sarcoma (FES). Interestingly, CD9 and FES have previously been associated with cell adhesion and tumor metastasis, with the effect of their expression seeming to be cell type-specific (Delfino et al., 2006; Huan et al., 2015; Kanda et al., 2009; Rappa et al., 2015). Of the two, only CD9 has been investigated in regards to SCLC specifically, and low expression levels were linked to increased motility and invasiveness (Funakoshi et al., 2003). The fact that RBM5 expression decreased CD9 expression was thus unexpected, as we hypothesized that RBM5 would promote a non-transformed state in SCLC cells. However, CD9 gene expression levels in our control were much lower than those in our clinical SCLC tumor samples (described below) (average of 6.6 FPKM in our cell line control and 109.1 FPKM in tumor samples), suggesting that although RBM5’s influence on CD9 may be statistically significant, it may not be physiologically relevant in our system.
Fig. 3.

Transcriptome analysis of GLC20 sublines. (A) RBM5 expression in control, T2 and C4 samples as determined by RNA-Seq. **p < 0.01. (B) Venn diagram demonstrating significantly differentially expressed genes between T2 and C4 compared to control, respectively, as determined by RNA-Seq. Number of genes in each group indicated in parenthesis. (C and D) FIDEA pathway analysis results for altered KEGG pathways in control vs T2 (C) and C4 (D) samples, respectively, from RNA-Seq transcriptome data. (E) GSAASeqSP blue-pink o’gram representing the expression levels of core enriched genes within the MSigDB Angiogensis Hallmark gene set in control and C4 (RNA-Seq data). Blue indicates low expression, whereas red indicates high expression levels. Genes listed in order of Rank Metric Score. (F) KRAS and SOX2 expression in control and C4 samples as determined by RNA-Seq.
On the other hand, two genes other than RBM5 were highly upregulated in both T2 and C4 compared to control; MKRN3 and ZNF85. Of note, the former has been previously associated with development, particularly the onset of puberty, which is in line with the pathway results presented below (Abreu et al., 2013).
Using two different pathway analysis programs, we went on to determine the functional implications of genes differentially expressed upon expression of RBM5. First, the Functional Interpretation of Differential Expression Analysis (FIDEA) (D'Andrea et al., 2013) program with the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (Kanehisa and Goto, 2000; Kanehisa et al., 2014) was used, thus differentially expressed genes are placed in known signaling pathways to determine if these pathways are significantly up- or downregulated. Analysis results are presented in Fig. 3C and D. Pathways in cancer was significantly downregulated in both T2 and C4 compared to control. Other transformation-related pathways were also downregulated upon RBM5 expression in T2 such as the cell cycle, small cell lung cancer and pancreatic cancer. The fact that these pathways were not also significantly changed in C4 may be due to the larger number of differentially expressed genes in this sample, which may mask the effect of RBM5 on these particular cancer-related pathways. In addition, both axon guidance and alcoholism were significantly altered in both T2 and C4. This is very interesting since axon guidance, as well as alcoholism, have been shown to play a substantial role in cancer development and progression (Chedotal et al., 2005; Forsyth et al., 2010).
To complement our KEGG pathway results, we used an additional pathway analysis program − the Gene Set Association Analysis for RNA-Seq with Sample Permutation (GSAASeqSP) program (Xiong et al., 2014) - as it takes into account inherent bias present in RNA-Seq data. In combination with the Broad Institute’s Molecular Signatures Database (MSigDB) Hallmark gene set (Liberzon et al., 2015), genes are grouped based on established biological processes, and enriched gene sets determined. We identified 12 and 17 gene sets with false discovery rates (FDR) at or below 10% in T2 and C4, respectively, compared to control. Over half of these T2 altered gene sets were common to C4, once again suggesting that any effect seen in C4 is not likely due to a clonal effect related to subclone establishment. Gene sets with FDR at or below 10% in control vs. C4 are presented in Table 1, along with their FDR in control vs. T2. Interestingly, many differentially expressed gene sets are important to development, which validates our experimental findings, as RBM5 was recently shown to be involved in myogenesis, spermatogenesis and neuronal development (Fushimi et al., 2008; Loiselle and Sutherland, 2014; O'Bryan et al., 2013). Furthermore, apoptosis and TNFα signaling, both pathways RBM5 has been previously associated with in Jurkat T lymphoblastoid cells (Rintala-Maki and Sutherland, 2004; Sutherland et al., 2000) and MCF-7 cells (Rintala-Maki et al., 2004) were altered in T2 and C4. Fig. 4 lists the core enriched genes in these gene sets and how their expression changed upon RBM5 expression in C4, highlighting the importance of RBM5 to these cellular functions.
Table 1.
Altered gene sets with FDR below 10% between control and C4 samples, as determined by GSAASeqSP analysis with the MSigDB Hallmark gene set using the samples’ RNA-Seq results. Ranked based on FDR value in control vs. C4 analysis. FDR value in control vs. T2 samples is also indicated for the given pathways. Italics indicates FDR values above 10%.
| Control vs. C4 |
Control vs. T2 |
|
|---|---|---|
| Gene Set | FDR | FDR |
| Hedgehog signaling | 0.000 | 0.033 |
| Angiogenesis | 0.000 | 0.067 |
| Apoptosis | 0.054 | 0.28 |
| Androgen response | 0.058 | 0.067 |
| Myogenesis | 0.064 | 0.183 |
| Estrogen response late | 0.064 | 0.033 |
| TNFα signaling via NF-κB | 0.070 | 0.075 |
| Coagulation | 0.071 | 0.115 |
| IL2 STAT5 signaling | 0.075 | 0.125 |
| UV response down | 0.076 | 0.226 |
| Apical surface | 0.078 | 1.000 |
| Cholesterol homeostasis | 0.080 | 0.113 |
| IL6 JAK STAT3 signaling | 0.083 | 0.147 |
| Estrogen response early | 0.087 | 0.057 |
| Inflammatory response | 0.094 | 0.432 |
| KRAS signaling up | 0.100 | 0.114 |
| Epithelial to mesenchymal transition | 0.100 | 0.073 |
Fig. 4.
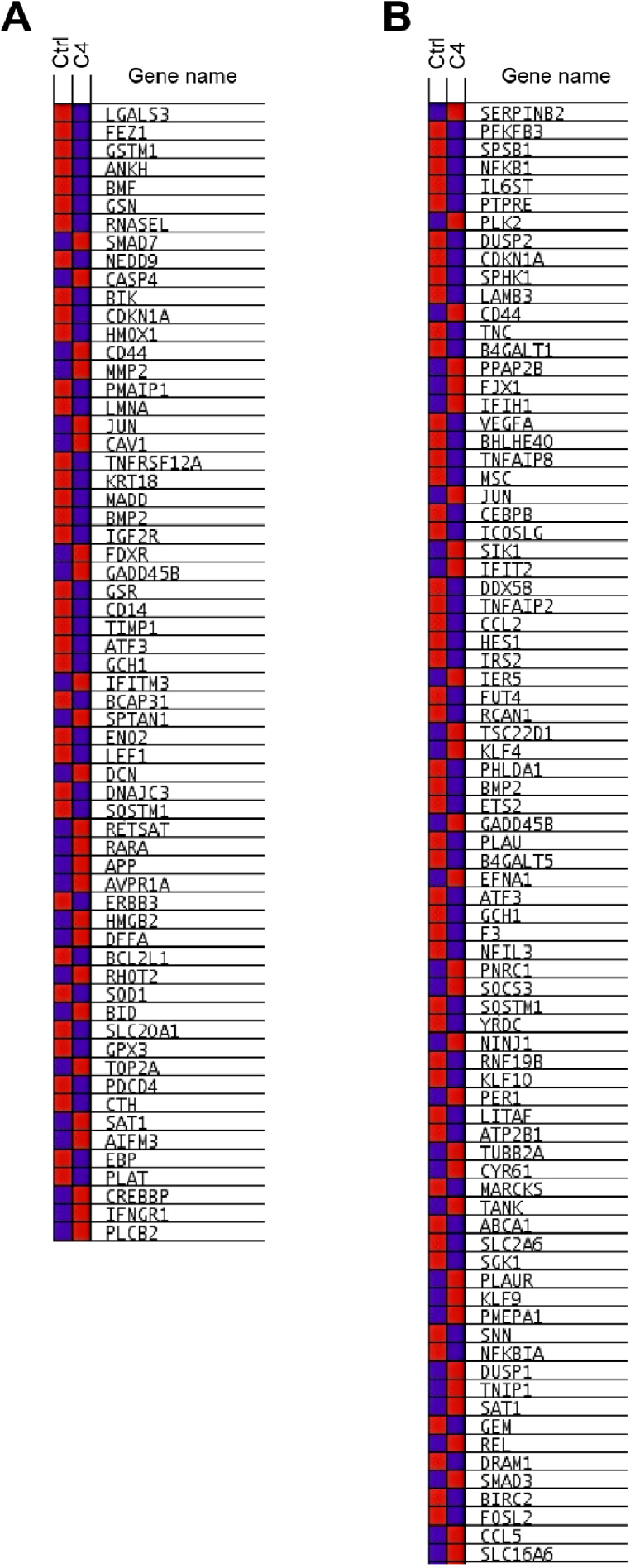
Expression changes of core enriched genes in RBM5-altered gene sets. GSAASeqSP blue-pink o’gram representing the expression changes of core enriched genes from the MSigDB Hallmark Apoptosis (A) and TNFα signaling via NFκB (B) gene sets in control vs. C4 samples (RNA-Seq data). Blue indicates low expression, whereas red indicates high expression. Genes listed in order of Rank Metric Score.
Other gene sets identified in this GSAASeqSP analysis are important to the transformed state, supporting our KEGG pathway results and providing additional insight into the role of RBM5 in SCLC cells. These gene sets (Table 1) include genes involved in (a) angiogenesis, where RBM5 expression (in T2 and C4) correlated with significantly decreased expression of pro-angiogenic factors, such as Vascular Endothelial Growth Factor A (VEGFA) (Fig. 3E), (b) epithelial-mesenchymal transition (EMT), and (c) the immune response. RBM5 may, therefore, not only promote a non-transformed state, but be important for identification and elimination of cells that adopt cancerous characteristics.
Taken together, these RNA-Seq pathway analysis results for the GLC20 sublines indicate that in SCLC cells, RBM5 expression is very important to the maintenance of the non-transformed state, particularly via regulation of cell death, angiogenesis, cell-cell adhesion and immune response pathways.
2.3. RBM5 regulates expression of SCLC-associated genes
To investigate the influence of RBM5 expression particularly on SCLC-associated pathways, we examined the expression of previously identified lung cancer-associated genes in our GLC20 sublines. In a report released by the National Cancer Institute (NCI) in June 2014, entitled Scientific Framework for Small Cell Lung Cancer, a list of 37 ‘Genes of Interest in SCLC’ was compiled. Interestingly, the expression of 43.2% of these ‘Genes of Interest in SCLC’ (16/37) significantly changed expression in C4, compared to control (Table 2), with 11 of these differentially expressed genes changing expression in a manner that would promote suppression of tumor growth upon RBM5 expression; BCL2, CCNE1, COBL, CREBBP, EPHA7, MED12L, MYCL1, RAB37, SLIT2, SMO and SOX2. Furthermore, 11 of the 21 non-differentially expressed genes had very low expression in the control and C4 (FPKM below one), and thus may not play an important role in GLC20 cells. The observation that RBM5 expression influences the expression levels of so many NCI-collated ‘Genes of Interest in SCLC’ highlights (1) the importance of RBM5 to SCLC, (2) the impact that RBM5 gene deletion may have on the development of lung cancer, and (3) the effect that re-establishment of RBM5 expression may have on SCLC tumors.
Table 2.
Expression of NCI ‘Genes of Interest in SCLC’ in control and C4 samples, as determined by RNA-Seq. Expression values are expressed as FPKM. Italics identify genes with non-significant differential expression between control and C4 and expression below one FPKM in both samples.
| Gene name | Control | C4 | p-value | Significance |
|---|---|---|---|---|
| BCL2† | 1.8307 | 0.2914 | 0.00005 | Yes |
| CCNE1† | 18.139 | 10.6106 | 0.00005 | Yes |
| CDK14‡ | 17.0371 | 19.8042 | 0.0083 | Yes |
| CDKN2A | 78.3537 | 89.0263 | 0.0378 | No |
| COBL† | 0.885007 | 0.011419 | 0.004 | Yes |
| CREBBP‡ | 5.47952 | 6.5709 | 0.0018 | Yes |
| DMBX1 | 0.048243 | 0.102193 | 1 | No |
| EP300 | 7.62326 | 7.82019 | 0.789641 | No |
| EPHA7‡ | 20.1031 | 26.6501 | 0.0001 | Yes |
| FGFR1 | 0.171331 | 0.212293 | 1 | No |
| GPR113 | 0.475767 | 0.661619 | 0.6777 | No |
| GPR133 | 0.003737 | 0 | 1 | No |
| GPR55 | 0.001965 | 0 | 1 | No |
| GRID1 | 0.553261 | 0.587185 | 0.6624 | No |
| LRRK2 | 0.068785 | 0.016732 | 1 | No |
| MED12L† | 3.18195 | 0.191165 | 0.00005 | Yes |
| MLL (KMT2A) | 11.1758 | 11.5143 | 0.5842 | No |
| MYCL† | 11.114 | 8.99515 | 0.0044 | Yes |
| NOTCH1‡ | 4.87757 | 6.22612 | 0.00005 | Yes |
| NOTCH2‡ | 3.44146 | 4.85108 | 0.00005 | Yes |
| NOTCH3 | 0.06277 | 0.11642 | 1 | No |
| PIK3CA | 9.39427 | 8.64243 | 0.1883 | No |
| PPEF2 | 0.037315 | 0.063875 | 1 | No |
| PRKD3 | 26.8136 | 28.0848 | 0.4054 | No |
| PTEN† | 17.1134 | 14.0056 | 0.0007 | Yes |
| PTPRD | 0 | 5.72938 | 1 | No |
| RAB37† | 0.660452 | 0.186656 | 0.00005 | Yes |
| RASGRF1 | 0.00182 | 0.006163 | 1 | No |
| RASGRF2 | 0.377979 | 0.006163 | 1 | No |
| RB1 | 6.22425 | 6.86606 | 0.1533 | No |
| RUNX1T1‡ | 5.11373 | 8.39718 | 0.00005 | Yes |
| SLIT2‡ | 5.19232 | 9.74325 | 0.00005 | Yes |
| SMO† | 5.93771 | 3.35434 | 0.00005 | Yes |
| SOX2† | 14.0653 | 10.8382 | 0.0003 | Yes |
| STK38 | 20.2447 | 18.0817 | 0.0533 | No |
| TP53 | 2.85615 | 3.40231 | 0.2023 | No |
| TRRAP | 13.9452 | 12.2657 | 0.0716 | No |
Indicates genes significantly downregulated in C4.
Indicates genes significantly upregulated in C4.
In addition to these NCI-collated ‘Genes of Interest in SCLC’, we examined the influence of RBM5 expression on Sex-determining region Y-box 2 (SOX2) and Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS), as overexpression of the former has been observed in many types of lung cancers and is linked with tumor initiation (Tam and Ng, 2014), and activation of the latter occurs in approximately 30% of smoking-associated lung adenocarcinomas (Unni et al., 2015). In our C4 samples, SOX2 and KRAS were significantly downregulated (Fig. 3F), suggesting once again the importance of RBM5 expression in lung tissue.
2.4. Alternative splicing accounts for a minority of the differential gene expression
RBM5 influences processes such as apoptosis and cell cycle arrest in a variety of cancer cell lines at least in part via the modulation of alternative splicing of key factors, such as NUMB and CASPASE 2 (CASP2) (Bechara et al., 2013; Fushimi et al., 2008). To see if RBM5 influences processes in SCLC via modulation of alternative splicing, we mined our RNA-Seq data for significant alternative splicing changes.
In the control vs. T2 group, 2,546 variants were significantly differentially expressed (5.12% of the 49,772 variants examined), with 10 genes showing a significant change from one alternative splice variant to another (Table 3). In the control vs. C4 group, 4,180 variants were differentially expressed, with 47 genes showing a significant change from one alternative splice variant to another (Table 4). The greater number of alternative splicing events occurring in the C4 subline, compared to T2, suggests an additive effect of RBM5 on alternative splicing, which was expected. There was no overlap, however, of splicing events between T2 and C4, potentially due to the stringent search parameters used, and no significant enrichment of these splicing events in any KEGG pathway or MSigDB Hallmark gene set. Furthermore, the number of significant alterations in splicing between control and T2 and C4, respectively, was much lower than expected, given the high number of differentially expressed genes (even considering the stringent search parameters). This suggests that modulation of alternative splicing is not the only means by which RBM5 influences important transformation-associated pathways in SCLC. RBM5 may, for instance, regulate gene expression levels by influencing transcription or stabilizing transcripts, as has been shown for other RNA-binding proteins, including the RBM5-related protein RBM10 (Guallar and Wang, 2014; Mueller et al., 2009). We therefore proceeded to carry out binding studies in order to identify targets with which RBM5 interacts, either directly or indirectly, and thereby gain a better understanding of how RBM5 influences the identified pathways.
Table 3.
Genes with at least one alternative splice variant significantly upregulated, and at least one alternative splice variant significantly downregulated between control and T2 (RNA-Seq results). Each row indicates expression levels of a different variant for the given gene. Arranged alphabetically, by gene.
| Control vs. T2 |
|||
|---|---|---|---|
| Gene name | Log2 (fold-change) | p-value | q-value |
| BAG6 | 0.633915 | 0.0002 | 0.00279 |
| -2.0923 | 0.0027 | 0.03018 | |
| EML1 | 1.18146 | 0.004 | 0.04069 |
| -0.321613 | 0.0019 | 0.02236 | |
| FERMT2 | -0.682011 | 0.0012 | 0.01589 |
| 0.326233 | 0.0039 | 0.04029 | |
| INF2 | -0.707866 | 5e-05 | 0.00106 |
| 1.40674 | 5e-05 | 0.00106 | |
| IQCE | -1.59194 | 0.0004 | 0.00645 |
| 0.571945 | 0.0045 | 0.04469 | |
| KCTD15 | 0.509451 | 0.0006 | 0.00906 |
| -0.932889 | 5e-05 | 0.00106 | |
| KIF2A | 1.13138 | 5e-05 | 0.00106 |
| -1.01307 | 5e-05 | 0.00106 | |
| LIG3 | 1.44254 | 0.0003 | 0.00434 |
| -0.360436 | 0.0007 | 0.01032 | |
| LIPA | 0.547552 | 0.0005 | 0.0078 |
| -0.676966 | 5e-05 | 0.00106 | |
| ZBTB14 | -1.18342 | 0.0025 | 0.02843 |
| 2.62976 | 0.001 | 0.01382 | |
Table 4.
Genes with at least one alternative splice variant significantly upregulated, and at least one alternative splice variant significantly downregulated between control and C4 (RNA-Seq results). Each row indicates expression levels of a different variant for the given gene. Arranged alphabetically, by gene.
| Control vs. C4 |
|||
|---|---|---|---|
| Gene | log2 (fold-change) | p-value | q-value |
| ADAM22 | 0.983 | 8E-04 | 0.007 |
| -0.691 | 0.006 | 0.0374 | |
| ADAT2 | -0.412 | 0.002 | 0.0155 |
| 0.885 | 1E-04 | 0.0011 | |
| AR | 0.444 | 5e-05 | 0.0006 |
| -0.979 | 0.002 | 0.0119 | |
| ASAP1 | -0.454 | 0.003 | 0.0219 |
| 0.722 | 5e-05 | 0.0006 | |
| ASPH | 1.693 | 1E-04 | 0.0011 |
| -0.679 | 0.005 | 0.0327 | |
| ATG16L1 | 0.718 | 5e-05 | 0.0006 |
| -0.648 | 0.003 | 0.0204 | |
| BCKDHB | 0.544 | 0.004 | 0.0242 |
| -0.531 | 2E-04 | 0.0016 | |
| BZW2 | -1.239 | 0.001 | 0.0091 |
| 0.281 | 0.002 | 0.0125 | |
| C14orf93 | -1.649 | 8E-04 | 0.0066 |
| 1.096 | 0.002 | 0.0149 | |
| C8orf59 | -0.413 | 0.004 | 0.0267 |
| 0.564 | 0.001 | 0.0098 | |
| CDK13 | 0.711 | 1E-04 | 0.0011 |
| -0.303 | 0.003 | 0.0201 | |
| CEP41 | 1.715 | 0.006 | 0.0395 |
| -0.482 | 5e-05 | 0.0006 | |
| CEP78 | -1.025 | 5e-05 | 0.0006 |
| 1.100 | 5e-05 | 0.0006 | |
| CTNNB1 | 0.498 | 0.004 | 0.0281 |
| -0.689 | 5e-05 | 0.0006 | |
| DMTF1 | -0.427 | 0.002 | 0.0155 |
| 1.026 | 0.001 | 0.0091 | |
| DST | -0.791 | 5e-05 | 0.0006 |
| 0.740 | 4E-04 | 0.0034 | |
| GGA1 | 1.047 | 8E-04 | 0.007 |
| -1.099 | 0.002 | 0.0161 | |
| 1.028 | 0.001 | 0.0091 | |
| HMGXB4 | 0.567 | 5e-05 | 0.0006 |
| -0.764 | 5e-05 | 0.0006 | |
| KIAA1958 | 1.630 | 1E-04 | 0.0011 |
| -1.107 | 4E-04 | 0.0034 | |
| LIMA1 | 1.434 | 0.003 | 0.0213 |
| -0.742 | 5e-05 | 0.0006 | |
| MAPKAP1 | 0.660 | 5e-05 | 0.0006 |
| -0.474 | 0.001 | 0.0105 | |
| MUM1 | 0.479 | 0.004 | 0.0248 |
| -0.875 | 5e-05 | 0.0006 | |
| MYO1B | 0.763 | 5e-05 | 0.0006 |
| 0.906 | 0.001 | 0.0098 | |
| -1.790 | 0.004 | 0.0273 | |
| NAP1L1 | -0.41 | 0.001 | 0.0109 |
| 0.404 | 0.002 | 0.0167 | |
| NUMB | -1.472 | 0.008 | 0.0499 |
| 0.951 | 0.005 | 0.0314 | |
| -0.516 | 8E-04 | 0.0066 | |
| PAXBP1 | -1.167 | 0.005 | 0.0319 |
| 1.058 | 5e-05 | 0.0006 | |
| PCBP2 | -1.724 | 0.002 | 0.0177 |
| 0.652 | 0.006 | 0.0356 | |
| PDE1C | -0.722 | 8E-04 | 0.007 |
| 0.911 | 5e-05 | 0.0006 | |
| PDHA1 | -0.269 | 0.007 | 0.0435 |
| 2.175 | 0.003 | 0.0195 | |
| PHF8 | 0.834 | 2E-04 | 0.0021 |
| -0.396 | 0.006 | 0.0374 | |
| PTK2 | 0.558 | 0.003 | 0.0231 |
| -0.666 | 5e-05 | 0.0006 | |
| RAC1 | 0.297 | 0.006 | 0.0389 |
| -1.588 | 5e-05 | 0.0006 | |
| RBBP5 | -1.255 | 5e-05 | 0.0006 |
| 0.472 | 0.002 | 0.017 | |
| RBFOX2 | 0.971 | 5e-05 | 0.0006 |
| -0.499 | 3E-04 | 0.0026 | |
| RPAP3 | -1.041 | 0.005 | 0.0309 |
| 0.308 | 0.005 | 0.0303 | |
| RTN1 | 0.501 | 4E-04 | 0.0034 |
| -2.439 | 3E-04 | 0.003 | |
| SLC29A1 | -0.849 | 5e-05 | 0.0006 |
| 0.395 | 0.005 | 0.0325 | |
| SLC9A6 | -1.393 | 5e-05 | 0.0006 |
| 0.492 | 0.006 | 0.0364 | |
| SSBP3 | 0.785 | 5e-05 | 0.0006 |
| -0.597 | 3E-04 | 0.003 | |
| SYT1 | 1.903 | 5e-05 | 0.0006 |
| -5.079 | 1E-04 | 0.0011 | |
| TPM1 | 1.473 | 5e-05 | 0.0006 |
| -0.922 | 0.004 | 0.0258 | |
| UGGT1 | 0.626 | 5e-05 | 0.0006 |
| -0.95 | 5e-05 | 0.0006 | |
| UPF3B | 0.927 | 5e-05 | 0.0006 |
| -1.231 | 5e-05 | 0.0006 | |
| ZBTB20 | -1.234 | 0.004 | 0.0261 |
| 0.746 | 6E-04 | 0.0055 | |
| ZNF260 | 0.424 | 2E-04 | 0.0021 |
| -0.933 | 0.001 | 0.0098 | |
| ZNF566 | 0.791 | 0.004 | 0.0273 |
| -0.804 | 5E-04 | 0.0043 | |
| ZNF655 | -1.174 | 0.005 | 0.034 |
| 0.690 | 0.007 | 0.0425 | |
2.5. RIP-Seq identified RBM5 RNA targets regulate RNA metabolism and cell cycle pathways, ZNRF3 being the most enriched gene
Although some studies have investigated RBM5’s binding motif (Bechara et al., 2013; Ray et al., 2013), the number of identified RNA targets that are directly bound by RBM5 is low. In cancer cell lines, only three directly bound RNA targets have been identified to date; Caspase-2 (CASP2) (Fushimi et al., 2008), the antisense transcript of FAS (FAS-AS1) (Sehgal et al., 2014), and Activation-induced cytidine deaminase (AID) (Jin et al., 2012). In mouse spermatid differentiation, 11 RNA targets have been identified (O'Bryan et al., 2013). In its capacity as a component of spliceosomal complexes (Hegele et al., 2012; Niu et al., 2012), RBM5 is also likely capable, however, of binding many important RNA targets indirectly (e.g., via another protein within the complex).
In order to identify direct and indirect RNA targets of the RBM5 protein in GLC20 cells, we used RNA Immunoprecipitation followed by next generation sequencing (RIP-Seq). RIP-Seq experiments were performed using our non-commercially available RBM5-specific LUCA-15UK antibody (Sutherland et al., 2000) because, as demonstrated in Fig. 5, the commercially available antibodies that were tested were not specific for RBM5. Two different negative controls were used in each of two RIP-Seq replicates, to ensure the validity of identified RBM5 targets. Firstly, a non-specific IgG IP in C4 cells (vs. LUCA-15UK IP in C4 cells), and secondly, LUCA-15UK IP in GLC20.pcDNA3 cells (vs. LUCA-15UK IP in C4 cells). Western blots showing successful IP of RBM5 for both replicates are presented in Fig. 6A.
Fig. 5.

RBM5 antibody testing. The same whole cell lysate from either GLC20 cells (wt) or GLC20.C4 RBM5 containing cells (C4) was loaded in alternate lanes and probed with the antibodies indicated. The LUCA-15-UK blot was probed with a 1:1000 antibody dilution, overnight at 4 °C, and exposed for 2 min. The upper Origene blot was probed with a 1:3000 antibody dilution, for 3 h at RT, and exposed for 60 min. The lower Origene blot was reprobed with a 1:500 antibody dilution, overnight at 4 °C, and exposed for 60 min. The upper Abnova blot was probed with a 1:500 antibody dilution, for 3 h at RT, and exposed for 60 min. The lower Abnova blot was reprobed with a 1:350 antibody dilution, overnight at 4 °C, and exposed for 60 min. The Abcam blot was probed with a 1:2500 antibody dilution, overnight at 4 °C, and exposed for 5 min. The Sp1 and Sp2 blots were probed with a 1:5000 antibody dilution, for 3 h at RT, and exposed for 1 min. All commercially available antibodies (therefore excluding Sp1 and Sp2 – a gift from Juan Valcárcel − and LUCA-15-UK) interacted with product around the same molecular weight as RBM5 even in GLC20 cells, making them unsuitable for use in RIP-Seq experiments. See Figure S3 full blots of loading controls.
Fig. 6.

RBM5 RIP-Seq optimization and quality control. (A) Raw Western Blot data demonstrating successful immunoprecipitation of RBM5. (B) Scatterplot representing expression of genes with FPKM < 5000 in control and RBM5 RIP samples, respectively. All Western Blot ladder sizes are in kilodaltons (kDa).
RNA from both RIP-Seq experiments was sequenced. The distribution of gene expression values in our control vs. RBM5 RIP samples is presented in Fig. 6B, for genes with FPKMs < 5000 (10 genes had FPKMs above 5000). This figure highlights the high level of gene expression in our RBM5 RIP samples, compared to the control, confirming the success of our technique. We identified 773 genes as RBM5 targets (see Materials & methods for exclusion details), a not unexpectedly large number since this technique identifies all RNA bound by a complex of which RBM5 is a part.
The most significant RBM5 targets included Zinc & ring finger 3 (ZNRF3), Suppressor of cancer cell invasion (SCAI), Growth factor receptor-bound protein 2 (GRB2), Zw10 kinetochore protein (ZW10), DEP domain containing (DEPDC1) and Activating transcription factor 2 (ATF2). None of these genes were detected in the control IP and all had FPKM values above 350 in the RBM5 RIP (ZNRF3 had the highest count with 1.23 × 107 FPKM, followed by SCAI with 2866.53 FPKM and GRB2 with 1482.74 FPKM). Interestingly, these six genes have all been shown to play important roles in the control of the cell cycle and proliferation. It is important to note that the previously identified direct RBM5 targets - CASP2, FAS and AID - were not identified in our RIP-Seq experiments. Lack of detection of these potentially direct targets could be a cell type-specific phenomenon, since neither FAS nor AID were expressed above 0.1 FPKM in our samples.
To determine the importance of these targets to cellular functions, pathway analysis was carried out on RIP-Seq identified RBM5 RNA targets. FIDEA analysis, using the KEGG database, identified only three significantly changed pathways; ‘Spliceosome’ (hsa03040) (with a Benjamini value of 1.37 × 10−4), ‘RNA transport’ (hsa03013) (1.53 × 10−3) and ‘Ribosome’ (hsa03010) (9.07 × 10−3). These results support our findings that RBM5 influences important pathways in SCLC via regulation of alternative splicing and other pre- and/or post-transcriptional processes. These results also help to confirm the success of our technique, since RBM5 was previously shown to be a key component of spliceosomal complexes (Hegele et al., 2012; Niu et al., 2012).
As with our RNA-Seq data, we used an additional pathway analysis program to support our results. For this RIP-Seq data, our second program was Cytoscape with the Reactome FI Network plugin, as the GSAASeq program is designed specifically for RNA-Seq data. Using Reactome, we found 68 pathways with an FDR < 8% (Table 5), many involved in gene expression and mRNA splicing/metabolism. Interestingly, there was an enrichment of RBM5 targets in the EGFR pathway (Fig. 7), as well as many cell cycle pathways and the ‘Apoptosis induced DNA fragmentation’ pathway (Fig. 8), supporting our RNA-Seq results and suggesting that RBM5 may play a direct role in regulating the cell cycle and apoptosis in SCLC cells.
Table 5.
RBM5 RIP-Seq results, showing top enriched pathways (FDRs below 8%), identified by Cytoscape Reactome FI Plugin tool using the Reactome Pathway Database.
| Reactome Pathway | FDR |
|---|---|
| Nonsense-Mediated Decay (NMD) | 5.88E-05 |
| NMD enhanced by the Exon Junction Complex | 5.88E-05 |
| Influenza Infection | 6.67E-05 |
| HATs acetylate histones | 7.14E-05 |
| Influenza Life Cycle | 7.69E-05 |
| Processing of Capped Intron-Containing Pre-mRNA | 8.33E-05 |
| GTP hydrolysis and joining of the 60S ribosomal subunit | 9.09E-05 |
| NMD independent of the Exon Junction Complex | 1.00E-04 |
| mRNA Splicing | 1.00E-04 |
| mRNA Splicing - Major Pathway | 1.00E-04 |
| Influenza Viral RNA Transcription and Replication | 1.05E-04 |
| Eukaryotic Translation Elongation | 1.11E-04 |
| L13a-mediated translational silencing of Ceruloplasmin expression | 1.25E-04 |
| 3' -UTR-mediated translational regulation | 1.25E-04 |
| Eukaryotic Translation Initiation | 1.67E-04 |
| Cap-dependent Translation Initiation | 1.67E-04 |
| Cell Cycle, Mitotic | 2.27E-04 |
| Formation of a pool of free 40S subunits | 2.38E-04 |
| Translation | 2.50E-04 |
| Attenuation phase | 2.61E-04 |
| Metabolism of proteins | 3.08E-04 |
| Translation initiation complex formation | 3.20E-04 |
| Ribosomal scanning and start codon recognition | 3.20E-04 |
| Chromatin modifying enzymes | 3.33E-04 |
| Chromatin organization | 3.33E-04 |
| SRP-dependent cotranslational protein targeting to membrane | 6.88E-04 |
| Cellular responses to stress | 6.97E-04 |
| Cell Cycle | 7.10E-04 |
| Activation of the mRNA upon binding of the cap-binding complex and eIFs, and subsequent binding to 43S | 7.33E-04 |
| Mitochondrial biogenesis | 7.35E-04 |
| Viral mRNA Translation | 7.59E-04 |
| Peptide chain elongation | 7.59E-04 |
| Eukaryotic Translation Termination | 7.59E-04 |
| HSF1-dependent transactivation | 9.14E-04 |
| Organelle biogenesis and maintenance | 1.00E-03 |
| Gene Expression | 1.00E-03 |
| Calnexin/calreticulin cycle | 1.84E-03 |
| Deadenylation of mRNA | 2.11E-03 |
| G2/M Transition | 2.21E-03 |
| Transcriptional activation of mitochondrial biogenesis | 2.53E-03 |
| Mitotic G2-G2/M phases | 2.81E-03 |
| N-glycan trimming in the ER and Calnexin/Calreticulin cycle | 4.26E-03 |
| EGFR downregulation | 4.67E-03 |
| Cellular response to heat stress | 6.91E-03 |
| ISG15 antiviral mechanism | 7.04E-03 |
| Antiviral mechanism by IFN-stimulated genes | 7.04E-03 |
| M Phase | 7.45E-03 |
| Formation of the ternary complex, and subsequently, the 43S complex | 1.07E-02 |
| Activation of gene expression by SREBF (SREBP) | 1.11E-02 |
| Deadenylation-dependent mRNA decay | 1.11E-02 |
| Regulation of PLK1 Activity at G2/M Transition | 1.12E-02 |
| Regulation of cholesterol biosynthesis by SREBP (SREBF) | 1.84E-02 |
| Cellular Senescence | 2.37E-02 |
| Mitotic Anaphase | 2.40E-02 |
| Mitotic Prometaphase | 2.51E-02 |
| Mitotic Metaphase and Anaphase | 2.55E-02 |
| Resolution of Sister Chromatid Cohesion | 3.48E-02 |
| Centrosome maturation | 4.44E-02 |
| Recruitment of mitotic centrosome proteins and complexes | 4.44E-02 |
| Loss of Nlp from mitotic centrosomes | 6.15E-02 |
| Loss of proteins required for interphase microtubule organization from the centrosome | 6.15E-02 |
| mRNA Splicing − Minor Pathway | 7.52E-02 |
| MicroRNA (miRNA) biogenesis | 7.79E-02 |
| Initiation of Nuclear Envelope Reformation | 7.79E-02 |
| Nuclear Envelope Reassembly | 7.79E-02 |
| Recycling of eIF2:GDP | 7.79E-02 |
| Apoptosis induced DNA fragmentation | 7.79E-02 |
| Activator of DNA fragmentation factor | 7.79E-02 |
Fig. 7.

RBM5 targets in the EGFR Signaling pathway. Network analysis results of RBM5 targets (identified by RIP-Seq) in a portion of the EGFR Signaling pathway. Purple indicates that the gene was identified as an RBM5 target.
Fig. 8.

RBM5 targets in the Apoptotic Execution Phase pathway. Pathway analysis results of RBM5 targets (identified by RIP-Seq) in a portion of the Reactome Apoptotic Execution Phase pathway. Purple indicates that the gene was identified as an RBM5 target.
It is interesting to note that of all the genes shown in our RNA-Seq experiments to experience a significant change in alternative splicing upon RBM5 expression in T2 or C4 (Table 3 and Table 4), only one, ArfGAP with SH3 Domain, Ankyrin Repeat and PH Domain (ASAP1), was identified as an RBM5 target in our RIP-Seq experiments. This suggests that the effect of RBM5 expression on alternative splicing largely results from downstream consequences of changes in RBM5 expression, as opposed to a targeted RBM5 interaction with the alternatively spliced transcript.
Taken together, our RNA-Seq and RIP-Seq pathway analyses revealed that RBM5 directly influences many processes involved in the maintenance of a non-transformed state, and this by means distinct from regulation of alternative splicing.
2.6. Functionally, RBM5 inhibits cell growth and sensitizes cells to cisplatin–mediated apoptosis
In other cancer cell lines, RBM5 has been shown to regulate the cell cycle and modulate responses to apoptogenic stimuli (Kobayashi et al., 2011; Oh et al., 2006; Rintala-Maki and Sutherland, 2004). Since we identified ‘Cell Cycle’ (Fig. 3C and D) and ‘Apoptosis’ (Table 1) as pathways likely influenced by the differentially expressed genes in T2 and C4, respectively, we decided to validate our sequencing data by carrying out functional studies.
To see if and how RBM5 affects the cell cycle in SCLC, we performed a proliferation and membrane integrity assay using our GLC20 cells and sublines. As shown in Fig. 9A, C4 had significantly decreased cell numbers, relative to the vector control, by day six, while membrane integrity was unaffected (Fig. 9B). This suggests that when RBM5 levels are high, such as in C4, RBM5 slows cell cycle progression in untreated SCLC cells, thereby promoting a non-transformed state, a result that supports our RNA-Seq findings.
Fig. 9.

Effect of RBM5 expression +/− cisplatin on cell proliferation and membrane integrity. (A and B) GLC20 sublines were left untreated, or exposed to either a saline control or 1.0 μM cisplatin and cell numbers (A) or membrane integrity (B) monitored every other day by cell counting using a hemocytometer. Average of three biological replicates carried out in technical triplicate with standard error is displayed. A two-way ANOVA was performed between pcDNA3 and the other sublines, with Bonferroni post-hoc analysis. (C) GLC20 subline membrane integrity was monitored after eight days of exposure to various concentrations of cisplatin. Results represent the average of three biological replicates performed in technical triplicate with standard error using the calculated average EC50 (calculated from Graphpad Prism 5, ‘non-linear fit − log(inhibitor) vs response (3 parameters)’). Graph represents the average EC50 of three biological replicates. One-way ANOVA was performed with Tukey post-hoc analysis, between sublines, with *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
In North America, due to the usual late stage diagnosis and consequent metastasis, SCLC tumors are not commonly resected, but rather treated with a combination of platinum-based agents such as cisplatin or carboplatin, and the topoisomerase inhibitor etoposide (Gaspar et al., 2012; Jackman and Johnson, 2005; Johnson et al., 2014; Travis et al., 2004). Initially, the patient may show a complete response to the treatment, but most will relapse as the cancer develops resistance (Jackman and Johnson, 2005). Understanding how SCLC cells develop resistance to these drugs is thus of great importance. We therefore included a platinum-based treatment in our functional analysis, and determined the effects of RBM5 expression on SCLC cells’ response to the drug. Cisplatin was our drug of choice since, as demonstrated in Fig. 2, GLC20 cells are already quite resistant to cisplatin, making them a good model for a drug sensitization study. Interestingly, RBM5 overexpression in the cisplatin-resistant NSCLC cell line A549 reduced resistance to cisplatin, manifesting as increased cisplatin-induced apoptosis (Li et al., 2012), thus we expected that RBM5 expression in our SCLC cell line would have a similar effect.
The GLC20 parental cell line and sublines were treated with 1 μM cisplatin, and cell proliferation and membrane integrity assessed. Both T2 and C4 cells showed significantly decreased cell numbers, relative to the vector control, following 10 days of cisplatin exposure (Fig. 9A), and a significant decrease in membrane integrity by day four (Fig. 9B). In addition, the IC50 for cisplatin in both of the RBM5 expressing sublines was significantly lower than the vector control (Fig. 9C).
To determine if the decreased membrane integrity observed in T2 and C4 upon cisplatin treatment was due to increased apoptosis, we examined apoptotic marker expression using fluorescence microscopy. When cells were untreated, no significant change in the level of cell death was observed between sublines (Fig. 10A and B), consistent with our membrane integrity results (Fig. 9B). Following four days of exposure to 5 μM cisplatin, however, significantly more cells were observed in both early and late stage apoptosis in the C4 cells, compared to the vector control (Fig. 10C and D). Furthermore, Poly(ADP-Ribose) Polymerase (PARP) cleavage was significantly increased in the C4 cells, following cisplatin treatment (Fig. 10E and F). (An insignificant increase in cisplatin-mediated apoptosis was observed in the T2 subline, by Western blot (Fig. 10E and F) and fluorescence microscopy (Fig. 10C and D)).
Fig. 10.

Effect of RBM5 expression +/− cisplatin on apoptosis. GLC20 sublines were left untreated (A, B) or treated with 5 μM cisplatin (C, D, E, F) and collected after four days for fluorescence microscopy (A, C) or PARP cleavage analysis (E, F). Average number of Live (only Hoechst/blue), Early Apoptosis (condensed Hoechst/blue and/or Annexin-V/green) and Late Apoptosis/Necrosis (7-AAD/Red) fluorescence microscopy events from three biological replicates, each with 10 different fields of view, with standard error, for the untreated cells (B) and 5 μM cisplatin (D). (E) A representative Western blot for PARP cleavage, in the cisplatin-treated samples, with (F) densitometric analysis of ‘percent 89kDa PARP cleavage product’ [(89kDa cleaved PARP/total PARP)x100], with standard error, from three biological replicates, using the AlphaEase FC, ‘1D-Multi’ analysis tool. One-way ANOVA was performed with Tukey post-hoc analysis, between sublines, with *p < 0.05 and ***p < 0.001. See Figure S4 for full blots of E.
These results suggest that RBM5 expression does in fact sensitize cells to cisplatin’s pro-apoptotic effects, being in line with our RNA-Seq and RIP-Seq data, which suggest that RBM5 influences the regulation of apoptotic pathways.
2.7. In cisplatin-treated GLC20 cells, RNA-Seq shows that 7% of the transcriptome is differentially expressed by RBM5, and that DSG2 and ATP11C are the most altered genes, potentially driving the observed downstream effects of RBM5 expression
Having demonstrated that RBM5 expression can significantly influence a SCLC cell line’s response to cisplatin, at least in terms of cell proliferation, membrane integrity and apoptosis, we decided to examine this influence on a more global level, by performing RNA-Seq on the cells treated with 5 μM cisplatin for four days.
We identified 1,797 differentially expressed genes in the control vs. T2 group (7% of genes examined), 1,225 in the control vs. C4 group (4.7%), with 457 genes being common to both groups. Fewer differentially expressed genes in C4 compared to T2 was an unexpected finding, based on our RNA-Seq data from the untreated samples, and was possibly due to the increased rate of apoptosis in the high RBM5 expressing cells, limiting the measurable effect on gene expression by RNA-Seq.
The most highly upregulated genes common to treated T2 and C4 cells were Protein Phosphatase 1 Regulatory Inhibitor Subunit 1A (PPP1R1A) and Makorin Ring Finger Protein 3 (MKRN3). MKRN3 has been shown to be important to development, particularly the onset of puberty (Abreu et al., 2013), which is in line with previously identified RBM5 functions, as described above.
The most highly downregulated genes common to treated T2 and C4 include (a) Translocase of Outer Mitochondrial Membrane 40 Like (TOMM40L), which is a component of the outer mitochondrial membrane translocase, and thus influences many mitochondrial processes (Humphries et al., 2005), (b) ATPase Phospholipid Transporting 11C (ATP11C), whose cleavage (or downregulation) is required for phosphatidylserine exposure and consequent phagocytosis during apoptosis (Segawa et al., 2014; Yabas et al., 2016), and (c) Desmoglein 2 (DSG2), a ubiquitously expressed cell adhesion protein upregulated in many epithelial-derived cancers (Brennan and Mahoney, 2009; Harada et al., 1996; Kurzen et al., 2003) and shown to promote cell cycle progression and apoptosis-resistance (Brennan et al., 2007; Gupta et al., 2015). These three genes thus play an important role in cell survival, and it may be via their downregulation that RBM5 slows SCLC growth and sensitizes them to cisplatin-mediated apoptosis. Interestingly, ATP11C and DSG2 are also significantly downregulated in untreated T2 and C4 cells, suggesting that cisplatin treatment may complement/enhance the effect of RBM5’s expression on SCLC.
FIDEA analysis using the KEGG database showed that no pathways were downregulated and two pathways were significantly enriched in our control vs. T2 group; ‘Axon guidance’ (Benjamini value of 2.02 × 10−6) and ‘Retrograde endocannabinoid signaling’ (Benjamini value of 4.22 × 10−4). In our control vs. C4 group, only ‘Axon guidance’ was significantly enriched (Benjamini value of 9.44 × 10−3). These results are very interesting since axon guidance is involved in the establishment of a transformed state (Chedotal et al., 2005), and was also significantly altered in our untreated T2 and C4 samples, suggesting cisplatin treatment may complement this particular RBM5 function. Absence of ‘Retrograde endocannabinoid signaling’ in the control vs. C4 group might explain the presence of fewer differentially expressed genes in the C4 vs. T2 group.
Using the GSAASeqSP program with the MSigDB Hallmark gene set, we identified 24 enriched gene sets with FDRs < 10% between cisplatin-treated control and T2, and 18 between control and C4, of which 16 were common between both (Table 6). It is important to note that only 12 MSigDB Hallmark gene sets were identified as significantly enriched in our untreated control vs. T2 samples, and 17 in untreated control vs. C4. The greater number of significantly changed pathways in the treated samples, but with fewer genes differentially expressed overall, suggests once again that cisplatin directs the effect of RBM5’s expression to particular genes. Noticeably, 10 of the 16 enriched pathways that were common to the T2 and C4 cisplatin treated sample sets were also enriched in the untreated C4 samples at a FDR of 10% or lower, demonstrating that cisplatin exposure did not totally alter the influence of RBM5 expression on the SCLC cell’s transcriptomes.
Table 6.
Altered gene sets with FDRs below 10% between both cisplatin-treated control and T2, and control and C4 samples, respectively, as determined by GSAASeq analysis with the MSiDB Hallmark gene set using the samples’ RNA-Seq results. Ranked based on FDR value in control vs. C4 analysis.
| Control vs. T2 |
Control vs. C4 |
|||
|---|---|---|---|---|
| Gene set | p-value | FDR | p-value | FDR |
| TGFβ signaling | 0.0 | 0.05 | 0.0 | 03 |
| Notch signaling | 0.2 | 0.07 | 0.0 | 0.04 |
| EMT | 0.0 | 0.00 | 0.0 | 0.04 |
| Myogenesis | 0.0 | 0.05 | 0.0 | 0.04 |
| Estrogen response early | 0.0 | 0.04 | 0.0 | 0.05 |
| Coagulation | 0.0 | 0.04 | 0.0 | 0.05 |
| Angiogenesis | 0.0 | 0.00 | 0.1 | 0.07 |
| Androgen response | 0.0 | 0.05 | 0.0 | 0.07 |
| UV response down | 0.0 | 0.05 | 0.0 | 0.08 |
| Apoptosis | 0.0 | 0.04 | 0.0 | 0.08 |
| Inflammatory response | 0.0 | 0.03 | 0.0 | 0.08 |
| Estrogen response late | 0.0 | 0.03 | 0.0 | 0.08 |
| KRAS signaling up | 0.0 | 0.04 | 0.0 | 0.08 |
| TNFα signaling via NFκB | 0.0 | 0.03 | 0.0 | 0.08 |
| IL2 STAT5 signaling | 0.0 | 0.04 | 0.0 | 0.08 |
| Apical junctions | 0.0 | 0.04 | 0.0 | 0.09 |
The ‘Apoptosis’ gene set was one of the significantly enriched pathways identified by GSAASeqSP in cisplatin-treated T2 and C4 samples, compared to control, which is in line with the functional work presented herein (Fig. 9 and Fig. 10). In cisplatin-treated T2 samples, 72 genes from this ‘Apoptosis’ gene set were significantly enriched compared to control, and 58 in C4. Of these genes, 35 were common between both groups (Fig. 11), suggesting that many of the same apoptotic pathways were significantly affected in both samples. T2 had 37 uniquely differentially expressed genes while C4 had 23, suggesting that a wider range of apoptotic processes were significantly affected by cisplatin in T2. Interestingly, SMAD7 was the top enriched gene common to both T2 and C4 cisplatin-treated samples (third most enriched gene in both samples). Since SMAD7 has been shown to sensitize lung cells to cisplatin-mediated apoptosis (Jeon et al., 2012), it may be an important mediator of RBM5’s sensitizing effect regarding cisplatin-induced promotion of apoptosis.
Fig. 11.

Expression of apoptosis-related genes in cisplatin treated samples. Adapted GSAASeqSP blue-pink o’gram representing the expression changes of core enriched genes from the MSigDB Hallmark Apoptosis gene set in cisplatin treated control vs. T2 and control vs. C4. Blue indicates decreased expression compared to control, whereas red indicates increased expression compared to control. Genes listed in alphabetical order.
To further investigate which specific apoptotic processes were enriched in our cisplatin-treated samples, we analyzed the enriched genes from the ‘Hallmark Apoptosis’ gene set, as identified by GSAASeqSP, using the Reactome plug-in for Cytoscape (as described for RIP-Seq samples) (Fig. 12). These results confirmed that a wider range of apoptotic processes were influenced in the T2, compared to the C4, population; the ‘Intrinsic pathway for apoptosis’ and the ‘Apoptotic execution’ phase were significantly enriched in both cisplatin-treated T2 and C4 samples, however ‘Caspase-8 activation’ and the ‘Extrinsic pathway’ were only significantly enriched in T2 (Fig. 13 and Fig. 14). These T2-specific pathways are part of the early steps of apoptosis, suggesting that many cells in the cisplatin-treated T2 population were beginning the apoptotic process, but had not yet fully committed to programmed cell death. The cisplatin-treated C4 population, however, had a significant enrichment of the ‘Intrinsic pathway for apoptosis’ subgroups ‘Activation of BH3-only proteins’ and ‘BH3-only proteins associate with and inactivate anti-apoptotic BCL-2 members’. These BH3-only proteins are essential for apoptosis (Shamas-Din et al., 2011).
Fig. 12.

Enriched ‘Programmed cell death’ pathways in cisplatin treated samples. Diagram illustrating the Reactome ‘Programmed cell death’ pathways enriched with an FDR below 10% in cisplatin-treated control vs. T2 (purple), control vs. C4 (blue) or control vs. T2 and C4 (orange).
Fig. 13.

Gene enrichment for apoptosis-related pathways in cisplatin-treated T2 samples. Reactome diagram for ‘Death receptor signaling’ (A) and ‘Caspase-8 activation by cleavage’ (B). Differentially expressed genes between cisplatin treated control vs. T2 are presented in purple.
Fig. 14.

‘Apoptotic execution phase’ gene enrichment in cisplatin-treated samples. Differentially expressed genes between cisplatin treated control vs. T2 (A) or control vs. C4 (B) are presented in purple in a portion of the Reactome ‘Apoptotic execution phase’ pathway.
This in-depth pathway analysis shows that relatively low levels of RBM5 are able to sensitize SCLC cells to cisplatin-mediated apoptosis by influencing changes in the expression of genes involved in early apoptosis events. At relatively higher levels of RBM5 expression, this effect on cell death levels is detectable via fluorescent microscopy (Fig. 10C). These results suggest that the level of RBM5 expression in lung tumors could predict response to cisplatin.
Taken together, our results show that RBM5 can functionally (a) influence cell cycle progression in untreated and cisplatin-treated SCLC cells (potentially via decreased DSG2 expression), and (b) sensitize cisplatin-treated SCLC cells to cisplatin-mediated apoptosis (potentially via decreased DSG2 and ATP11C expression, as well as increased SMAD7 expression). These findings reinforce the importance of RBM5 expression to SCLC cisplatin-sensitivity and highlight the potential of RBM5 as a response marker for this chemotherapy treatment in SCLC. In fact, a recent publication suggested that RBM5 gene therapy might be considered for NSCLC and that RBM5 might be a predictive marker to indicate the potential success of using cisplatin on a particular lung cancer (Li et al., 2012): our results support this suggestion and extend it to SCLC.
It is important to note that RBM5 expression was recently shown to increase autophagy levels in a NSCLC cell line (Su et al., 2016), therefore, we also examined the expression of autophagy markers BCL2, NF-κB, LC-3, LAMP1 and BECLIN1 in the RNA-Seq data from our cisplatin-treated C4 cells. Only NF-κB, however, was significantly differentially expressed compared to control; thus, autophagy does not seem to be a mechanism by which RBM5 expression influences membrane integrity in cisplatin-treated SCLC cells.
2.8. In patient samples, RNA-Seq shows that RBM5 expression is reduced by 50% in tumors and that similar pathways are disrupted
To determine if the RBM5-influenced pathways that we identified in vitro were also influenced in vivo, we carried out RNA-Seq on primary tissue specimens. SCLC tumor resection is not routinely carried out in North America, but we did obtain two fresh frozen paired non-tumor and SCLC specimens. Transcriptome sequencing and analysis were performed on all four specimens.
We identified 14,346 and 14,119 genes differentially expressed between both non-tumor and tumor pairs, respectively, thus slightly more than half of the genes studied. Interestingly 12,116 (∼85%) of these differentially expressed genes were common to both paired groups, suggesting that, contrary to what one might expect in a tumor, a fairly conserved mechanism is involved in the evolution of SCLC, a favorable feature for treatment development.
RBM5 expression was very similar in both non-tumor specimens, and decreased by half in the corresponding tumor specimens; from 40.43 FPKM to 18.55 FPKM in the first patient specimens, and from 39.00 FPKM to 22.30 FPKM in the second patient specimens. This significant decrease of approximately 50% suggests that loss of heterozygosity (LOH) may be the cause of decreased RBM5 expression in SCLC. In fact, LOH of a portion of 3p21.3 close to RBM5 has been observed in over 95% of SCLC and 70% of NSCLC (Ji et al., 2005; Kok et al., 1997; Lerman and Minna, 2000; Sutherland et al., 2010; Wei et al., 1996; Wistuba et al., 2000). It is important to note that the RBM5 expression values in the non-tumor samples are very similar to that of T2 (58.26 FPKM), thus lending physiological relevance to our in vitro model.
Using FIDEA with the KEGG database, 34 pathways were shown to be significantly differentially regulated in both tumor specimens compared to their respective non-tumor control (Table 7). GSAASeqSP with MSigDB Hallmark gene set also showed that many gene sets were enriched; 18 in the first patient sample, and 26 in the second, with 15 gene sets being common between both sample pairs (Table 8). Differentially expressed pathways, as determined by both FIDEA and GSAASeqSP, included almost all of the RBM5-influenced pathways identified above, including many transformation-associated pathways, notably ‘Pathways in cancer’, ‘Cell cycle’, ‘Small cell lung cancer’, ‘Axon guidance’, ‘p53 signaling’, ‘TNFα signaling via NFĸB’ and ‘Apoptosis’. This high level of correlation between significantly differentially expressed pathways in our SCLC patient samples and the RBM5 expressing cell lines supports, once again, our conclusion that RBM5 plays a role influencing pathways that are very important to the transformed state of SCLC cells.
Table 7.
Significantly altered pathways between both patient SCLC tumor/non-tumor pairs as determined by FIDEA analysis with KEGG database (using the samples’ RNA-Seq results). Pathways are listed based on significance of enrichment in the first patient sample.
| Pathway | Non-tumor/tumor pair 1 (Benjamini value) | Non-tumor/tumor pair 2 (Benjamini value) |
|---|---|---|
| Pathways in cancer | 1.23E-12 | 6.36E-14 |
| Cell cycle | 9.75E-10 | 1.99E-10 |
| Regulation of actin cytoskeleton | 3.03E-06 | 3.06E-07 |
| Focal adhesion | 3.03E-06 | 5.50E-08 |
| Chronic myeloid leukemia | 2.60E-05 | 7.09E-10 |
| p53 signaling pathway | 2.60E-05 | 2.23E-06 |
| Fc gamma R-mediated phagocytosis | 2.64E-05 | 3.95E-06 |
| Neurotrophin signaling pathway | 1.11E-04 | 3.68E-10 |
| Colorectal cancer | 1.64E-04 | 5.82E-08 |
| Bacterial invasion of epithelial cells | 1.82E-04 | 1.22E-06 |
| Pancreatic cancer | 1.82E-04 | 1.22E-06 |
| Phosphatidylinositol signaling system | 2.81E-04 | 4.93E-04 |
| Prostate cancer | 3.58E-04 | 1.58E-06 |
| Small cell lung cancer | 6.00E-04 | 3.74E-04 |
| Axon guidance | 1.04E-03 | 1.16E-06 |
| Leukocyte transendothelial migration | 1.37E-03 | 3.74E-04 |
| Acute myeloid leukemia | 1.73E-03 | 1.27E-05 |
| Renal cell carcinoma | 1.79E-03 | 3.74E-04 |
| Inositol phosphate metabolism | 2.29E-03 | 1.39E-02 |
| Ubiquitin mediated proteolysis | 4.01E-03 | 6.36E-04 |
| Salmonella infection | 4.20E-03 | 1.68E-02 |
| Adherens junction | 4.20E-03 | 9.09E-06 |
| DNA replication | 4.20E-03 | 1.39E-02 |
| T cell receptor signaling pathway | 7.53E-03 | 2.78E-05 |
| mTOR signaling pathway | 8.57E-03 | 4.60E-04 |
| Base excision repair | 1.69E-02 | 1.26E-02 |
| Glioma | 1.72E-02 | 1.31E-04 |
| Shigellosis | 1.76E-02 | 2.25E-05 |
| B cell receptor signaling pathway | 1.78E-02 | 8.53E-04 |
| MAPK signaling pathway | 1.78E-02 | 4.77E-02 |
| ErbB signaling pathway | 1.97E-02 | 1.57E-05 |
| Endometrial cancer | 2.21E-02 | 1.57E-05 |
| N-Glycan biosynthesis | 3.89E-02 | 1.23E-02 |
| Chemokine signaling pathway | 4.69E-02 | 1.35E-04 |
Table 8.
Altered gene sets with FDRs below 1% in both patient SCLC tumor/non-tumor pairs, as determined by GSAASeqSP analysis with the MSigDB Hallmark gene set (using the samples’ RNA-Seq results). Pathways are listed based on FDR in the first patient sample.
| Non-tumor/tumor pair 1 |
Non-tumor/tumor pair 2 |
|||
|---|---|---|---|---|
| Gene set | p-value | FDR | p-value | FDR |
| TNFα signaling via NFĸB* | 0.0 | 0.0 | 0.0 | 0.0 |
| IL6 JK STAT3 signaling* | 0.0 | 0.0 | 0.0 | 0.0 |
| G2M checkpoint | 0.0 | 0.0 | 0.0 | 0.0 |
| Estrogen response late* | 0.0 | 0.0 | 0.0 | 0.0 |
| Interferon γ response | 0.0 | 0.0 | 0.0 | 0.0 |
| E2F targets | 0.0 | 0.0 | 0.0 | 0.0 |
| EMT* | 0.0 | 0.0 | 0.0 | 0.0 |
| Inflammatory response* | 0.0 | 0.0 | 0.0 | 0.0 |
| KRAS signaling up* | 0.0 | 0.0 | 0.0 | 0.0 |
| Pancreas β cells* | 0.0 | 0.0 | 0.0 | 0.0 |
| UV response down* | 0.0 | 0.01 | 0.0 | 0.0 |
| Interferon α response* | 0.0 | 0.01 | 0.0 | 0.01 |
| IL2 STAT5 signaling* | 0.0 | 0.01 | 0.0 | 0.0 |
| Apoptosis* | 0.0 | 0.01 | 0.0 | 0.0 |
| Coagulation* | 0.0 | 0.01 | 0.0 | 0.0 |
Beside a gene set name indicates it’s enrichment in untreated control vs. C4 samples, at an FDR < 10%.
3. Conclusion
Lung cancer is the leading cause of cancer-related deaths worldwide. The most aggressive form of lung cancer is SCLC, with a staggering 95% of diagnosed patients succumbing to the disease (Govindan et al., 2006). This high mortality rate clearly demonstrates the need for more effective screening techniques and treatment options. Our results show, for the first time, that RBM5 expression is important to the maintenance of the non-transformed state of lung cells, and that this is accomplished via direct regulation of the cell cycle and apoptosis, and indirect regulation of overall cell death, angiogenesis, and cell adhesion. Functional studies confirmed that RBM5 expression in SCLC (1) slowed the cell cycle, and (2) decreased membrane integrity, partially via increased apoptosis when cells were treated with the chemotherapy agent cisplatin. Therefore, decreased RBM5 expression, as is observed in 95% of SCLC, may be a critical step in the establishment of this disease. In a clinical setting, downregulation of RBM5 expression could be a novel biomarker for the determination of SCLC risk. Therapeutic options involving RBM5 and/or direct targets, or pathways altered by RBM5 expression, may also be very fruitful avenues to pursue. Furthermore, due to the significant sensitization RBM5 expression had on cisplatin-treated samples, RBM5 expression could be a valuable predictive aid for assessing how a patient may respond to this chemotherapy. Ultimately, this work demonstrates, for the first time, the importance of RBM5 to SCLC. Our results present a stepping-off point for additional targeted functional work.
4. Materials & methods
4.1. Cell culture
SCLC GLC20 cells were a kind gift from the late Dr. Charles Buys from the University of Groningen (Groningen, Netherlands). GLC20 cells and sublines pcDNA3, T2 and C4 were maintained in RPMI 1640 (Gibco, Life Technologies) supplemented with 10% fetal bovine serum (FBS, Gibco) (termed complete media), with the addition of 0.1 mg/mL G418/Geneticin (Gibco) for pcDNA3, T2 and C4 sublines. Cells were maintained at 37 °C with 5% CO2 in a humidified chamber.
4.2. Southern blotting
Genomic DNA was isolated by spooling, following overnight incubation in Tail Buffer (1% SDS, 0.1 M NaCl, 0.1 M EDTA, 0.05 M Tris, pH8), treated with 50 μg/ml of RNase A (Amersham Biosciences) and cleaned using Qiagen Genomic-tip 100/G columns, following the manufacturer’s instructions. The Southern blot was prepared and probed as previously described (Rintala-Maki and Sutherland, 2009). The RBM5 probe was prepared using the PROSTAR HF single tube RT-PCR System (Stratagene), labelling with radioactive phosphate, and cleaning with a G25 sepharose column, following manufacturer’s instructions.
4.3. GLC20 apoptosis profiles
GLC20 cells were diluted 1:2 in a 24 well plate 24 h before treatment. The following day, cells were treated with cisplatin (Sigma-Aldrich) dissolved in dimethyl sulfoxide (DMSO) and/or etoposide (Sigma-Aldrich) dissolved in DMSO. Cells were then incubated for varying amounts of time, as shown in Fig. 2, at 37 °C in a 5% CO2 humidified incubator.
4.4. Western blotting
Primary antibodies used were rabbit anti-RBM5 LUCA-15-UK (non-commercially available) (1:2,500 or 1:5,000) (Sutherland et al., 2000), rabbit anti-human PARP (1:1,500–1:2,000, C2-10: BD Pharmingen) and mouse anti-α-tubulin primary antibody (1:10,000, sc-8035, Santa Cruz Biotechnology, Inc.). Secondary antibodies used were goat anti-mouse HRP-conjugated secondary antibody (1:10,000, sc-2005; Santa Cruz Biotechnologies Inc.) or goat anti-rabbit HRP-conjugated secondary antibody (1:10,000, sc-2004; Santa Cruz Biotechnologies Inc.). Protein was exposed using Amersham ECL Western blotting detection reagents (GE-Healthcare) to Amersham Hyperfilm (GE-Healthcare). Film was then developed using a SRX-101A medical film processor (Konica Minolta Medical and Graphic Inc.).
4.5. Establishment of stable RBM5-expressing GLC20 sublines
GLC20 sublines were established following DMRIE-C transfection with pcDNA3 or pcDNA3.RBM5, and stable selection with G418 at 1.0 mg/ml in soft agar for 120 days. Detailed protocol is as follows; Cells were passaged 1:2, 24–48 h prior to transfection. 10 ml of cell culture (∼ 2 × 106 cells) were used in each transfection, with 24 μl DMRIE-C (Life Technologies) and 8 μg total DNA (pcDNA3 or pcDNA3.RBM5). DMREI-C and DNA were incubated for ∼30 min prior to cell addition. The cell/DMREI-C/DNA mix was then incubated at 37 °C and 5% CO2 for 4 h, and the transfection terminated by adding serum. Transfected cells were selected using G418, at a concentration of 1.0 mg/ml, a concentration previously determined to kill all untransfected cells by seven days. Following transfection, a clonal population of cells was established following plating in soft agar, as previously described (Longthorne and Williams, 1997). Four empty vector transfected clones and five RBM5 transfected clones were picked following 36 days of G418 selection in soft agar. After 120 days of continuous growth in G418 (following the first dilution of 10 ml of stably transfected cells, the selection reagent concentration was reduced from 1.0 to 0.1 mg/ml), only two of the five RBM5-transfected clones survived. One of these, designated C4, as well as one empty vector control clone, were used in subsequent studies. A second transfection was carried out in order to generate a pooled population of RBM5-transfected cells (eventually designated T2).
4.6. RNA extraction
GLC20 RNA samples were isolated using Tri-Reagent (BioCan Scientific). For RNA templates used in PCR, reverse transcription was performed as previously described (Loiselle and Sutherland, 2014). In regards to the Ontario Tumour Bank (OTB) tissue samples, 20 mg of tissue was cut with a sterile blade from fresh frozen tissue specimens that had been stored at −80 °C. The 20 mg tissue piece was transferred to a Bessman Tissue Pulverizer (VWR) that had been placed in liquid nitrogen for 10 min. Once the pulverizer was secured, a hammer was used to smash the tissue until it obtained a powder-like consistency. Powdered tissue was transferred to a solution containing Buffer RLT (Qiagen) and 0.14 mM β-mercaptoethanol, and homogenized using a Polytron PT 1300 D Homogenizer (Kinematica). The lysate was centrifuged for 3 min at 17000 x g to pellet tissue matter that did not homogenize. Supernatant was transferred to an Allprep DNA/RNA/Protein Mini Kit spin column (Qiagen) and RNA was extracted according to manufacturer’s instructions. It is important to note that RNA was extracted from two 20 mg pieces of tissue, and combined.
4.7. PCR
RBM5 genomic DNA PCR was performed using Gen1E2Fc (exon 2: 5′-CTTCAGTGGGACAATGGGTTCAGA-3′) and Gen2E3I2R (exon 3/intron 2: 5′-CCACTACGCTCTGTTCTACTCACTCTGCCA-3′) primers, with the following PCR amplification program 95 °C 5 min, [95 °C 30 s/65 °C 30 s/68 °C 2 min] (40 cycles), 68 °C 10 min. RBM5 RT-PCR was performed using LU15(2) (exon 4) and LU15(3) (exon 8) primers, as previously described (Sutherland et al., 2000). GAPDH was amplified using GAPDH-F (exon 6: 5′ AACACAGTCCATGCCATCAC 3′) and GAPDH-R (exon 7: 5′ TCCACCACCCTGTTGCTGTA), with an annealing temperature of 58−59 °C and 25–35 amplification cycles. PCR and product visualization were carried out as previously described (Loiselle and Sutherland, 2014). Approximate primer locations shown in Fig. 1G.
4.8. RNA-Sequencing and analyses
Extracted RNA was sent to the Donnelly Sequencing Centre (Toronto, Canada) for Illumina TruSeq stranded mRNA library preparation with Ribozero depletion, followed by paired-end high throughput sequencing using the Illumina HiSeq 2500 platform.
All four untreated GLC20 subline samples were multiplexed together during sequencing, as were the four cisplatin-treated samples. It is important to note that there were some differences between sample preparation and sequencing for cisplatin treated and untreated RNA-Seq samples; (1) untreated samples were DNase treated prior to sequencing, (2) reads were 100 bp for untreated samples and 125 bp for cisplatin treated samples, and (3) untreated samples were sequenced in duplicate (multiplexed on each of two paired-end lanes), while treated samples were not sequenced in duplicate. Due to the longer read length for the cisplatin treated samples and overall higher output, however, similar depth was achieved for treated and untreated samples. Nonetheless, we only ran analysis within the treated and untreated groups, and compared the outputs, so as to not potentially introduce sequencing biases into our analyses. All four OTB specimens (two paired tumor and non-tumor specimens) were also multiplexed and sequenced in duplicate.
Transcriptome sequencing data quality was verified by FastQC (Babraham Bioinformatics, http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). All specimens had quality score distributions over all sequences above 37 (on a phred 33 quality scale). Primers and adapters used for sequencing were then removed using cutadapt version 1.4.2 (Martin, 2011) using a quality cut-off of 26, as recommended (Del Fabbro et al., 2013). Following trimming, data quality was verified using FastQC. Trimmed reads were analyzed using the Tuxedo suite tools as follows: (1) TopHat 2.0.11 (Kim et al., 2013) was used to align reads to the human reference genome USCS hg19, (2) Cufflinks 2.1.1 (Trapnell et al., 2010) was used to assemble the mapped reads and obtain FPKM values for each investigated gene and isoform, (3) Cuffdiff 2.1.1 (Trapnell et al., 2013) was used to investigate differential expression, and (4) CummeRbund (Trapnell et al., 2010) was used for visualization. Samtools (Li et al., 2009) and Picard (http://broadinstitute.github.io/picard/) were used to assess mapping quality. For untreated and treated GLC20 subline RNA-Seq samples, all samples had a very good percentage of overall mapped reads and concordantly mapped paired reads, with small standard deviations, permitting comparison between samples; in untreated samples 94.06% ± 0.44% (SD) reads mapped, with 89.18% ± 0.28% being concordant for both forward and reverse reads, and in the cisplatin treated samples 94.51% ± 0.87% reads were mapped, with 91.73 ± 1.07 of paired reads mapping concordantly. Values were slightly lower for OTB patient specimens, as expected, with 89.84 ± 3.08% reads mapped and 86.06 ± 2.87% concordant.
Prior to differential expression analysis, gene expression levels in both control samples (parental GLC20 and GLC20.pcDNA3) were compared to see if they were sufficiently similar to be combined into one experimental group; more replicates provides greater power and accuracy in RNA-Seq experiments (Liu et al., 2014). In fact, gene expression levels in both samples were very highly correlated (r = 0.9699 in untreated samples and r = 0.9813 in cisplatin treated samples), and thus both controls were combined into one experimental group.
Pathway enrichment was investigated using the FIDEA (Functional Interpretation of Differential Expression Analysis) program (D'Andrea et al., 2013) with the KEGG database (Kanehisa and Goto, 2000; Kanehisa et al., 2016). Pathway analysis was also performed using GSAASeqSP (Gene Set Association Analysis for RNA-Seq with Sample Permutation) (Xiong et al., 2014) with the Molecular Signatures Database (MSigDB) Hallmark gene set collection (Liberzon et al., 2015). Since each program uses a different algorithm in their calculations, and each gene set/functional interaction pathway set is curated separately, with different foci, a broad view of affected cellular processes affected is gained, and results can be compared between programs to identify the most robust changes.
For analysis of alternative splicing events in our RNA-Seq data, stringent parameters were used in order to identify alternative splicing events: (1) the expression of at least one alternative splice variant had to be significantly upregulated, and (2) the expression of at least one other alternative splice variant had to be significantly downregulated.
4.9. RNA immunoprecipitation followed by next generation sequencing (RIP-Seq)
The basic RIP technique was carried out as previously described (Jain et al., 2011). RNA associated with the immunoprecipitated samples was sequenced by the Donnelly Sequencing Centre. Illumina TruSeq Stranded Total RNA library preparation, including Ribozero Gold depletion, was performed along with random priming. Samples were sequenced in duplicate on the Illumina HiSeq 2500 platform, with each run being paired-ended, 125 bp reads.
RIP-Seq results were analyzed using the Tuxedo Pipeline (Trapnell et al., 2012), with the same quality control steps used for RNA-Seq analysis. Using the FPKM and log2-fold change values generated by Cuffdiff, the following inclusion criteria were used to distinguish RBM5 targets from IP contaminants: the RBM5 RIP sample target had to have (1) an FPKM value greater than one, (2) a log2-fold change greater than one, and (3) a positive log2-fold change (meaning it was more highly expressed than in the control RIP).
4.10. Cell counting, cell growth and cell death assays
Cells were plated at 10,000 per well in 96-well flat-bottom plates at a density of 50 cells/μL, in triplicate wells per treatment. Cells were left for 24 h at 37 °C with 5% CO2 in a humidified chamber. After 24 h, the cells were treated with the following conditions: left untreated, saline (0.9% NaCl in H2O) control, 1.0 μM cisplatin in saline for 0, 2, 4, 6, 8 and 10 days, or 0.1, 0.5, 10.0 and 100.0 μM cisplatin for 4 and 8 days. Cisplatin (Sigma) was prepared, as previously described (Hall et al., 2014), in saline at a concentration of 1.0 mg/mL. The cells were then counted every other day by transferring cells to a 96-well Vee-bottom plate and subjecting them to centrifugation at 500 x g for 5 min at 21 °C. The supernatant was discarded and cells treated with 0.25% Trypsin-EDTA for 10 min at 37 °C with 5% CO2 in a humidified chamber. Complete media was added to cells and they were subjected to centrifugation at 500 x g for 5 min 21 °C. Cells were resuspended in complete media and counted in a 1:1 ratio of cells in complete media and 0.2% nigrosin, using a hemocytometer.
Live cells were counted as cells with intact membranes, characterized by a lack of blue/purple nigrosin within the cells, and dead cells were counted as cells without intact membranes, characterized by the presence of blue/purple nigrosin within the cell. Live cell counts were used to monitor cell growth, relative to day 0 counts, and the average of biological triplicates was plotted. A two-way ANOVA was performed with Bonferroni post-hoc analysis, comparing all subline cell growth to the pcDNA3 subline, calculated using Graphpad Prism 5. Percent intact membrane by nigrosin was calculated using the following equation: percent intact membrane by nigrosin = number of live cells divided by (number of live cells + number of dead cells) x 100.
Percent intact membrane by nigrosin for the 1.0 μM cisplatin values was also made relative to saline controls, and the average of biological triplicates was plotted. A two-way ANOVA was performed with Bonferroni post-hoc analysis, comparing all subline results to the pcDNA3 subline, calculated using Graphpad Prism 5. Day eight cisplatin results were made relative to the saline control for the calculation of EC50 values. EC50 values were calculated using ‘log (inhibitor) vs. response (three parameters)’ on Graphpad Prism 5, and the average of biological triplicates was plotted with the saline control represented at 10−10 M on the graphs. A one-way ANOVA was performed with Tukey post-hoc analysis, comparing all sublines to the pcDNA3 subline.
4.11. Apoptosis assays − Fluorescent microscopy
GLC20 cells and sublines were counted and 2.0 × 106 cells were plated in T75 flasks at a cell density of 50 cells/μL. After 24 h incubation at 37 °C with 5% CO2 in a humidified chamber, cells were either exposed to 5.0 μM cisplatin or left untreated for 4 days. A fraction of cells was collected by centrifugation at 149 x g for 7 min at 21 °C and supernatant was discarded, followed by centrifugation at 5,900 x g for 2 min at 21 °C, with supernatant discarded, and the pellet stored at −80 °C for PARP cleavage analysis by Western Blot.
Another fraction of cells was used for apoptosis analysis by fluorescent microscopy. These cells were subjected to centrifugation at 149 x g for 7 min at 21 °C, supernatant was discarded, and the cells were treated with 0.25% Trypsin-EDTA for 10 min at 37 °C with 5% CO2 in a humidified chamber, to obtain a single cell suspension. Cells were then washed two times in complete media and centrifuged at 500 x g for 5 min at 21 °C, with changes in media between each centrifugation, then counted in a 1:1 ratio of cells in complete media and 0.2% nigrosin, using a hemocytometer. 0.5 × 106 cells were resuspended at a 1000 cell/μL density in cold PBS for fluorescence analysis. If fewer than 0.5 × 106 cells were counted, cells were adjusted to a smaller volume at a 1000 cell/μL density in cold PBS.
Before staining, cells were washed three times in cold PBS at 5,900 x g for 2 min at 21 °C, with changes in cold PBS between centrifugations, then resuspended in Annexin V binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2) at a density of 1000 cells/μL. Cells were triple stained with 7-aminoactinomycin D (7AAD) in DMSO, Annexin V-AlexaFlour® 488 conjugated, and Hoescht 33342 Nucblue® Live Cell Stain ReadyProbe Reagent (all Life Technologies) at 0.02 mg/mL, 5 μL/100 μL, and 1 drop/500 μL concentrations, respectively, at 21 °C for 15 min in the dark. Cells were then washed three times in cold Annexin V binding buffer at 5,900 x g for 10 min at 21 °C, then resuspended in 100 μL of cold PBS.
Samples were loaded into Cytospin™ columns (Symport, VWR International) that were pre-loaded with a microscope slide (VistaVision, VWR International). Cells were centrifuged onto the microscope slides at 500 rpm for 2 min 21 °C in a Shandon Cytospin™ 4 cytocentrifuge (Thermo Scientific), rotor # 4127 0806 59930093. Samples on slides were air-dried, then fixed with 4% paraformaldehyde (Sigma-Aldrich) in PBS at 21 °C in the dark for 10 min. Slides were washed sequentially in three changes of PBS, then air-dried. 90% glycerol (Sigma-Aldrich) in PBS was added to the samples and a No. 1 coverslip (VWR) was placed on top of the sample. The cover slip was sealed with nail polish and the slides stored at 4 °C in the dark until visualized (∼24 h later).
Cells were visualized using an Olympus 1 × 73 Microscope (Olympus Life Sciences). Fluorphores were excited using Lumen Dynamics Xcite 120 LED (Lumen Dynamics), Olympus LED PS and LBM laser systems (Olympus Life Sciences). Fluorescence emission spectra were captured for Hoescht 33342 (Ex: 250 nm/Em: 461 nm), AlexaFlour® 488 (Ex: 495 nm/Em: 519 nm), and 7-AAD (Ex: 546 nm/Em: 647 nm). In addition to fluorphores, cell morphology was observed using phase contrast (images not included). Images of the stained cells were captured using the Olympus DP80 camera (Olympus Life Sciences) and CellSens Dimensions imaging software (Olympus Life Sciences). Images obtained that were later counted underwent no post-production adjustments. Images presented underwent post-production adjustments using the ‘Adjust Display’ function in the CellSens Dimensions imaging software. In brief, background colour intensities were excluded from the images using the histogram tool. Colour threshold intensity was then increased. These changes were applied to all the images and at the same intensities.
Exposure times and gain were made constant during the imaging of each biological replicate. Although exposure times varied, the Hoechst 33342 stain was exposed for roughly 250 milliseconds (ms), the Annexin V- Alex-Fluor 488 stain was exposed for roughly 2 seconds (s) with a gain of 2X, and the 7-AAD stain was exposed for roughly 800 ms. All images were taken using a 40X objective lens. ‘Live’ events were defined as cells with visually uncondensed nuclei, stained with Hoechst 33342, and lacking green Annexin V stain or red 7-AAD stain. ‘Early Apoptosis’ events were defined as cells that had either or both green Annexin V stain (indicative of phosphatidylserine flipping) and condensed nuclei (condensed blue Hoechst 33342 staining). Lastly, ‘Late Apoptosis/Necrosis’ was defined as the loss of membrane integrity, which was indicated by the presence of the red stain of 7-AAD (made purple/pink in images). Three biological replicates were performed. For each biological replicate, the events were totalled between ten fields of view counted. A minimum of 300 events was counted per biological replicate. Values were then transformed to a percentage of the total number of events that were counted, for each biological replicate. The average of the three biological replicates was presented and one-way ANOVA was performed between the sublines for each defined event. Tukey post-hoc analysis was done, comparing all the sublines to the pcDNA3 subline.
Declarations
Author contribution statement
Julie J. Loiselle: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Wrote the paper.
Justin G. Roy: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data.
Leslie C. Sutherland: Conceived and designed the experiments; Analyzed and interpreted the data.
Funding statement
J.J.L. was supported by an Alexander Graham Bell Canada Graduate Scholarship from the Natural Sciences and Engineering Research Council of Canada (NSERC). The project was supported by funding to L.S. from NSERC grant #9043429 and the Northern Ontario Heritage Fund Corporation (NOHFC): Youth Internship Program. Support was also provided to L.C.S. from the Northern Cancer Foundation (NCF) and the Northeastern Ontario Cancer Therapeutics Research Initiative (CTRI).
Competing interest statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
Acknowledgments
The authors thank N. Rintala-Maki for characterization of the GLC20 cell line, B. Koenderink for RBM5 antibody analysis, S. Tessier for help with RNA-Seq sample preparation and S. Hunt for extraction of RNA from the primary tissue specimens. Biological materials were provided by the Ontario Tumor Bank, which is funded by the Ontario Institute for Cancer Research.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- Abreu A.P., Dauber A., Macedo D.B., Noel S.D., Brito V.N., Gill J.C., Cukier P., Thompson I.R., Navarro V.M., Gagliardi P.C. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N. Engl. J. Med. 2013;368:2467–2475. doi: 10.1056/NEJMoa1302160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeloni D. Molecular analysis of deletions in human chromosome 3p21 and the role of resident cancer genes in disease. Brief. Funct. Genomic. Proteomic. 2007 doi: 10.1093/bfgp/elm007. [DOI] [PubMed] [Google Scholar]
- Bechara E.G., Sebestyen E., Bernardis I., Eyras E., Valcarcel J. RBM5, 6, and 10 Differentially Regulate NUMB Alternative Splicing to Control Cancer Cell Proliferation. Mol. Cell. 2013;52:720–733. doi: 10.1016/j.molcel.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Brennan D., Hu Y., Joubeh S., Choi Y.W., Whitaker-Menezes D., O'Brien T., Uitto J., Rodeck U., Mahoney M.G. Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J. Cell. Sci. 2007;120:758–771. doi: 10.1242/jcs.03392. [DOI] [PubMed] [Google Scholar]
- Brennan D., Mahoney M.G. Increased expression of Dsg2 in malignant skin carcinomas: A tissue-microarray based study. Cell Adh. Migr. 2009;3:148–154. doi: 10.4161/cam.3.2.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedotal A., Kerjan G., Moreau-Fauvarque C. The brain within the tumor: new roles for axon guidance molecules in cancers. Cell Death Differ. 2005;12:1044–1056. doi: 10.1038/sj.cdd.4401707. [DOI] [PubMed] [Google Scholar]
- D'Andrea D., Grassi L., Mazzapioda M., Tramontano A. FIDEA: a server for the functional interpretation of differential expression analysis. Nucleic Acids Res. 2013;41:W84–W88. doi: 10.1093/nar/gkt516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Fabbro C., Scalabrin S., Morgante M., Giorgi F.M. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0085024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfino F.J., Stevenson H., Smithgall T.E. A growth-suppressive function for the c-fes protein-tyrosine kinase in colorectal cancer. J. Biol. Chem. 2006;281:8829–8835. doi: 10.1074/jbc.M507331200. [DOI] [PubMed] [Google Scholar]
- Forsyth C.B., Tang Y., Shaikh M., Zhang L., Keshavarzian A. Alcohol stimulates activation of Snail, epidermal growth factor receptor signaling, and biomarkers of epithelial-mesenchymal transition in colon and breast cancer cells. Alcohol Clin. Exp. Res. 2010;34:19–31. doi: 10.1111/j.1530-0277.2009.01061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi T., Tachibana I., Hoshida Y., Kimura H., Takeda Y., Kijima T., Nishino K., Goto H., Yoneda T., Kumagai T. Expression of tetraspanins in human lung cancer cells: frequent downregulation of CD9 and its contribution to cell motility in small cell lung cancer. Oncogene. 2003;22:674–687. doi: 10.1038/sj.onc.1206106. [DOI] [PubMed] [Google Scholar]
- Fushimi K., Ray P., Kar A., Wang L., Sutherland L.C., Wu J.Y. Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc. Natl. Acad. Sci. U S A. 2008;105:15708–15713. doi: 10.1073/pnas.0805569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar L.E., McNamara E.J., Gay E.G., Putnam J.B., Crawford J., Herbst R.S., Bonner J.A. Small-cell lung cancer: prognostic factors and changing treatment over 15 years. Clin. Lung Cancer. 2012;13:115–122. doi: 10.1016/j.cllc.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Govindan R., Page N., Morgensztern D., Read W., Tierney R., Vlahiotis A., Spitznagel E.L., Piccirillo J. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J. Clin. Oncol. 2006;24:4539–4544. doi: 10.1200/JCO.2005.04.4859. [DOI] [PubMed] [Google Scholar]
- Guallar D., Wang J. RNA-binding proteins in pluripotency, differentiation, and reprogramming. Front. Biol. (Beijing) 2014;9:389–409. doi: 10.1007/s11515-014-1326-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A., Nitoiu D., Brennan-Crispi D., Addya S., Riobo N.A., Kelsell D.P., Mahoney M.G. Cell cycle- and cancer-associated gene networks activated by Dsg2: evidence of cystatin A deregulation and a potential role in cell-cell adhesion. PLoS One. 2015;10 doi: 10.1371/journal.pone.0120091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M.D., Telma K.A., Chang K.E., Lee T.D., Madigan J.P., Lloyd J.R., Goldlust I.S., Hoeschele J.D., Gottesman M.M. Say no to DMSO: dimethylsulfoxide inactivates cisplatin, carboplatin, and other platinum complexes. Cancer Res. 2014;74:3913–3922. doi: 10.1158/0008-5472.CAN-14-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H., Iwatsuki K., Ohtsuka M., Han G.W., Kaneko F. Abnormal desmoglein expression by squamous cell carcinoma cells. Acta Derm. Venereol. 1996;76:417–420. doi: 10.2340/0001555576417420. [DOI] [PubMed] [Google Scholar]
- Hegele A., Kamburov A., Grossmann A., Sourlis C., Wowro S., Weimann M., Will C.L., Pena V., Luhrmann R., Stelzl U. Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell. 2012;45:567–580. doi: 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- Huan J., Gao Y., Xu J., Sheng W., Zhu W., Zhang S., Cao J., Ji J., Zhang L., Tian Y. Overexpression of CD9 correlates with tumor stage and lymph node metastasis in esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2015;8:3054–3061. [PMC free article] [PubMed] [Google Scholar]
- Humphries A.D., Streimann I.C., Stojanovski D., Johnston A.J., Yano M., Hoogenraad N.J., Ryan M.T. Dissection of the mitochondrial import and assembly pathway for human Tom40. J. Biol. Chem. 2005;280:11535–11543. doi: 10.1074/jbc.M413816200. [DOI] [PubMed] [Google Scholar]
- Jackman D.M., Johnson B.E. Small-cell lung cancer. Lancet. 2005;366:1385–1396. doi: 10.1016/S0140-6736(05)67569-1. [DOI] [PubMed] [Google Scholar]
- Jain R., Devine T., George A.D., Chittur S.V., Baroni T.E., Penalva L.O., Tenenbaum S.A. RIP-Chip Analysis: RNA-Binding Protein Immunoprecipitation-Microarray (Chip) Profiling. In: Nielsen H., editor. RNA. Humana Press; 2011. pp. 247–263. [DOI] [PubMed] [Google Scholar]
- Jeon W.K., Hong H.Y., Seo W.C., Lim K.H., Lee H.Y., Kim W.J., Song S.Y., Kim B.C. Smad7 sensitizes A549 lung cancer cells to cisplatin-induced apoptosis through heme oxygenase-1 inhibition. Biochem. Biophys. Res. Commun. 2012;420:288–292. doi: 10.1016/j.bbrc.2012.02.151. [DOI] [PubMed] [Google Scholar]
- Ji L., Minna J.D., Roth J.A. 3p21.3 tumor suppressor cluster: prospects for translational applications. Future. Oncol. 2005;1:79–92. doi: 10.1517/14796694.1.1.79. [DOI] [PubMed] [Google Scholar]
- Jin W., Niu Z., Xu D., Li X. RBM5 promotes exon 4 skipping of AID pre-mRNA by competing with the binding of U2AF65 to the polypyrimidine tract. FEBS Lett. 2012;586:3852–3857. doi: 10.1016/j.febslet.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Johnson D.H., Schiller J.H., Bunn P.A., Jr. Recent clinical advances in lung cancer management. J. Clin. Oncol. 2014;32:973–982. doi: 10.1200/JCO.2013.53.1228. [DOI] [PubMed] [Google Scholar]
- Kanda S., Miyata Y., Kanetake H., Smithgall T.E. Downregulation of the c-Fes protein-tyrosine kinase inhibits the proliferation of human renal carcinoma cells. Int. J. Oncol. 2009;34:89–96. [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Ishida J., Musashi M., Ota S., Yoshida T., Shimizu Y., Chuma M., Kawakami H., Asaka M., Tanaka J. p53 transactivation is involved in the antiproliferative activity of the putative tumor suppressor RBM5. Int. J. Cancer. 2011;128:304–318. doi: 10.1002/ijc.25345. [DOI] [PubMed] [Google Scholar]
- Kok K., Naylor S.L., Buys C.H. Deletions of the short arm of chromosome 3 in solid tumors and the search for suppressor genes. Adv. Cancer Res. 1997;71:27–92. doi: 10.1016/s0065-230x(08)60096-2. [DOI] [PubMed] [Google Scholar]
- Kok K., van den Berg A., Veldhuis P.M., van der Veen A.Y., Franke M., Schoenmakers E.F., Hulsbeek M.M., van der Hout A.H., de Leij L., van de Ven W. A homozygous deletion in a small cell lung cancer cell line involving a 3p21 region with a marked instability in yeast artificial chromosomes. Cancer Res. 1994;54:4183–4187. [PubMed] [Google Scholar]
- Kurzen H., Munzing I., Hartschuh W. Expression of desmosomal proteins in squamous cell carcinomas of the skin. J. Cutan. Pathol. 2003;30:621–630. doi: 10.1034/j.1600-0560.2003.00122.x. [DOI] [PubMed] [Google Scholar]
- Lerman M.I., Minna J.D. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer Res. 2000;60:6116–6133. [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Wang K., Zhang J., Zhao L., Liang H., Shao C., Sutherland L.C. The 3p21.3 tumor suppressor RBM5 resensitizes cisplatin-resistant human non-small cell lung cancer cells to cisplatin. Cancer Epidemiol. 2012;36:481–489. doi: 10.1016/j.canep.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhou J., White K.P. RNA-seq differential expression studies: more sequence or more replication? Bioinformatics. 2014;30:301–304. doi: 10.1093/bioinformatics/btt688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiselle J.J., Sutherland L.C. Differential downregulation of Rbm5 and Rbm10 during skeletal and cardiac differentiation. In Vitro Cell. Dev. Biol. Anim. 2014;50:331–339. doi: 10.1007/s11626-013-9708-z. [DOI] [PubMed] [Google Scholar]
- Longthorne V.L., Williams G.T. Caspase activity is required for commitment to Fas-mediated apoptosis. EMBO J. 1997;16:3805–3812. doi: 10.1093/emboj/16.13.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal. 2011;17:10–12. [Google Scholar]
- Mueller C.F., Berger A., Zimmer S., Tiyerili V., Nickenig G. The heterogenous nuclear riboprotein S1-1 regulates AT1 receptor gene expression via transcriptional and posttranscriptional mechanisms. Arch. Biochem. Biophys. 2009;488:76–82. doi: 10.1016/j.abb.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Network T.C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Z., Jin W., Zhang L., Li X. Tumor suppressor RBM5 directly interacts with the DExD/H-box protein DHX15 and stimulates its helicase activity. FEBS Lett. 2012;586:977–983. doi: 10.1016/j.febslet.2012.02.052. [DOI] [PubMed] [Google Scholar]
- O'Bryan M.K., Clark B.J., McLaughlin E.A., D'Sylva R.J., O'Donnell L., Wilce J.A., Sutherland J., O'Connor A.E., Whittle B., Goodnow C.C. RBM5 Is a Male Germ Cell Splicing Factor and Is Required for Spermatid Differentiation and Male Fertility. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J.J., Boctor B.N., Jimenez C.A., Lopez R., Koegel A.K., Taschereau E.O., Phan D.T., Jacobsen S.E., Slamon D.J. Promoter methylation study of the H37/RBM5 tumor suppressor gene from the 3p21.3 human lung cancer tumor suppressor locus. Hum. Genet. 2008;123:55–64. doi: 10.1007/s00439-007-0449-5. [DOI] [PubMed] [Google Scholar]
- Oh J.J., Koegel A.K., Phan D.T., Razfar A., Slamon D.J. The two single nucleotide polymorphisms in the H37/RBM5 tumour suppressor gene at 3p21.3 correlated with different subtypes of non-small cell lung cancers. Lung Cancer. 2007;58:7–14. doi: 10.1016/j.lungcan.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J.J., Razfar A., Delgado I., Reed R.A., Malkina A., Boctor B., Slamon D.J. 3p21.3 tumor suppressor gene H37/Luca15/RBM5 inhibits growth of human lung cancer cells through cell cycle arrest and apoptosis. Cancer Res. 2006;66:3419–3427. doi: 10.1158/0008-5472.CAN-05-1667. [DOI] [PubMed] [Google Scholar]
- Oh J.J., West A.R., Fishbein M.C., Slamon D.J. A candidate tumor suppressor gene, H37, from the human lung cancer tumor suppressor locus 3p21.3. Cancer Res. 2002;62:3207–3213. [PubMed] [Google Scholar]
- Palmieri F. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol. Aspects Med. 2013;34:465–484. doi: 10.1016/j.mam.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S., Ross K.N., Lander E.S., Golub T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Rappa G., Green T.M., Karbanova J., Corbeil D., Lorico A. Tetraspanin CD9 determines invasiveness and tumorigenicity of human breast cancer cells. Oncotarget. 2015;6:7970–7991. doi: 10.18632/oncotarget.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintala-Maki N.D., Abrasonis V., Burd M., Sutherland L.C. Genetic instability of RBM5/LUCA-15/H37 in MCF-7 breast carcinoma sublines may affect susceptibility to apoptosis. Cell Biochem. Funct. 2004;22:307–313. doi: 10.1002/cbf.1106. [DOI] [PubMed] [Google Scholar]
- Rintala-Maki N.D., Sutherland L.C. LUCA-15/RBM5, a putative tumour suppressor, enhances multiple receptor-initiated death signals. Apoptosis. 2004;9:475–484. doi: 10.1023/B:APPT.0000031455.79352.57. [DOI] [PubMed] [Google Scholar]
- Rintala-Maki N.D., Sutherland L.C. Identification and characterisation of a novel antisense non-coding RNA from the RBM5 gene locus. Gene. 2009;445:7–16. doi: 10.1016/j.gene.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Segawa K., Kurata S., Yanagihashi Y., Brummelkamp T.R., Matsuda F., Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344:1164–1168. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- Sehgal L., Mathur R., Braun F.K., Wise J.F., Berkova Z., Neelapu S., Kwak L.W., Samaniego F. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia. 2014;28:2376–2387. doi: 10.1038/leu.2014.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamas-Din A., Brahmbhatt H., Leber B., Andrews D.W. BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- Shao C., Zhao L., Wang K., Xu W., Zhang J., Yang B. The tumor suppressor gene RBM5 inhibits lung adenocarcinoma cell growth and induces apoptosis. World J. Surg. Oncol. 2012;10:160. doi: 10.1186/1477-7819-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit E.F., de Vries E.G., Timmer-Bosscha H., de Leij L.F., Oosterhuis J.W., Scheper R.J., Weening J.J., Postmus P.E., Mulder N.H. In vitro response of human small-cell lung-cancer cell lines to chemotherapeutic drugs; no correlation with clinical data. Int. J. Cancer. 1992;51:72–78. doi: 10.1002/ijc.2910510115. [DOI] [PubMed] [Google Scholar]
- Su Z., Wang K., Li R., Yin J., Hao Y., Lv X., Li J., Zhao L., Du Y., Li P. Overexpression of RBM5 induces autophagy in human lung adenocarcinoma cells. World J. Surg. Oncol. 2016;14:57. doi: 10.1186/s12957-016-0815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland L.C., Edwards S.E., Cable H.C., Poirier G.G., Miller B.A., Cooper C.S., Williams G.T. LUCA-15-encoded sequence variants regulate CD95-mediated apoptosis. Oncogene. 2000;19:3774–3781. doi: 10.1038/sj.onc.1203720. [DOI] [PubMed] [Google Scholar]
- Sutherland L.C., Wang K., Robinson A.G. RBM5 as a putative tumor suppressor gene for lung cancer. J. Thorac. Oncol. 2010;5:294–298. doi: 10.1097/JTO.0b013e3181c6e330. [DOI] [PubMed] [Google Scholar]
- Tam W.L., Ng H.H. Sox2: masterminding the root of cancer. Cancer Cell. 2014;26:3–5. doi: 10.1016/j.ccr.2014.06.024. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis W.D., Brambilla E., Muller-Hermelink H.K., Harris C.C. World Health Organization Classification of Tumours. IARC Press; Lyon, France: 2004. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. [Google Scholar]
- Unni A.M., Lockwood W.W., Zejnullahu K., Lee-Lin S.Q., Varmus H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. Elife. 2015;4:e06907. doi: 10.7554/eLife.06907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M.H., Latif F., Bader S., Kashuba V., Chen J.Y., Duh F.M., Sekido Y., Lee C.C., Geil L., Kuzmin I. Construction of a 600-kilobase cosmid clone contig and generation of a transcriptional map surrounding the lung cancer tumor suppressor gene (TSG) locus on human chromosome 3p21.3: progress toward the isolation of a lung cancer TSG. Cancer Res. 1996;56:1487–1492. [PubMed] [Google Scholar]
- Wistuba I.I., Behrens C., Virmani A.K., Mele G., Milchgrub S., Girard L., Fondon J.W., III, Garner H.R., McKay B., Latif F. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res. 2000;60:1949–1960. [PubMed] [Google Scholar]
- Xiong Q., Mukherjee S., Furey T.S. GSAASeqSP: a toolset for gene set association analysis of RNA-Seq data. Sci. Rep. 2014;4:6347. doi: 10.1038/srep06347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabas M., Jing W., Shafik S., Broer S., Enders A. ATP11C Facilitates Phospholipid Translocation across the Plasma Membrane of All Leukocytes. PLoS One. 2016;11:e0146774. doi: 10.1371/journal.pone.0146774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.