

Abstract
Background
Recent advances in next-generation sequencing have revolutionized genomic research. 16S rRNA amplicon sequencing using paired-end sequencing on the MiSeq platform from Illumina, Inc., is being used to characterize the composition and dynamics of extremely complex/diverse microbial communities. For this analysis on the Illumina platform, merging and quality filtering of paired-end reads are essential first steps in data analysis to ensure the accuracy and reliability of downstream analysis.
Results
We have developed the Merging and Filtering Tool (MeFiT) to combine these pre-processing steps into one simple, intuitive pipeline. MeFiT invokes CASPER (context-aware scheme for paired-end reads) for merging paired-end reads and provides users the option to quality filter the reads using the traditional average Q-score metric or using a maximum expected error cut-off threshold.
Conclusions
MeFiT provides an open-source solution that permits users to merge and filter paired end illumina reads. The tool has been implemented in python and the source-code is freely available at https://github.com/nisheth/MeFiT.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-016-1358-1) contains supplementary material, which is available to authorized users.
Keywords: 16S rRNA, microbiome, paired-end sequencing, quality filtering, pre-processing
Background
Since its inception, next-generation sequencing (NGS) techniques have transformed the way scientists extract multifaceted biological information from complex systems, fostering research in the fields of human disease, environmental science, evolutionary science, etc. [1] The exploration of bacterial ecology by sequencing the small-subunit 16S rRNA gene, or more commonly, portions thereof, is being used as a gold-standard technique for various human/environmental microbiome studies [2–6]. Thus, the predictable conservation and variability of different portions of the prokaryotic 16S rRNA gene have been exploited to provide high resolution identification and quantification of the bacterial source [6]. This strategy for phylogenetic analysis and quantification has proven to be significantly more efficient than more traditional cloning and sequencing or RT-PCR based approaches. These massively parallel, cost-effective, and high-throughput sequencing technologies can now produce up to 15Gb of genomic data in one run [7]. The sheer volume and complexity of data generated by these NGS systems necessitates the development of bioinformatics tools that streamline the downstream analyses.
The Illumina platforms provide paired-end sequencing, in which a DNA sequence is read from both ends up to a specified read length. Depending on the read length selected, currently up to 300 bases, a target DNA segment that is longer than the sum of the forward and reverse reads would result in a gap of missing sequence between them, and a shorter target segment will result in an overlap between the reads. Moreover, since quality tends to degrade towards the ends of the reads, reliable merging of overlapping paired-end reads can results in a combined DNA sequence that might permit bioinformatics correction of these 3′-end sequencing errors and yield higher quality sequence output.
In paired-end sequencing, accurate merging of the forward and reverse reads is a crucial first step that affects the results of a plethora of downstream analyses, especially, including but not limited to microbial taxonomic profiling. Various tools; e.g., SHERA [8], FLASH [9], PANDAseq [10], COPE [11], and others, have been developed for merging paired end data. These tools generally apply sequence alignments to attempt to identify the best overlap between the paired-end reads. Thus, they resolve mismatching bases by considering their quality scores, with the higher quality base simply replacing one with poorer quality. Recently, a new method was proposed – CASPER, context-aware scheme for paired-end reads [12]. CASPER uses the traditional quality-score based method to resolve mismatches, except when the difference in quality scores is not significant, relies on k-mer-based contexts surrounding the mismatch to make the decision.
Another important aspect of sequence data that significantly impacts downstream analysis is read quality. Sequences of poor quality (base calling errors, small insertions/deletions) need to be identified and removed prior to analysis to minimize sources of false positivity. Each NGS platform provides some quality control measures, and others have been developed for filtering of these sequencing artifacts [13–18]. Most pipelines follow a QC protocol that filters reads based on their Phred, or other equivalent quality (Q) scores. Thus, reads of a specified length with greater than or equal to a specified, and often quite arbitrary average quality score are labeled as high-quality reads, and others with lower average quality scores are labeled low quality and removed from further analysis [18]. However, Phred scores are logarithmically related to the error probability of base-calling, which means that an average Q-score of the read is not necessarily a good indicator of the expected accuracy/error within it [19]. For example, consider two reads of length 100 nucleotides – read1 with 90 nt with Q-score of 40 and 10 nt with Q-score of 2 (average Q score ~36), and read2 with 100 nt with Q-scores of 25 (average Q score ~25)). However, the number of miscalls or maximum expected error (MEE) in read1 will be ~6; i.e. 10 bases each with a probability of being incorrect of 0.63. In contrast, the MEE of read2 is 0.316 (100 * 0.00316). Thus, despite its lower average Q-score, read2 has lower error probability, likely has many fewer sequence errors, and is for most purposes a higher quality read than read1.
The focus of research in our lab has been on applying high-throughput technologies to characterize the genome, transcriptome and proteome of the human microbiome and of the host-microbial interactions. We are leading bioinformatics analysis on multiple human microbiome projects including the Vaginal Human Microbiome Project (VaHMP) [20, 21], the Multi-Omics Microbiome Study: Pregnancy Initiatives (MOMS-PI) [22], oral and gut microbiome in neonates [23]. Each project involves the development of specialized computational methodologies and tools for analysis of large data sets generated by NGS sequencing on Illumina platforms. The accuracy of 16S rRNA microbial profiling is highly dependent on the accuracy of pre-processing of sequencing data. Thus, we have developed a strategy entitled MeFiT (Me rging and Fi ltering T ool) that efficiently merges overlapping paired-end sequence reads from the Illumina MiSeq™ sequencing platform and quality filters them using the MEE measure outlined above. MeFiT invokes a version of CASPER [12] for merging paired-end reads and extends it by including careful quality-filtering. We provide users the option to quality filter the amplicons using the traditional average Q-score metric or using a maximum expected error cut-off threshold. We have also optimized MeFiT with the additional functionality of appending non-overlapping paired end reads; i.e., paired end reads from amplicons of greater than 600 bases which do not overlap are not discarded and are used to accurately determine the taxon from which they were derived.
Most life science researchers lack an extensive IT-infrastructure to support data analysis of the scale required for projects employing high throughput NGS technologies. These investigators often cobble together a variety of software packages and pipelines to perform their analyses, often leading to a lack of quality control and computational efficiency. Herein, we describe our efforts to develop a fast and accurate in-house decision support tool for use as a first pre-processing step to obtain clean high-quality data for several ongoing projects generating microbiome profiles from targeting 16S rRNA sequencing using paired end sequencing technology provided by the Illumina MiSeq platform. The MeFiT pipeline combines the essential first steps in analysis of paired-end sequencing data; i.e., merging of overlapping reads and quality filtering of the reads. It is freely available (https://github.com/nisheth/MeFiT) and can be easily and intuitively used by other groups dealing with similar challenges.
Implementation
The MeFiT pipeline
The MeFiT pipeline performs following two steps: (1) merging of overlapping paired-end sequences, and (2) filtering data for quality (Fig. 1). The pipeline accepts the forward and reverse reads files in the FastQ format and automates the merging and quality filtering of overlapping and non-overlapping (optional) reads. It also generates an extensive report on the quality and merging statistics.
Fig. 1.
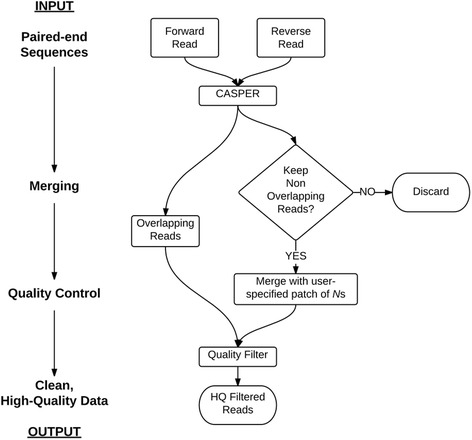
The MeFiT pipeline
Merging of paired-end reads
MeFiT invokes a version of the CASPER algorithm (see REF [7]) to merge the forward and reverse reads generated in paired-end sequencing. Briefly, the first step of the CASPER algorithm is to identify the best possible overlap region, with the least number of mismatching bases, between the forward and reverse reads. Any mismatches are then resolved by relying on the difference in their quality scores, with a lower quality base being replaced by the one with the higher score. Where the difference is not significant, CASPER makes partial decisions on k-mer-based contexts around the mismatch to make a final decision for resolving the mismatch. The user, as needed, can modify the default CASPER parameters implemented into MeFit.
Non-overlapping reads
As outlined above, 2 × 300 base PE reads will not overlap if the target amplicon is greater than 600 base pairs. Although targeted 16S amplicons are generally selected to be smaller than 600 base pairs, some bacterial taxa, including unknown taxa, occasionally exhibit target sequences exceeding the expected size. Most existing analysis packages tend to discard these sequences, resulting in the gross underrepresentation of some taxa. MeFiT permits the user to specify to retain (default: discard) non-overlapping reads by linking the forward and reverse reads with a user-specified patch of Ns. MeFiT reverse complements the reverse read and appends it to the 3′ end of the forward linked by a string of Ns (default: 15) with a assigned Phred quality scores of 2, which are subsequently ignored during the filtering step and calculation of quality statistics. Thus, taxa with unexpectedly large target amplicons are retained in the analysis.
Quality filtering
As outlined briefly above, we were unsatisfied with the use of a simple average Q score to identify good and bad reads. Thus, the program permits the user to select either the average read Phred-quality score or Q score, or the maximum expected error (MEE) as a percent of read length (meep score) (Koparde VN, et. al. MEEPTOOLS: A maximum expected error based FASTQ read filtering and trimming tool, submitted). The maximum expected error (MEE) within a read is the sum of error probabilities of each base, given by –
where, P is the error probability and Q is the Phred-quality score (represented as ASCII character) of the i th base of a read. The meep-score is then computed from the MEE of a read as the percentage of its read length. Thus, a meep cut-off of 1% permits a maximum error of 1 in a read of length 100 nt.
Results and discussion
With the advent and availability of Illumina’s MiSeq system, capable of generating 2 × 300 base paired-end reads and up to 15Gb per run of 600 base-pair sequence information, it has become a go-to platform for targeted gene sequencing, metagenomics, small genome sequencing, etc. Accurate merging and quality control of raw MiSeq data is essential before any downstream analysis. Inaccurate merging or the presence of low-quality reads (unreliable base-calls) result in an increase in the false negatives and false positives in subsequent analyses. The MeFiT pipeline was developed to perform the two prerequisite steps – merging and quality filtering of the paired end reads, to obtain high-quality data prior to subsequent analyses.
As the first step of the pipeline, MeFiT uses CASPER to merge the forward and reverse reads. As previously reported, CASPER, with its novel mismatch-resolving algorithm, showed a high level of accuracy when compared to COPE, FLASH and PANDAseq. [12] Although CASPER is not the fastest approach computationally, as expected from a k-mer based approach, the higher accuracy and robustness of the software compensates for this weakness.
One of the drawbacks of most current merging tools is that paired end reads that do not merge properly are not retained, possibly leading to the erroneous elimination of these reads from downstream analysis. Also, the target DNA fragment may be longer than the sequenced read length resulting in a gap between the forward and reverse reads. For example, in 16 S rRNA-based taxonomic profiling, a targeted region of a known (or unknown) bacterial taxon may exceed the 600 base length of the 2 × 300 base paired end sequencing of the MiSeq platform. Such forward and reverse reads will not overlap, and discarding them as most analysis protocols do results in the loss of valuable information. Considering a typical paired-end sequencing experiment of 2 × 300 bp reads, one can sequence the 16S hypervariable regions up to ~540 bp in length (with a 20 bp forward and reverse primers on average and a minimum of 20 bp overlapping region). Our analysis of the lengths of hypervariable regions (V1–V3, V2–V4, V3–V5, V6–V9) of sequences in the SILVA database (16S, SSU Ref NR, v119) [24], extracted using V-Xtractor [25], showed that a significant fraction of taxa have hypervariable regions that would likely result in amplicons of greater than 540 bp. The most impacted taxa are the Firmicutes and Proteobacteria, which are abundant in gut microbiome samples (Additional file 1: Table S1). A similar investigation of the STIRRUPS database, which targets largely taxa of relevance to the female human urogenital tract, showed that ~1% of bacteria have V1–V3 regions in excess of 580 bp when the primer sequences are included [26]. We believe that retaining such non-overlapping high-quality data will improve the accuracy of downstream phylogeny and taxonomy analysis. The MeFiT pipeline addresses the above-mentioned shortcoming of other tools and provides users with the flexibility to append forward/reverse reads where no overlap was identified by CASPER.
The merged amplicons (overlapping and non-overlapping) are subjected to a quality control filter. Mefit invokes two alternative quality assessment metrics: the average Q-score, or the meep-score. Amplicons that pass the user-defined filtering criteria (average Q-score above threshold, or meep-score below threshold) are saved as High Quality in FastQ format for further downstream analysis. Finally, MeFiT generates a detailed quality-statistics output for each processed sample that provides the total number of reads, the number of overlapping reads, the number of non-overlapping reads, the average amplicon length, the average Q-score, the average meep-score in amplicons, the number of high-quality amplicons, the average read length in high-quality amplicons, the average Q-score in high-quality amplicons, the average meep-score in high-quality amplicons, and the percent of overlapping reads in high-quality amplicons. Additional file 1: Table S2 shows the detailed statistics report for 4 samples (randomly selected in-house samples) processed through the MeFiT pipeline. Filtering amplicons for quality using an average Q-score cutoff (threshold: 20) retains > 99% reads for each sample, in contrast, filtering for quality using meep cutoff (threshold: 1) only results in 83–0% high-quality amplicons. This clearly indicates that quality filtering using average Q-scores result in amplicons being labeled high-quality in spite of having higher error probabilities.
To demonstrate the utility of MeFiT, we generated a 2 × 300 bp simulated dataset from V1-V3 region of the 16S gene of six reference species: Lactobacillus crispatus, L. iners, Prevotella bivia, and Gardnerella vaginalis, which have V1-V3 amplicons varying from 471–519 bp; and Clostridium josui and Campylobacter rectus which have amplicons of over 595 bp. The simulated dataset, generated using Grinder [27], consisted of 120,000 paired-end reads of 2 × 300 read length configurations, 20,000 from each of the six species. Sequencing errors were introduced using a modified 4th degree polynome Illumina error model 3e-3 + 1.8e-9 *i^4 [28]. MeFiT was run on this dataset, saving the non-overlapping reads appending them with a patch of 15 Ns. The merged amplicons were filtered using a meep cut-off of 1. As seen in Additional file 1: Table S3, paired-end reads simulated from species with shorter V1-V3 regions (less than 540 bp) result in overlapping amplicons, while those simulated from C. josui and Campylobacter rectus are non-overlapping and the standard applications of most analyses would simply discard the reads from the two taxa with longer V1-V3 regions, resulting in a biased community profile. OTU clustering or taxonomic classification, on a set of only high-quality overlapping amplicons will result in a community profile composed of only the four species with shorter V1-V3 amplicons. However, accurate identification and community abundance profiles are obtained if high quality non-overlapping amplicons are retained (Additional file 1: Figure S1).
Some of the common bioinformatics tools for microbiome analysis; including mothur [29, 30], QIIME [31] and USEARCH [32] have their own implementations for merging paired-end reads. The standard operating procedure for analyzing 16S rRNA sequences generated using Illumina’s MiSeq platform, demonstrated by mothur [30], suggests using the ‘make.contigs’ command for preprocessing reads, and subsequently discards reads that cannot be assembled into contigs. Similarly, QIIME and USEARCH [33] have implementations that merge and quality filter overlapping paired-end reads. However, these steps are not a part of the standard analysis pipelines, resulting in the need for users to incorporate these preprocessing steps in their custom pipelines to obtain high-quality data for analysis. CASPER outperforms majority of merging tools in terms of sensitivity and specificity [12]. Characterizing a mock bacterial community [34] using high-quality data obtained by MeFiT resulted in the identification of fewer spurious OTUs compared to that obtained by quality control steps implemented in mothur (see Additional file 1, Additional file 2). Table 1 shows an overview of features of MeFiT compared to QIIME, mothur and USEARCH. We have developed a facile pipeline that takes sample-specific raw reads as input and provides a set of high-quality amplicons that can be directly fed into softwares/tools to perform OTU analysis, taxonomic classification, functional profiling and other microbiome analysis.
Table 1.
Overview of features of MeFiT compared to other popular tools
| Merging | Quality Filtering | ||||
|---|---|---|---|---|---|
| Single Step | Tool | Merge non-overlapping paired-end reads | AvgQ | Expected Error | |
| MeFiT | ✔ | CASPER | ✔ | ✔ | ✔ |
| Qiime v1.9.1 | ✗ | fastq-join/SeqPrep | ✗ | ✔ | ✗ |
| mothur v1.37 | ✗ | make.contigs | ✗ | ✔ | ✗ |
| usearch v8.0 | ✗ | fastq_mergepairs | ✗ | ✗ | ✔ |
Conclusions
The MeFiT pipeline combines open-source merging tool CASPER to a quality filtering step that automates the primary steps of analyzing overlapping paired-end sequencing data. Our pipeline reduces chances of human error due to minimal input requirements. Due to its simplified implementation and configuration, it can be easily incorporated into other analyses pipelines.
Availability and requirements
Project name: MeFiT
Project home page: https://github.com/nisheth/MeFiT
Operating system(s): UNIX-based platform
Programming language: Python v2.7
Other requirements: CASPER (http://best.snu.ac.kr/casper); numpy (http://www.numpy.org/); HTSeq (http://www-huber.embl.de/users/anders/HTSeq/doc/install.html).
License: Apache 2.0
Any restrictions to use by non-academics: Apache 2.0
Acknowledgements
We gratefully acknowledge the technical and philosophical assistance and advice of our colleagues MG Serrano, NC Asmussen, SW Norris and other members of the Vaginal Microbiome Consortium (VMC) at VCU. We especially thank K Hendricks-Munoz and P Xu for providing the datasets for development and testing purposes.
Funding
This work was partially funded by the NIH Common Fund Human Microbiome Project (HMP) program through grant 8U54HD080784 to G Buck, J Strauss, and K Jefferson.
Authors’ contributions
HIP and NS participated in the design of the study. HIP, VNK, SPB implemented the software, performed the analysis. HIP, GB, NS wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- CASPER
Context-aware scheme for paired-end reads
- COPE
Connecting overlapped pair-end Reads
- DNA
deoxyribonucleic acid
- FLASH
Fast length adjustment of short reads
- MEE
Maximum expected error
- MeFiT
Merging and filtering tool
- MOMS-PI
Multi-omic microbiome study-pregnancy initiative
- NGS
Next-generation sequencing
- OTU
Operational taxonomic unit
- PEAR
Paired-end read merger
- QIIME
Quantitative insights into microbial ecology
- RNA
Ribonucleic ACid
- RT-PCR
Reverse transcriptase polymerase chain reaction
- SHERA
Shortread error-reducing aligner
- SSU rRNA
Small subunit ribosomal RNA
- STIRRUPS
Species-level Taxon Identification of rDNA Reads using a USEARCH Pipeline Strategy
- VaHMP
Vaginal human microbiome project
Additional files
Supplementary methods, tables and figures. (DOCX 505 kb)
OTU-analysis commands. (TXT 6 kb)
Contributor Information
Hardik I. Parikh, Email: parikhhi@vcu.edu
Vishal N. Koparde, Email: vnkoparde@vcu.edu
Steven P. Bradley, Email: bradleysp@vcu.edu
Gregory A. Buck, Email: gabuck@vcu.edu
Nihar U. Sheth, Email: nsheth@vcu.edu
References
- 1.Mardis ER. Next-generation DNA, sequencing methods. Ann Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 2.Hyman RW, Fukushima M, Diamond L, Kumm J, Giudice LC, Davis RW. Microbes on the human vaginal epithelium. Proc Natl Acad Sci U S A. 2005;102:7952–7957. doi: 10.1073/pnas.0503236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci U S A. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao Z, Tseng CH, Pei Z, Blaser MJ. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci U S A. 2007;104:2927–2932. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Illumina: 16S Metagenomics Studies with the MiSeq System. Application Note: Microbial Genomics. http://www.illumina.com/content/dam/illumina-marketing/documents/products/appnotes/appnote_16s_sequencing.pdf. Accessed 17 Sept 2016.
- 8.Rodrigue S, Materna AC, Timberlake SC, Blackburn MC, Malmstrom RR, Alm EJ, Chisholm SW. Unlocking short read sequencing for metagenomics. PLoS One. 2010;28:e11840. doi: 10.1371/journal.pone.0011840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;14:13–31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lui B, Yuan J, Yiu SM, Li Z, Xie Y, Chen Y, Shi Y, Zhang H, Li Y, Lam TW, Luo R. COPE: an accurate k-mer-based pair-end reads connection tool to facilitate genome assembly. Bioinformatics. 2012;28:2870–2874. doi: 10.1093/bioinformatics/bts563. [DOI] [PubMed] [Google Scholar]
- 12.Kwon S, Lee B, Yoon S. CASPER: context-aware scheme for paired-end reads from high-throughput amplicon sequencing. BMC Bioinformatics. 2014;15(Suppl 9):S10. doi: 10.1186/1471-2105-15-S9-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solex/Illumina FASTQ variants. Nucleic Acids Res. 2009;38:1767–1771. doi: 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez-Alcantara A, Ballesteros E, Rojas FM, Koshinsky H, Fofanov VY, Havlak P, Fofanov Y. PIQA: pipeline for Illumina G1 genome analyzer data quality assessment. Bioinformatics. 2009;25:2438–2439. doi: 10.1093/bioinformatics/btp429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, Nekrutenko A, Galaxy Team Manipulation of FATSQ data with Galaxy. Bioinformatics. 2010;26:1783–1785. doi: 10.1093/bioinformatics/btq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox MP, Peterson DA, Biggs PJ. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinfomratics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgar RC. Average Q score. http://www.drive5.com/usearch/manual/avgq.html. Accessed 17 September 2016.
- 20.Fettweis JM, Alves JP, Borzelleca JF, Brooks JP, Friedline CJ, Gao Y, Gao X, Girerd P, Harwich MD, Hendricks SL, Jefferson KK, Lee V, Mp H, Neale MC, Puma FA, Reimers MA, Rivera MC, Roberts SB, Serrano MG, Sheth N, Silberg JL, Voegtly L, Prom-Wormley EC, Xie B, York TP, Cornelissen CN, Strauss III JF, Eaves LJ, Buck GA. The Vaginal Microbiome: Disease, genetics and the environment. Nature Proceedings. 2011.
- 21.Fettweis JM, Brooks JP, Serrano MG, Sheth NU, Girerd PH, Edwards DJ, Strauss JF, Vaginal Microbiome Consortium. Jefferson KK, Buck GA. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiology. 2014;160:2272–2282. doi: 10.1099/mic.0.081034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Integrative HMP (iHMP) Research Network Consortium The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe. 2014;10:276–289. doi: 10.1016/j.chom.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hendrics-Munoz KD, Xu J, Parikh HI, Xu P, Fetweis JM, Kim Y, Louie M, Buck GA, Thacker LR, Sheth NU. Skin-to-skin care and the development of the preterm infant oral microbiome. Am J Perinatol. 2015 [epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 24.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res. 2013;41(D1):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartmann M, Howes CG, Abarenkov K, Mohn WW, Nilsson RH. V-Xtractor: an open-source, high-throughtput software tool to identify and extract hypervariable regions of small subunit (16S/18S) ribosomal RNA gene sequences. J Microbiol Methods. 2010;83:250–253. doi: 10.1016/j.mimet.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Fettweis JM, Serrano MG, Sheth NU, Mayer CM, Glascock AL, Brooks JP, Jefferson KK. Vaginal Microbiome Consortium (additional members), Buck GA. Species-level classification of the vaginal microbiome. BMC Genomics. 2012;13(Suppl 8):S17. doi: 10.1186/1471-2164-13-S8-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angly FE, Willner D, Rohwer F, Hugenholtz P, Tyson GW. Grinder: a versatile amplicon and shotgun sequence simulator. Nucleic Acids Res. 2012;40:e94. doi: 10.1093/nar/gks251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korbel JO, Abyzov A, Mu XJ, Carriero N, Cayting P, Zhang Z, Snyder M, Gerstein MB. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009;10:R23. doi: 10.1186/gb-2009-10-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schloss PD, Westcott SL, Ryabin T, HAll JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stress B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nature. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 33.Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics. 2015;31(21):3476–3482. doi: 10.1093/bioinformatics/btv401. [DOI] [PubMed] [Google Scholar]
- 34.Allen HK, Bayles DO, Looft T, Trachsel J, Bass BE, Alt DP, Bearson SMD, Nicholson T, Casey TA. Pipeline for amplifying and analyzing amplicons of the V1-V3 region of the 16S rRNA gene. BMC Res Notes. 2016;9:380. doi: 10.1186/s13104-016-2172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]