

Abstract
Background
Staphylococci are a major constituent of the nasal microbiome and a frequent cause of hospital-acquired infection. Antibiotic surgical prophylaxis is administered prior to surgery to reduce a patient’s risk of postoperative infection. The impact of surgical prophylaxis on the nasal staphylococcal microbiome is largely unknown. Here, we report the species present in the nasal staphylococcal microbiome and the impact of surgical prophylaxis revealed by a novel culture independent technique. Daily nasal samples from 18 hospitalised patients, six of whom received no antibiotics and 12 of whom received antibiotic surgical prophylaxis (flucloxacillin and gentamicin or teicoplanin +/− gentamicin), were analysed by tuf gene fragment amplicon sequencing.
Results
On admission to hospital, the species diversity of the nasal staphylococcal microbiome varied from patient to patient ranging from 4 to 10 species. Administration of surgical prophylaxis did not substantially alter the diversity of the staphylococcal species present in the nose; however, surgical prophylaxis did impact on the relative abundance of the staphylococcal species present. The dominant staphylococcal species present in all patients on admission was Staphylococcus epidermidis, and antibiotic administration resulted in an increase in species relative abundance. Following surgical prophylaxis, a reduction in the abundance of Staphylococcus aureus was observed in carriers, but not a complete eradication.
Conclusions
Utilising the tuf gene fragment has enabled a detailed study of the staphylococcal microbiome in the nose and highlights that although there is no change in the heterogeneity of species present, there are changes in abundance. The sensitivity of the methodology has revealed that the abundance of S. aureus is reduced to a low level by surgical prophylaxis and therefore reduces the potential risk of infection following surgery but also highlights that S. aureus does persist.
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-016-0210-1) contains supplementary material, which is available to authorized users.
Keywords: tuf gene, Staphylococcus, S. aureus, S. epidermidis, Nose, Surgical prophylaxis, Antibiotics
Background
Staphylococci are ubiquitous commensals of the human skin and mucous membranes and are a major cause of hospital-acquired infections. Culture independent studies using the 16S rRNA gene have identified staphylococci as a major constituent of the nasal microbiome in both healthy individuals [1, 2] and hospitalised patients [3]. The staphylococcus genus consists of 52 species and 28 subspecies; however, culture independent studies using the 16S rRNA gene have only been able to identify Staphylococcus aureus from the staphylococcus genus, due to high sequence similarity of the other staphylococcal species. Other taxonomy genes including hsp60, sodA, rpoB, gap and tuf have been shown to be more discriminative than 16S rRNA gene at identifying staphylococcus species [4]. The tuf gene encodes the elongation factor Tu protein, which is involved in peptide chain formation. DNA sequencing of the tuf gene on a panel of reference strains has shown that it can discriminate all staphylococcal species and identification is comparable to MALDI-TOF [5].
In surgical patients, postoperative infection such as surgical site infections (SSI) and prosthetic joint infections (PJI) cause a substantial increase in morbidity and mortality. Staphylococci are the most common causative agent, with 20% of SSI cases caused by S. aureus [6] and 30–43% of PJI cases caused coagulase negative staphylococci (CNS) [7]. Antibiotic surgical prophylaxis is administered to surgical patients to reduce the risk of postoperative infection. The only culture-dependent study of the impact of antibiotic surgical prophylaxis on the nasal microbiota [8] reported that antibiotics did impact on staphylococcal diversity but that the effect was patient specific. However, a major limitation of the study was the culture-based methodology.
The aim of this study was to investigate the nasal staphylococcal microbiome during antibiotic prophylaxis using tuf gene fragment amplicon sequencing, a novel culture independent methodology.
Methods
Study design, population, nasal sampling and swab processing
Patients over the age of 18 years were recruited within 24 h of hospitalisation, to either one of the surgical or cardiology wards at the Heart of England NHS Foundation Trust via written consent. Patients were excluded if hospitalised for greater than 24 h or if the surgical procedure prohibited nasal sampling pre- and/or postoperatively. Ethical approval for the study was gained from the NRES Committee West Midlands (08/H1206/133). On recruitment, a nasal sample was taken using a flocked transport eswab (Sterilin, UK) within 24 h of admission, and prior to the administration of any antibiotics. Subsequently, nasal samples were taken daily, with all the patients that received prophylactic antibiotics receiving them after the first and prior to the second sample being taken.
Nasal sampling was carried out by a standardised procedure, with all samples being taken by rotating the swab three times applying a medium pressure to each nostril in turn, and then placing in the transport medium, and transported back to the laboratory to be processed. Swabs were vortexed for 10 s in the 1 ml liquid Amies transport media, 200 μl used for conventional culture and 800 μl stored at −80 °C for tuf gene fragment amplicon sequencing.
For all recruited patients, clinical and demographic data was collected including age, sex, antibiotic prophylaxis received, previous antibiotic usage and previous hospital admission in the last 6 months.
Staphylococcal species identified by conventional culture
Staphylococci were cultured on selective media and identified to species level as described previously [8]. In brief, S. aureus and coagulase negative staphylococci (CNS) isolates were preliminary identified by colony morphology from culture media. Multiple representative isolates of S. aureus and CNS were selected based on the total colony count of each bacterium using the following criteria: >50 colonies five isolates, between 10 and 50 three isolates, and <10 colonies one isolate. Isolates were speciated by PCR and MALDI-TOF (Bruker Daltonik GmbH, Bremen, Germany).
Tuf gene fragment amplicon sequencing
Frozen nasal samples were thawed, a 400-μl volume added to lysing matrix B beads (MP Biomedicals UK, Cambridge, UK) and homogenised at 6500 rpm for 30 s using the MagNA lyser instrument (Roche Applied Science, Penzberg, Germany). Samples were then centrifuged at 3000 rpm for 2 min, and the supernatants containing DNA material stored at −20 °C.
Amplicon sequencing targeted a 412-bp staphylococcus specific region of the tuf gene using previously published primers [9], with the inclusion of a heterogeneity spacer, Illumina adapter and linker [10] (see Additional file 1: Table S1). The initial PCRs of the tuf gene fragment were performed using the aforementioned primers in triplicate and pooled. Subsequently, samples were cleaned up, and an index PCR performed to add sample specific indices and Illumina sequence adapters using the Nextera XT index kit, as described previously [11]. DNA was quantified by Qubit 2.0 flurometer (Invitrogen, California, USA), normalised to 0.1 ng/μl and pooled to create amplicon library, which was quantified by qPCR using the Library Quantification Kit for Illumina (Kapa Biosciences, Woburn, MA, USA). For sequencing, PhiX Control library (v3) (Illumina) was added to the amplicon library at 20%. Library pool was sequenced using paired-end 300 cycles V3 Illumina Miseq reagent kit on an Illumina Miseq sequencer (Illumina, San Diego, CA, USA).
Illumina Miseq data processing and analysis
Initial quality filtering of raw sequence reads, construction of contigs and subsampling of 5000 representative sequences per sample were carried out in Mothur. Quality filtering was carried out using strict criteria of no ambiguous bases and no N bases; also, a minimum (412 bp) and maximum (426 bp) length of contig was applied. De novo analysis, using a 99% clustering similarity to form OTUs and assignment of staphylococcal species using a collated reference database of 37 staphylococcal species (see Additional file 2: Table S2) [5] at 97% similarity, was carried out in QIIME. The relative sequence abundance of each staphylococcus species composing the nasal staphylococcus microbial community was calculated for each sample. The presence of a staphylococcal species was determined using a >0.1% sequence abundance cut-off.
For calculation of alpha and beta diversity, de novo analysis using a 97% clustering similarity to form OTUs, and assignment of staphylococcal species using a collated reference database of 37 staphylococcal species at 97% similarity, was carried out in QIIME, and unassigned sequences were removed. Rarefaction was performed a depth at 1100 sequence reads.
Results
Study population
Eighteen patients were included in the study, six of which had received no antibiotics, six who received flucloxacillin (1 g) and gentamicin (160 mg) and six received teicoplanin (400 mg) +/− gentamicin (160 mg) (three patients received teicoplanin alone and three patients received teicoplanin in combination with gentamicin). No patients received nasal decolonisation treatment (mupirocin and chlorhedixine) prior to surgery, as all patients were MRSA negative by routine diagnostic screening. The patient demographic comprised of four males and 14 females, with age ranging from 52 to 91 years (median 81 years) (Table 1). A total of 94 swabs were taken, with a range of 3–7 days for each patient (mean = 5) (Table 1).
Table 1.
Demographic summary of study participants
| Antibiotic surgical prophylaxis received | Antibiotic dosage | Patient study ID | Age (years) | Sex |
S. aureus nasal carriage detected by conventional culture on admission |
Surgical speciality | Surgical procedure | Sample days |
|---|---|---|---|---|---|---|---|---|
| No antibiotics | 25 | 79 | Female | No | Orthopaedic | None | 5 | |
| 99 | 52 | Male | Yes | Cardiology | Angioplasty | 4 | ||
| 101 | 83 | Female | No | Cardiology | Angioplasty | 4 | ||
| 105 | 63 | Male | Yes | Cardiology | Angioplasty | 5 | ||
| 111 | 68 | Female | Yes | Cardiology | Angioplasty | 5 | ||
| 113 | 79 | Female | No | Cardiology | Angioplasty | 3 | ||
| Flucloxacillin (1 g) and gentamicin (160 mg) | Single dose IV on induction, and 2 further doses of flucloxacillin (1 g) post op at 6-h intervals | 34 | 87 | Female | No | Gamma Nail | 7 | |
| 36 | 87 | Female | No | Orthopaedic | Gamma Nail | 7 | ||
| 42 | 88 | Female | Yes | Orthopaedic | Dynamic Hip Screw | 7 | ||
| 51 | 82 | Female | No | Orthopaedic | Dynamic Hip Screw | 5 | ||
| 56 | 72 | Female | No | Orthopaedic | Open Reduction of internal fracture of wrist and K wires | 7 | ||
| 61 | 88 | Male | Yes | Orthopaedic | Total knee replacement | 5 | ||
| Orthopaedic | ||||||||
| Teicoplanin (400 mg) +/− gentamicin (160 mg) | Single dose IV on induction | 35b | 80 | Female | No | Orthopaedic | Hemiarthroplasty | 7 |
| 37b | 91 | Female | Yes | Orthopaedic | Hemiarthroplasty | 7 | ||
| 65a | 76 | Female | No | Orthopaedic | Total hip replacement | 4 | ||
| 86b | 71 | Male | Yes | Orthopaedic | Total hip replacement | 4 | ||
| 90a | 81 | Female | Yes | Orthopaedic | Hemiarthroplasty | 5 | ||
| 93a | 89 | Female | Yes | Orthopaedic | Hemiarthroplasty | 4 |
aTeicoplanin alone
bTeicoplanin and gentamicin
Tuf gene fragment amplicon sequencing
A total of 8,896,680 sequence reads were initially analysed from 94 samples, with the number of sequence reads ranging from 1084 to 600,830 per sample (median 71,991). Following quality filtering, 72 (76.6%) samples had >5000 sequence reads, and for these samples, 5000 representative sequence reads were subsampled; for the remaining samples, all the sequences were analysed (see Additional file 3: Table S3). After subsampling, 419,765 sequence reads remained to be analysed. Of the total sequence reads, 383,471 (91.4%) were assigned a staphylococcal species. Twenty-two staphylococcal species were identified in total, with 14 species identified from admission and subsequent samples and eight only on subsequent sampling during hospitalisation. All eight species identified from post-admission samples were present at <2% relative sequence abundance, with three species <0.1% relative abundance. Staphylococcus epidermidis (235,985, 56.2% of sequence reads) was the most dominant species. A total of 36,294 sequence reads were classified as unassigned. A BLAST search of unassigned sequences showed that the majority (85%) of unassigned sequences were from the staphylococcus genus, but species could not be assigned as identity was below the 97% threshold. A minority (15%) of unassigned sequences were identified as species of other Gram-positive bacteria including Gemella haemolysans and Abiotrophia defectiva.
Investigation of the composition of the nasal staphylococcal microbiome in patients on admission
Fourteen staphylococcal species were identified by tuf gene fragment amplicon sequencing in the 18 nasal samples on admission (Table 2). S. epidermidis was the dominant species of the nasal staphylococcal microbiome and was carried by 100% of patients on admission (sequence abundance ranging from 0.74 to 94.60%). Thirteen patients were S. aureus nasal carriers on admission with sequence abundance ranging from 0.10 to 98.14%. S taphylococcus capitis and Staphylococcus hominis were present in over 75% of patients on admission, at median relative sequence abundance of 1.49 and 0.80%, respectively. The remaining staphylococci identified were present at low relative sequence abundance with medians <1.2%. The composition and diversity of the nasal staphylococcal microbiome varied from patient to patient on admission, with a range of 4 to 10 species present (average 6.5 species).
Table 2.
Frequency of Staphylococcus species identified on admission and relative sequence abundance in study patients (n = 18)
| tuf fragment amplicon sequencing | ||||
|---|---|---|---|---|
| Staphylococcal species | Number of patients species was identified in | Percentage of patients species was identified in (%) | Relative sequence abundance | |
| Median (%) | Range (%) | |||
| S. aureus | 13 | 72.2 | 13.00 | (0.21–98.14) |
| S. auricularis | 4 | 22.2 | 0.54 | (0.44–19.16) |
| S. capitis | 16 | 88.9 | 1.49 | (0.14–22.42) |
| S. cohnii | 5 | 27.8 | 0.98 | (0.10–2.60) |
| S. epidermidis | 18 | 100.0 | 46.86 | (0.74–94.60) |
| S. haemolyticus | 7 | 38.9 | 0.76 | (0.10–3.58) |
| S. hominis | 14 | 77.8 | 0.80 | (0.24–47.86) |
| S. kloosii | 1 | 5.6 | 0.33 | (0.33–0.33) |
| S. lugdunensis | 8 | 44.4 | 0.24 | (0.10–41.46) |
| S. pasteuri | 7 | 38.9 | 0.57 | (0.12–1.04) |
| S. pettenkoferi | 7 | 38.9 | 1.18 | (0.14–4.69) |
| S. saccharolyticus | 4 | 22.2 | 1.05 | (0.12–6.34) |
| S. simulans | 3 | 16.7 | 0.22 | (0.14–0.26) |
| S. warneri | 11 | 61.1 | 0.53 | (0.12–2.80) |
On a sample by sample basis, the staphylococcal species identified from nasal samples by conventional culture and tuf gene fragment amplicon sequencing were compared. In 28.3% of instances, from the 18 samples, the same species was identified using both techniques, with 76.5% of these at a sequence abundance of >10% when analysed by tuf gene fragment amplicon sequencing. A significant proportion (70.0%) of species were only identified by tuf gene amplicon sequencing. Of the species with <10% sequence abundance, only 9.0% were also identified with conventional culture, with Staphylococcus warneri detected at the lowest sequence abundance (0.36%). There were only two instances of staphylococcal species being identified by conventional culture only (Staphylococcus lugdunensis in patient 86 and Staphylococcus haemolyticus patient 56). On admission, tuf gene fragment amplicon sequencing identified 13 patients with S. aureus carriage, compared to only 9 by conventional culture.
Impact of antibiotic surgical prophylaxis on the nasal staphylococcal microbiome on alpha and beta species diversity
All samples were rarefied to a depth of 1100 sequence reads, as rarefaction curves of the Shannon index reached a plateau at a sequencing depth of less than 1100 reads (data not shown). Ninety nasal samples were analysed by alpha and beta diversity; four samples were excluded due to insufficient sequence reads depth. The alpha diversity of all 90 samples, measured by the Shannon index, ranged from 0.115 to 2.193 (median 1.094). On admission and throughout hospitalisation, the alpha diversity of nasal samples varied between individual patients irrespective of antibiotic administration. The extent of variation in alpha diversity between nasal samples taken from an individual patient varied, with a narrow diversity range in some patients (e.g. 1.517 to 1.917 Shannon index, patient 51) to a wide diversity range in other patients (0.726 to 2.193 Shannon index, patient 37). There was no significant difference in the alpha diversity between nasal samples taken on each sample day from patients in the three antibiotic regimens. Beta diversity was measured by the Bray Curtis dissimilarity. No substantial shift in the diversity of the nasal staphylococcal microbiome was observed after the administration of antibiotics when compared to admission sample (Table 3). Only one patient who received flucloxacillin and gentamicin (patient 51) had Bray Curtis dissimilarity greater than 0.5 for all four sample days compared to admission (0.77, 060, 0.65 and 0.78).
Table 3.
The beta diversity of the nasal staphylococcal microbiome between admission and other sample days after administration of antibiotic surgical prophylaxis in individual study patients, measured by the Bray Curtis dissimilarity distance
| Antibiotic surgical prophylaxis received | Patient study ID | Beta diversity (Bray Curtis dissimilarity) Sample day |
|||||
|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | 7 | ||
| No antibiotics | 25 | 0.24 | 0.42 | # | 0.29 | ||
| 99 | 0.12 | 0.04 | 0.09 | ||||
| 101 | 0.55 | 0.35 | 0.54 | ||||
| 105 | 0.01 | 0.09 | 0.02 | 0.37 | |||
| 111 | 0.53 | 0.09 | 0.06 | 0.35 | |||
| 113 | 0.06 | 0.20 | |||||
| Flucloxacillin (1 g) and gentamicin (160 mg) | 34 | 0.23 | 0.21 | 0.20 | 0.20 | 0.21 | # |
| 36 | 0.35 | 0.10 | 0.11 | 0.13 | 0.14 | 0.41 | |
| 42 | 0.25 | 0.19 | 0.66 | 0.33 | 0.70 | 0.54 | |
| 51 | 0.42 | 0.25 | 0.34 | ||||
| 56 | 0.03 | 0.11 | 0.07 | 0.26 | # | 0.17 | |
| 61 | 0.77 | 0.60 | 0.65 | 0.78 | |||
| Teicoplanin (400 mg) +/− gentamicin (160 mg) | 35b | 0.21 | 0.33 | 0.33 | 0.24 | # | 0.17 |
| 37b | 0.30 | 0.06 | 0.22 | 0.32 | 0.40 | 0.20 | |
| 65a | 0.08 | 0.08 | 0.06 | ||||
| 86b | 0.10 | 0.18 | 0.23 | ||||
| 90a | 0.06 | 0.08 | 0.05 | 0.36 | |||
| 93a | 0.36 | 0.49 | 0.29 | ||||
aTeicoplanin alone
bTeicoplanin and gentamicin
#Beta diversity was not calculated for sample as excluded due to insufficient read depth (<1100)
Impact of antibiotic surgical prophylaxis on the relative abundance of species present in the nasal staphylococcal microbiome
To determine the impact of antibiotic surgical prophylaxis administration on the relative abundance of staphylococcal species present in the nose, samples from days 1 and 3 were compared. In patients who received no antibiotics, only minor changes in the relative sequence abundance of staphylococcus species occurred, with the majority of changes being an increase or decrease of <10% of a species (Fig. 1). More substantial changes in relative sequence abundance occurred in patients who received antibiotics. An increase in the sequence abundance of S. epidermidis, ranging from 6.7 to 47%, occurred in 10 out of 12 patients that received antibiotics. Of the nine S. aureus nasal carriers that received antibiotic surgical prophylaxis, eight had a reduced sequence abundance of S. aureus after the administration of antibiotics ranging from 0.2 to 57.4%. Although S. aureus relative sequence abundance was reduced, nasal carriage persisted. Six of the nine S. aureus carriers identified by amplicon sequencing were also identified by conventional culture. After the administration of antibiotics, S. aureus became undetectable by conventional culture in two patients but was still detectable by amplicon sequencing at low relative sequence abundance. A small reduction in the sequence abundance of S. capitis, S. haemolyticus and S. hominis was noted in the majority of patients who received antibiotics.
Fig. 1.
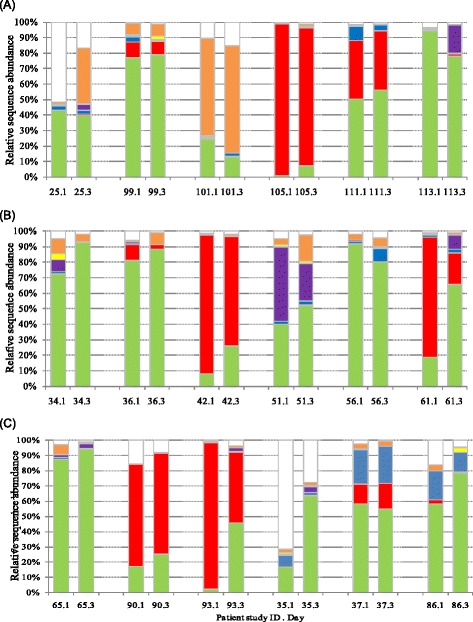
Relative sequence abundance of nasal staphylococcal microbiome on day 1 and day 3 of 18 patients who received antibiotic surgical prophylaxis regimen: a no antibiotics, b flucloxacillin and gentamicin, and c teicoplanin +/− gentamicin. Colours indicate S. aureus (red), S. epidermidis (green), S. capitis (blue), S. hominis (purple), S. haemolyticus (yellow), other staphylococci (orange), unassigned sequences (white). Number.number designation on x-axis represents the patient ID and sample day, with day 1 being pre-antibiotic treatment and day 3 being post-antibiotic treatment. Teicoplanin alone (patient study IDs 65, 90 and 93), teicoplanin and gentamicin (patient study IDs 35, 37 and 86)
Discussion
Application of a novel culture independent technique using the tuf gene has provided a more in-depth, detailed insight into the diversity of the nasal staphylococcal microbiome, and the impact of antibiotic surgical prophylaxis. Previous culture-dependent studies were restricted by their limit of detection, resulting in them being unable to reveal the true species diversity, especially at low abundance [12–14].
The novel use of the tuf gene in this culture independent study, as opposed to the more widely used 16S rRNA gene, has enabled the study of the nasal staphylococcal microbiome at species level [1–3, 12, 15–22]. Studies utilising the 16S rRNA gene have been unable to identify staphylococci at species level due to the high gene sequence similarity of 16S rRNA gene between staphylococcal species and thus described staphylococci either as Firmicutes staphylococcus genus, only identifying S. aureus to species level. Kaspar et al. [12] recently demonstrated this limitation, with only S. aureus being identified by both 16S rRNA and culture, with nine other staphylococcus species including S. epidermidis and S. captis only being identified to species level by culture.
The use and type of antibiotic agents used for surgical prophylaxis has changed over the last 20 years due to the increase in antibiotic resistance and changing practices. Most guidance on surgical prophylaxis now recommends the use of either a single dose of antibiotics or continuation for less than 24 h [23]. However, there are no studies that have used culture independent methods to investigate the impact of surgical prophylaxis on the human microbiome. A recent study looking at longer courses of antibiotics (5–10 days) on the salivary and faecal microbiome found very similar results to the current study in the salivary microbiome with it remaining relatively stable following antibiotics, whilst large changes were observed in the faecal microbiome [24]. The authors hypothesised that these differences may be due either to the pharmacokinetics of the antibiotics, or possibly that the populations in the salivary microbiome are more intrinsically resilient. Whilst the nasal microbiome is different to that of the salivary microbiome, it is still exposed to some of the same stresses such as changes in temperature, oxygen and physical disturbance and may mean that these populations are intrinsically more resilient.
The most significant change in the nasal staphylococcal microbiome after the administration of antibiotics was the increase in abundance of S. epidermidis, and the reduction but not eradication of S. aureus in nasal carriers. Within this study population of orthopaedic patients undergoing surgery involving implants, both these findings are clinically significant. In orthopaedic surgery, nasal carriage of S. aureus has been identified as the most important risk factor of developing SSI, with a relative risk of 8.9 [25]. Staphylococci are the leading cause of PJI with CNS causing 30–43% of infections, predominantly S. epidermidis, and S. aureus causing 12–23% [7]. Although PJIs occur in <1% of operations, the infections are difficult to treat involving additional surgery and prolonged antibiotic therapy, with a high morbidity. Other staphylococcal species present in the nose, for example S. capitis and S. lugdunensis, although less pathogenic than S. aureus and S. epidermidis, have been reported to cause PJI [26, 27], and infective endocarditis [28, 29].
Molecular methods for screening of S. aureus nasal carriage have been demonstrated to have a higher sensitivity than culture [30, 31]. Data from this study supports these findings, with only nine patients being identified as nasal carriers by culture as opposed to 13 by sequencing on admission. Due to the increased sensitivity of molecular methods, questions have been raised about false positives compared to culture methods; however, as this study is able to look at S. aureus relative abundance, it is able to provide evidence to demonstrate that it is not due to false positives.
S. aureus nasal carriage relative abundance was reduced to a low level by antibiotic surgical prophylaxis, which might be associated with a reduced risk of SSI, but it was not eradiated. Our finding may explain the higher S. aureus SSI rate, observed by Bode and colleagues, in patients who received surgical prophylaxis alone (8.4%), compared to patients who received prophylaxis in combination with S. aureus decolonisation with mupirocin and chlorhexidine prior to surgery (3.6%) [32].
This is the first culture independent study to use an alternative taxonomy gene to study the nasal staphylococcal microbiome in detail during surgical prophylaxis. The main limitation of the study is the small patient population analysed and its demographic bias of female patients; further investigation is required to determine the significance of these changes.
Conclusions
Tuf gene fragment amplicon sequencing has enabled a detailed culture independent study of the impact of surgical prophylaxis on the nasal staphylococcal microbiome during surgical prophylaxis. As the new technique discriminates to staphylococcus species level, it could be used to study staphylococci rich microbial communities and expand our knowledge of staphylococcal diversity and the potential risk of infection in at risk patient groups.
Acknowledgements
This work was supported by the Heart of England NHS Foundation Trust, Public Health England and University of Birmingham.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Availability of data and materials
The raw sequence files supporting the findings of this article are available in the NCBI Sequence Read Archive (SRA) under the BioProject ID PRJNA324352, Biosamples SAMN05241085 to SAMN05241178.
Authors’ contributions
KJH, PMH and CLM designed the study. CLM and STC carried out the amplicon sequencing. NJL advised on the bioinformatics analysis, and CLM performed the bioinformatics analysis. CLM, KJH, NJL and PMH participated in the interpretation of results. CLM wrote the manuscript with input from the other authors. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All subjects signed written consent to participate in this study. This study was approved by NRES Committee West Midlands (08/H1206/133).
Abbreviations
- CNS
Coagulase negative staphylococci
- MALDI-TOF
Matrix-assisted laser desorption/ionisation time of flight
- OTU
Operational taxonomic unit
- PCR
Polymerase chain reaction
- PJI
Prosthetic joint infections
- QIIME
Quantitative insights into microbial ecology
- SSI
Surgical site infections
Additional files
Primer sequences used in the initial PCR step of tuf gene fragment amplicon sequencing. (DOC 35 kb)
Accession numbers of 37 species included in reference database. (DOC 27 kb)
Staphylococcal relative sequence abundance for all patient samples. (XLSX 23 kb)
References
- 1.Wos-Oxley ML, Plumeier I, Von EC, Taudien S, Platzer M, Vilchez-Vargas R, Becker K, Pieper DH. A poke into the diversity and associations within human anterior nare microbial communities. ISME J. 2010;4:839–851. doi: 10.1038/ismej.2010.15. [DOI] [PubMed] [Google Scholar]
- 2.Lemon KP, Klepac-Ceraj V, Schiffer HK, Brodie EL, Lynch SV, Kolter R. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. MBio. 2010;1:e00129–10. doi: 10.1128/mBio.00129-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank DN, Feazel LM, Bessesen MT, Price CS, Janoff EN, Pace NR. The human nasal microbiota and Staphylococcus aureus carriage. PLoS One. 2010;5:e10598. doi: 10.1371/journal.pone.0010598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghebremedhin B, Layer F, Konig W, Konig B. Genetic classification and distinguishing of Staphylococcus species based on different partial gap, 16S rRNA, hsp60, rpoB, sodA, and tuf gene sequences. J Clin Microbiol. 2008;46:1019–1025. doi: 10.1128/JCM.02058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergeron M, Dauwalder O, Gouy M, Freydiere AM, Bes M, Meugnier H, Benito Y, Etienne J, Lina G, Vandenesch F, Boisset S. Species identification of staphylococci by amplification and sequencing of the tuf gene compared to the gap gene and by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Eur J Clin Microbiol Infect Dis. 2011;30:343–354. doi: 10.1007/s10096-010-1091-z. [DOI] [PubMed] [Google Scholar]
- 6.Mangram AJ, Horan TC, Pearson ML, Silver LC, Jarvis WR. Guideline for Prevention of Surgical Site Infection, 1999. Centers for Disease Control and Prevention (CDC) Hospital Infection Control Practices Advisory Committee. Am J Infect Control. 1999;27:97. doi: 10.1016/S0196-6553(99)70088-X. [DOI] [PubMed] [Google Scholar]
- 7.Zimmerli W, Trampuz A, Ochsner PE. Prosthetic-joint infections. N Engl J Med. 2004;351:1645–1654. doi: 10.1056/NEJMra040181. [DOI] [PubMed] [Google Scholar]
- 8.McMurray CL, Hardy KJ, Verlander NQ, Hawkey PM. Antibiotic surgical prophylaxis increases nasal carriage of antibiotic-resistant staphylococci. J Med Microbiol. 2015;64:1489–1495. doi: 10.1099/jmm.0.000182. [DOI] [PubMed] [Google Scholar]
- 9.Heikens E, Fleer A, Paauw A, Florijn A, Fluit AC. Comparison of genotypic and phenotypic methods for species-level identification of clinical isolates of coagulase-negative staphylococci. J Clin Microbiol. 2005;43:2286–2290. doi: 10.1128/JCM.43.5.2286-2290.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2:6–2. doi: 10.1186/2049-2618-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Illumina. 2013. 16S metagenomic sequencing library preparation. https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf.
- 12.Kaspar U, Kriegeskorte A, Schubert T, Peters G, Rudack C, Pieper DH, Wos-Oxley M, Becker K. The culturome of the human nose habitats reveals individual bacterial fingerprint patterns. Environ. Microbiol. 2015; 18:2130-2142. [DOI] [PubMed]
- 13.Rasmussen TT, Kirkeby LP, Poulsen K, Reinholdt J, Kilian M. Resident aerobic microbiota of the adult human nasal cavity. APMIS. 2000;108:663–675. doi: 10.1034/j.1600-0463.2000.d01-13.x. [DOI] [PubMed] [Google Scholar]
- 14.Alvarez AS, Remy L, Allix-Beguec C, Ligier C, Dupont C, Leminor O, Lawrence C, Supply P, Guillemot D, Gaillard JL, Salomon J, Herrmann JL. Patient nostril microbial flora: individual-dependency and diversity precluding prediction of Staphylococcus aureus acquisition. Clin Microbiol Infect. 2013;20:70–78. doi: 10.1111/1469-0691.12208. [DOI] [PubMed] [Google Scholar]
- 15.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camarinha-Silva A, Jauregui R, Pieper DH, Wos-Oxley ML. The temporal dynamics of bacterial communities across human anterior nares. Environ Microbiol Rep. 2012;4:126–132. doi: 10.1111/j.1758-2229.2011.00313.x. [DOI] [PubMed] [Google Scholar]
- 17.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan M, Pamp SJ, Fukuyama J, Hwang PH, Cho DY, Holmes S, Relman DA. Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell Host. Microbe. 2013;14:631–640. doi: 10.1016/j.chom.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biswas K, Hoggard M, Jain R, Taylor MW, Douglas RG. The nasal microbiota in health and disease: variation within and between subjects. Front Microbiol. 2015;9:134. doi: 10.3389/fmicb.2015.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu CM, Price LB, Hungate BA, Abraham AG, Larsen LA, Christensen K, Stegger M, Skov R, Andersen PS. Staphylococcus aureus and the ecology of the nasal microbiome. Sci Adv. 2015;1:e1400216. doi: 10.1126/sciadv.1400216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, Beck JM, Curtis JL, Huffnagle GB. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio. 2015;6:e00037–15. doi: 10.1128/mBio.00037-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camarinha-Silva A, Jauregui R, Chaves-Moreno D, Oxley AP, Schaumburg F, Becker K, Wos-Oxley ML, Pieper DH. Comparing the anterior nare bacterial community of two discrete human populations using Illumina amplicon sequencing. Environ Microbiol. 2014;16:2939–2952. doi: 10.1111/1462-2920.12362. [DOI] [PubMed] [Google Scholar]
- 23.Bratzler DW, Dellinger EP, Olsen KM, Perl TM, Auwaerter PG, Bolon MK, Fish DN, Napolitano LM, Sawyer RG, Slain D, Steinberg JP, Weinstein RA. Clinical practice guidelines for antimicrobial prophylaxis in surgery. Am J Health Syst Pharm. 2013;70:195–283. doi: 10.2146/ajhp120568. [DOI] [PubMed] [Google Scholar]
- 24.Zaura E, Brandt BW, Teixeira de Mattos MJ, Buijs MJ, Caspers MP, Rashid MU, Weintraub A, Nord CE, Savell A, Hu Y, Coates AR, Hubank M, Spratt DA, Wilson M, Keijser BJ, Crielaard W. Same exposure but two radically different responses to antibiotics: resilience of the salivary microbiome versus long-term microbial shifts in feces. MBio. 2015;6:e01693–15. doi: 10.1128/mBio.01693-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalmeijer MD, van Nieuwland-Bollen E, Bogaers-Hofman D, de Baere GA. Nasal carriage of Staphylococcus aureus is a major risk factor for surgical-site infections in orthopedic surgery. Infect Control Hosp Epidemiol. 2000;21:319–323. doi: 10.1086/501763. [DOI] [PubMed] [Google Scholar]
- 26.Harris LG, El-Bouri K, Johnston S, Rees E, Frommelt L, Siemssen N, Christner M, Davies AP, Rohde H, Mack D. Rapid identification of staphylococci from prosthetic joint infections using MALDI-TOF mass-spectrometry. Int J Artif Organs. 2010;33:568–574. doi: 10.1177/039139881003300902. [DOI] [PubMed] [Google Scholar]
- 27.Peel TN, Cole NC, Dylla BL, Patel R. Matrix-assisted laser desorption ionization time of flight mass spectrometry and diagnostic testing for prosthetic joint infection in the clinical microbiology laboratory. Diagn Microbiol Infect Dis. 2015;81:163–168. doi: 10.1016/j.diagmicrobio.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 28.Selton-Suty C, Celard M, Le MV, Doco-Lecompte T, Chirouze C, Iung B, Strady C, Revest M, Vandenesch F, Bouvet A, Delahaye F, Alla F, Duval X, Hoen B. Preeminence of Staphylococcus aureus in infective endocarditis: a 1-year population-based survey. Clin Infect Dis. 2012;54:1230–1239. doi: 10.1093/cid/cis199. [DOI] [PubMed] [Google Scholar]
- 29.Petti CA, Simmon KE, Miro JM, Hoen B, Marco F, Chu VH, Athan E, Bukovski S, Bouza E, Bradley S, Fowler VG, Giannitsioti E, Gordon D, Reinbott P, Korman T, Lang S, Garcia-de-la-Maria C, Raglio A, Morris AJ, Plesiat P, Ryan S, Doco-Lecompte T, Tripodi F, Utili R, Wray D, Federspiel JJ, Boisson K, Reller LB, Murdoch DR, Woods CW. Genotypic diversity of coagulase-negative staphylococci causing endocarditis: a global perspective. J Clin Microbiol. 2008;46:1780–1784. doi: 10.1128/JCM.02405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy K, Price C, Szczepura A, Gossain S, Davies R, Stallard N, Shabir S, McMurray C, Bradbury A, Hawkey PM. Reduction in the rate of methicillin-resistant Staphylococcus aureus acquisition in surgical wards by rapid screening for colonization: a prospective, cross-over study. Clin Microbiol Infect. 2010;16:333–339. doi: 10.1111/j.1469-0691.2009.02899.x. [DOI] [PubMed] [Google Scholar]
- 31.Conterno LO, Shymanski J, Ramotar K, Toye B, Van WC, Coyle D, Roth VR. Infect Control Hosp Epidemi. 2007;28:1134–1141. doi: 10.1086/520099. [DOI] [PubMed] [Google Scholar]
- 32.Bode LG, Kluytmans JA, Wertheim HF, Bogaers D, Vandenbroucke-Grauls CM, Roosendaal R, Troelstra A, Box AT, Voss A, Van DT,I, Van BA, Verbrugh HA, Vos MC. Preventing surgical-site infections in nasal carriers of Staphylococcus aureus. N Engl J Med. 2010;362:9–17. doi: 10.1056/NEJMoa0808939. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw sequence files supporting the findings of this article are available in the NCBI Sequence Read Archive (SRA) under the BioProject ID PRJNA324352, Biosamples SAMN05241085 to SAMN05241178.