

Key Clinical Message
Glucose‐6‐phosphate (G6PD) deficiency is the most common human enzyme defect, often presenting with neonatal jaundice and/or acute hemolytic anemia, triggered by oxidizing agents. G6PD deficiency is an X‐linked, hereditary disease, mainly affecting men, but should also be considered in females with an oxidative hemolysis.
Keywords: Acute hemolytic anemia, Glucose‐6‐phosphate deficiency, X‐linked inheritance
Introduction
Glucose‐6‐phosphate (G6PD) deficiency is the most common human enzyme defect which affects more than an estimated 400 million people worldwide 1, 2, 3. The most frequent clinical manifestations of G6PD deficiency are neonatal jaundice and acute hemolytic anemia, often triggered by oxidative stress 1, 3 from infection and exposure to medication and certain foods (e.g., fava beans) 3, 4. Some G6PD‐deficient variants cause chronic hemolysis, leading to congenital nonspherocytic anemia 1, 3.
The G6PD enzyme catalyzes the first step in the pentose phosphate pathway, oxidizing glucose‐6‐phosphate to 6‐phosphogluconolactone and reducing nicotinamide adenine dinucleotide phosphate (NADP) to NADPH 1, 3. NADPH is crucial in producing reduced glutathione in the erythrocyte cytoplasm for protecting hemoglobin against oxidative damage. In case of G6PD deficiency, the presence of oxidizing agents leads to oxidation of the sulfhydryl bridges between parts of the hemoglobin molecule, thereby decreasing the solubility of hemoglobin which causes hemolysis 2.
The inheritance of G6PD deficiency shows a typical X‐linked pattern 1, 3, 4, in which mainly men are affected, and females, in case being heterozygous, have less severe clinical manifestations 1, 2, 3. Here, we report two girls with compound heterozygote G6PD deficiency, one of them presented with a severe hemolytic anemia. We discuss the etiology of G6PD deficiency in females and emphasize the importance to consider G6PD deficiency in acute hemolytic anemia, not only in males, but also in females.
Case Presentation
A healthy 5‐year‐old girl was referred to our hospital for jaundice and fatigue after eating fava beans suggestive of G6PD deficiency. Her past health was normal. She was the first of three children of healthy, nonconsanguineous Iraqi parents. The family history revealed a paternal aunt who was not allowed to eat fava beans. Physical examination showed tachycardia, anemic sclera, and mild jaundice. Complete blood count showed a picture of a hemolytic anemia (Hb 5.4 g/dL, MCV 82 fl, reticulocytes 104/1000 RBC, schistocytes, and bite cells) with elevated LDH (912 U/L, normal 313–618 U/L) and total bilirubin (1.4 mg/dL, normal 0.2–1.3 mg/dL), and lowered haptoglobin (10.4 mg/dL, normal 26–185 mg/dL). The girl recovered after red blood cell transfusion. No further hemolytic episode occurred.
Spectrophotometric analysis revealed deficiency of G6PD (0.1 U/g Hb) indicating a severe G6PD deficiency (class II according to the World Health Organization classification). DNA analysis showed two mutations: the G6PD Mediterranean variant (c.563C>T (p.Ser188Phe)) and the G6PD Chatham variant (c.1003G>A (p.Ala335Thr)), both in compound heterozygous state.
Although her father and her 6‐yearold sister never had symptoms of hemolysis, family screening with spectrophotometric analysis revealed that both were severely affected (respectively, 0.1 U/g Hb and 0.0 U/g Hb, normal range on this age 4.1–7.9 U/g Hb). Enzyme deficiency was not confirmed in her mother and her brother (respectively, 6.0 U/g Hb and 8.7 U/g Hb). DNA analysis was performed. The father was hemizygous for the c.563C>T (p.Ser188Phe) mutation, while the mother was heterozygous for the c.1003G>A (p.Ala335Thr) mutation, but more surprisingly, her sister turned out to have the same mutations in compound heterozygosity. DNA analysis was not performed in the brother (Fig. 1).
Figure 1.
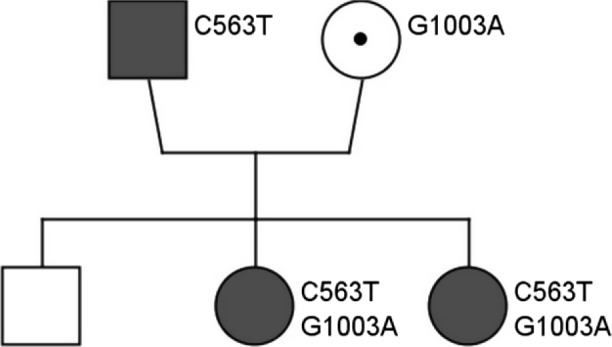
Family tree. DNA analysis was performed. Father: G6PD Mediterranean variant (c.563C>T (p.Ser188Phe)) in hemizygosity. Mother: G6PD Chatham variant (c.1003G>A (p.Ala335Thr)) in heterozygosity. Both girls had both mutations in compound heterozygosity.
Discussion
Males are hemizygous for the G6PD gene and can have normal gene expression or be G6PD deficient. Females, who have two copies of the G6PD gene on each X chromosome (Xq28), can have normal gene expression, be heterozygous or rarely homozygous for a mutation or compound heterozygous for two mutations on the G6PD gene 1. Heterozygous females have, in general, less severe clinical manifestations than G6PD‐deficient males 1, 2, 3, but the mean red blood cell enzyme activity depends on the degree of inactivation of one of the X chromosomes (lyonization) 4, and may be normal, mildly to moderate reduced, and grossly deficient. Although very rare, in some populations, in which the frequency of the G6PD‐deficient allele is very high 1, mainly in (sub) tropical regions 3, females can be homozygous 1 or compound heterozygous. They present with the same clinical manifestations as hemizygous males.
More than 400 variants of the G6PD enzyme (isoenzymes) have been identified. Almost all of the variants are missense point mutations causing single amino acid substitutions 3. The World Health Organization has categorized the mutant enzymes into five classes according to the degree of enzyme deficiency and hemolysis. The most common variants are noted in Table 1. G6PD‐B is the most common normal isoenzyme (wild type). The G6PD‐A is the most common variant, associated with mild to moderate hemolysis, while the G6PD Mediterranean variant is the most abnormal variant found in Caucasians and is classically associated with favism (hemolysis after ingestion of broad beans) 5.
Table 1.
Classification of G6PD variants
| Class | G6PD activity | Hemolysis | Most common variants |
|---|---|---|---|
| I | <10% | Chronic | G6PD San Diego |
| II | <10% | Acute | G6PD Mediterranean, G6PD Chatham |
| III | 10–60% | Acute | G6PD‐A−, G6PD Canton |
| IV | 60–150% | Absent | G6PD‐B, G6PD‐A+ |
| V | Increased | G6PD Verona | G6PD Verona |
Prevalence of hemizygous G6PD‐deficient males in Iraq is 6.1% 6. Corresponding prevalence in females can be predicted by the Hardy–Weinberg formula p2 + 2pq + q2 = 1 (2pq = heterozygous and q2 = homozygous state in the female population) 7. According to this formula, the prevalence of heterozygous G6PD‐deficient Iraqi females would be 11.5% versus 0.37% for homozygous females. Compound heterozygosity, in which two different heterozygous mutations on the G6PD gene are found, each on another chromosome, is even rarer. In Baghdad, Iraq, three polymorphic variants constituted more than 80% of G6PD‐deficient variants among males, namely: the Mediterranean (74.3%), Chatham (5%), and A‐ (2%) 8. The sisters discussed are compound heterozygous for two class II variants, Mediterranean, and Chatham, which can cause acute severe hemolysis.
Diagnosing G6PD deficiency can be challenging as normal results of a peripheral blood smear and spectrophotometric analysis don't exclude G6PD deficiency. In the acute setting of a hemolytic anemia, the presence of red blood cell anomalies, such as spherocytes and bite cells on a peripheral blood smear (Fig. 2) are suggestive of oxidative hemolysis, that is, G6PD deficiency. Several screening tests for G6PD deficiency are available, for example, the fluorescent spot test, the ascorbate–cyanide test, and the dye reduction test. The definitive test requires quantitation of the enzyme and can be performed by spectrophotometric analysis, which directly measures the rate of NADPH formation (proportional to the G6PD activity) 9 through its characteristic absorption peak at wavelength of 340 nm 10. The red cell activity is expressed in international units (IU) per gram of hemoglobin. The standardized normal reference value of the G6PD activity is established by taking the median G6PD activity value of a group of healthy male controls. In our laboratory, the reference for normal values at room temperature is in the range of 4.1–7.9 U/g Hb (newborns 50% higher). However, after severe hemolysis, young red blood cells and reticulocytes have much higher G6PD activity (up to 5x); the analysis can be false negative. In those cases or after red blood cell transfusion, tests need to be repeated after approximately 2–3 months when the red blood cell mass is repopulated with cells of all ages. Carrier state in females is difficult to determine, because of the normal enzyme activity in the unaffected X chromosome. In those cases, it is sometimes necessary to perform polymerase chain reaction (PCR) tests to reveal the genetic abnormality.
Figure 2.
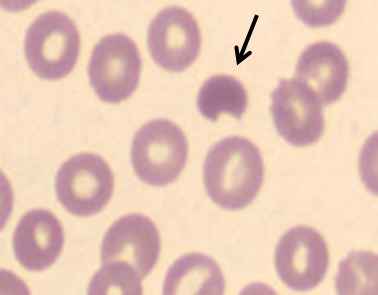
Bite cells, often seen during a hemolytic crisis in G6PD deficiency.
Conclusion
The red cells of millions of people are deficient in the enzyme G6PD. This X‐linked hereditary deficiency is mainly affecting males, but should also be considered in women presenting with a hemolytic anemia, even when family history is negative and the G6PD level is normal at presentation.
Conflict of Interest
The authors have no conflict of interests to disclose.
References
- 1. Cappellini, M. D. , and Fiorelli G.. 2008. Glucose‐6‐phosphate dehydrogenase deficiency. Lancet 371:64–74. [DOI] [PubMed] [Google Scholar]
- 2. Ronquist, G. , and Theodorsson E.. 2007. Inherited, non‐spherocytic haemolysis due to deficiency of glucose‐6‐phosphate dehydrogenase. Scand. J. Clin. Lab. Invest. 67:105–111. [DOI] [PubMed] [Google Scholar]
- 3. Mehta, A. , Mason P. J., and Vulliamy T. J.. 2000. Glucose‐6‐phosphate dehydrogenase deficiency. Baillieres Best Prac. Res. Clin. Haematol. 13:21–38. [DOI] [PubMed] [Google Scholar]
- 4. Lim, F. , Vulliamy T., and Abdalla S. H.. 2005. An Ashkenazi Jewish woman presenting with favism. J. Clin. Pathol. 58:317–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beutler, E. 1994. G6PD deficiency. Blood 84:3613–3636. [PubMed] [Google Scholar]
- 6. Hilmi, F. A. , Al‐Allawi N. A., Rassam M., Al‐Shamma G., and Al‐Hashimi A.. 2002. Red cell glucose‐6‐phosphate dehydrogenase phenotypes in Iraq. East. Mediterr. Health J. 8:42–48. [PubMed] [Google Scholar]
- 7. Chan, D. K. 2008. Glucose‐6‐phosphate dehydrogenase deficiency: correlation between the genotype, biochemistry and phenotype. Ann. Acad. Med. Singapore 37(Suppl. 12):81–83. [PubMed] [Google Scholar]
- 8. Al‐Musawi, B. M. , Al‐Allawi N., Abdul‐Majeed B. A., Eissa A. A., Jubrael J. M., and Hamamy H.. 2012. Molecular characterization of glucose‐6‐phosphate dehydrogenase deficient variants in Baghdad city ‐ Iraq. BMC Blood Disord. 12:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. WHO Working Group . 1989. Glucose‐6‐phosphate dehydrogenase deficiency. Bull. World Health Organ. 67:601–611. [PMC free article] [PubMed] [Google Scholar]
- 10. Beutler, E. 1984. Red cell metabolism: a manual of biochemical methods, 3rd ed. Grune & Strattan, New York. [Google Scholar]