

Abstract
The relationship between exposure to ultraviolet (UV) radiation and skin cancer urges the need for extra photoprotection, which is presently provided by widespread commercially available sunscreen lotions. Apart from having a large absorption cross section in the UVA and UVB regions of the electromagnetic spectrum, the chemical absorbers in these photoprotective products should also be able to dissipate the excess energy in a safe way, i.e. without releasing photoproducts or inducing any further, harmful, photochemistry. While sunscreens are tested for both their photoprotective capability and dermatological compatibility, phenomena occurring at the molecular level upon absorption of UV radiation are largely overlooked. To date, there is only a limited amount of information regarding the photochemistry and photophysics of these sunscreen molecules. However, a thorough understanding of the intrinsic mechanisms by which popular sunscreen molecular constituents dissipate excess energy has the potential to aid in the design of more efficient, safer sunscreens. In this review, we explore the potential of using gas-phase frequency- and time-resolved spectroscopies in an effort to better understand the photoinduced excited-state dynamics, or photodynamics, of sunscreen molecules. Complementary computational studies are also briefly discussed. Finally, the future outlook of expanding these gas-phase studies into the solution phase is considered.
Keywords: frequency- and time-resolved spectroscopy, ultrafast dynamics, photophysics, sunscreens, bottom-up approaches, increasing molecular complexity
1. Introduction
Despite efforts to raise awareness towards both the correct use of sunscreens and the risks of excessive sun exposure, skin cancer cases have risen in recent years [1–3]. According to the World Health Organization, 2–3 million non-melanoma and 132 000 melanoma skin cancers occur per year worldwide (as of 2003), which translates to one in every three cancers diagnosed being a skin cancer [1]. In the UK alone, 12 800 people were diagnosed with malignant melanoma (the most serious type of skin cancer) in 2010, rising to 14 509 new cases reported in 2013 [3]. The cost of skin cancer treatment in England in 2008 was estimated to be in the range of £106–112 million and is predicted to increase to approximately £180 million by 2020 [4]. The increase in skin cancer incidence is thought to be, in part, related to a cultural tendency for more sun exposure, as well as the increased use of sunbeds and, importantly, the inadequate use of sunscreen lotions [3,5].
Like all other types of cancer, skin cancer is a complex problem, the causes of which are not yet fully understood. Nevertheless, ultraviolet (UV) radiation has been consistently shown to be a carcinogen, involved in both direct and indirect DNA damage [6–10], despite its essential role in maintaining plant and animal life on the Earth [11]. UV radiation is classified according to wavelength as UVA (400–315 nm), UVB (315–280 nm) or UVC (280–100 nm), as represented in figure 1 [13]. While the amount of UVC that reaches the Earth is negligible [12], as it is absorbed by the ozone layer in the stratosphere [14], the amount of UVA and UVB radiation at the Earth's surface (approx. 220 µW cm−2 on a cloudless day, globally) is chemically significant [15,16].
Figure 1.

Wavelength regions of UVA (green), UVB (blue) and UVC (violet) radiation in the standard solar spectrum (black line) and the solar spectrum at the Earth's surface, after atmosphere effects (red line). The inset shows the total solar spectrum, with the UV region highlighted (grey shading). Solar spectra raw data obtained from the National Renewable Energy Laboratory (NREL) website [12]. (Online version in colour.)
Both UVA and UVB are capable of causing erythema (sunburn), which in itself is thought to be triggered by DNA damage. It is estimated, however, that approximately 1000 times more UVA radiation is necessary to cause the same level of damage as compared with UVB [17]. Despite being more prominent at ground level (as it is not absorbed as efficiently as UVB by the atmosphere) [18,19], UVA radiation is considered to be less carcinogenic than UVB radiation, because it does not interact with DNA as extensively [20–23]. UVB radiation can be directly absorbed by DNA [21], producing highly mutagenic photolesions, such as cyclobutane pyrimidine dimers and pyrimidine 6–4 photoproducts [20,24]. Even though these errors in the DNA sequence can be repaired by excision repair pathways [20], those which fail to be repaired will result in UV signature mutations and, potentially, carcinogenesis [25,26]. UVB is, therefore, considered to be the most significant type of radiation in the induction of skin cancers [27,28].
While there are natural protection mechanisms against UV-induced DNA damage (vide infra), the prevalence of skin cancer cases makes it clear that artificial enhancement of these mechanisms through man-made sunscreens is necessary. The active agents present in current sunscreens can be categorized into two main groups: physical blockers and chemical absorbers [29]. Physical blockers are substances which reflect or scatter UVA/UVB, such as titanium dioxide and zinc oxide nanoparticles (which, however, also absorb UV radiation) [30]. These species are usually regarded as inert and non-toxic [31], but they are also used in other industries as photocatalysts (for example, in water treatment) [29]. Photocatalytic activity could potentially present a problem in sunscreen formulations and, therefore, there are certain methods designed to reduce it. However, the effectiveness of these methods has yet to be established and their implications on sunscreen safety have not been explored [29]. While this is a considerable problem that requires addressing, this review will focus on the chemical absorbers: molecules which absorb UVA/UVB radiation and thus supplement the skin's own sunscreen molecules, such as melanin pigments.
The chemical absorbers commonly used in the sunscreen industry fit roughly into the seven categories shown in figure 2 (alongside representative sunscreen filter molecules): these are (i) para-aminobenzoate derivatives, (ii) cinnamate derivatives, (iii) salicylate derivatives, (iv) anthranilate derivatives, (v) camphor derivatives, (vi) dibenzoyl methane derivatives, and (vii) benzophenone derivatives [29,32,33]. The conjugated systems present in all these molecules allow for strong UV absorption [34–36]. However, no single molecule will provide photoprotection across the entire UVA and UVB range, so chemical absorbers are usually used in conjunction with each other [37] in order to ensure that the sunscreen complies with established regulations, such as sun protecting factor (SPF) values and UVA/UVB absorption ratios [38,39].
Figure 2.

(a–g) The seven main categories of most sunscreen active absorbing ingredients and representative examples of each.
Sunscreen formulations are tested for their photoprotective capability by measuring how long they delay the appearance of erythema in treated skin when compared with an untreated sample [40,41]. In addition, and as with any other cosmetic product, sunscreen lotions are tested for dermatological compatibility, in terms of their allergenic and photoallergenic potential [42,43]. It has been reported that organic sunscreen filters can act as photosensitizers, i.e. induce an allergic reaction after absorption of UV radiation; however, the exact mechanisms of photoallergy are still unknown [43].
Particularly in the context of sunscreen development, it is also informative to analyse the mechanisms responsible for photoprotection in natural sunscreens. In much the same way that human skin requires melanin pigments to provide protection against UV damage [44], other living organisms also have an intrinsic need for photoprotection and hence have developed their own photoprotective mechanisms [45,46]. In plant species, UVB radiation was found to have a number of damaging effects, such as growth inhibition, disruption to photosynthesis and transpiration, and general damage to DNA, proteins and membranes [47,48]. In addition, studies have found that plants which lack the ability to efficiently produce certain phenolic molecules, such as sinapate esters and flavonoids (figure 3), are more vulnerable to UVB-induced damage [49]. These molecules are, therefore, believed to provide photoprotection to plant species [50,51]. For example, in Arabidopsis thaliana (Brassicaceae), sinapate esters were found to play a major role in the attenuation of the adverse effects caused by excess UVB radiation [52]. The intrinsic photoprotective properties of these molecules may be instructive towards the design of the next generation of artificial sunscreens; hence, understanding their photodynamics is of particular relevance. As such, a discussion of these chemical absorbers will also feature herein.
Figure 3.

Two main types of plant sunscreen: (a) sinapate derivatives and flavonoids, of which (b) flavonol is an example.
The phenomena, occurring at a molecular level in synthetic and natural sunscreen species upon absorption of UV radiation, which are overlooked by the current sunscreen testing methods, could not only lead to photoproducts but also result in long-lived excited states, both of which have the potential to trigger photosensitivity [53]. Unravelling the photodynamics occurring in sunscreen molecules upon UV absorption will provide insight into their photochemistry and photophysics. This information may then help to identify and assess potentially harmful relaxation pathways and aid a guided development of a new generation of more efficient sunscreens.
Upon absorption of UV radiation, a sunscreen will be photoexcited to a higher energy and potentially reactive electronic state [54]. Over time, the system will relax back to the ground (lowest energy) electronic and vibrational state via a number of photochemical/photophysical processes [55,56], such as intramolecular vibrational redistribution (IVR) [57], internal conversion (IC) [58], intersystem crossing (ISC) [58], fluorescence [59] or phosphorescence [60], as summarized in figure 4, where only the photophysical processes are shown for simplicity. Some of these processes may induce damage to the skin if, for example, they result in the production of free radicals or other harmful photoproducts [29,61]. An ideal chemical absorber for a sunscreen lotion should not only strongly absorb UVA/UVB, but should also be capable of dissipating the excess energy gained via mechanisms that cause no chemical change to occur, reforming the molecule in its original state without forming any potentially harmful species.
Figure 4.
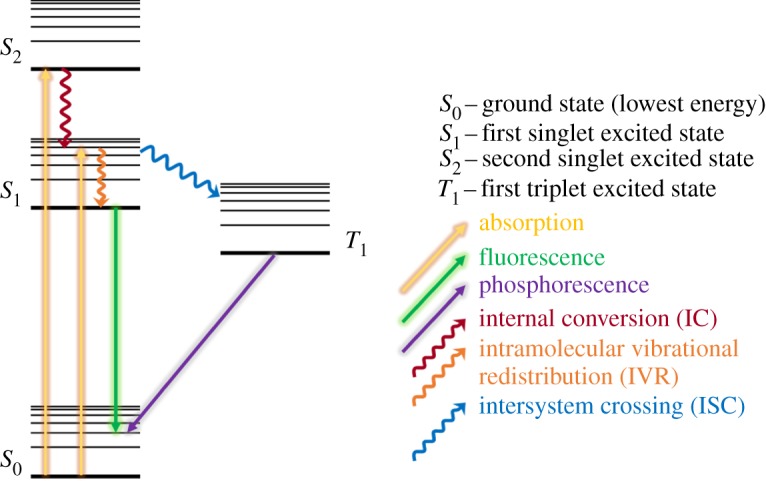
Simplified Jablonski diagram showing some of the possible photophysical processes undergone by a sunscreen molecule after UV absorption. (Online version in colour.)
The more efficient the mechanism by which the molecule returns to its ground electronic state, the less the chance of undesirable photochemistry taking place [62]. The fastest of these relaxation mechanisms typically occur within femtoseconds (fs, 10−15 s) or picoseconds (ps, 10−12 s) [63]: the timescales of nuclear motion and the breaking (or creating) of chemical bonds. However, other photophysical processes (fluorescence, for example) can take place on the nanosecond (ns, 10−9 s) timescale, while phosphorescence can take up to seconds [64]. In these cases, the excited states will persist long enough that there is a higher chance for detrimental photochemistry to occur. An in-depth study on these excited-state decay mechanisms has the potential to unveil any harmful photochemistry that might not be detectable at a macroscopic level if, for example, any long-lived excess energy facilitates damage to DNA.
In the remainder of this review, we will explore the spectroscopic techniques that have recently been utilized in gas-phase studies to initiate this stepwise approach towards unravelling the photochemistry and photophysics of sunscreen filter molecules, as well as providing a synopsis of the contemporary information gathered on this subject to date. These studies provide the initial ground work for an exciting field of research where much is yet to be unravelled, which we briefly expand on in the Summary and outlook.
2. Methodologies: frequency- and time-resolved spectroscopy in the gas phase
The advances in laser technology over the past few decades [65] have allowed for the development of a plethora of spectroscopic techniques [66] which enable one to experimentally obtain information on electronic structure and excited-state dynamics which could not be gathered previously. However, owing to the molecular complexity of sunscreen molecules, performing frequency- and time-resolved spectroscopic measurements on them often yields rather intricate spectra and dynamics which may not be unambiguously analysed. Therefore, it is advantageous to employ a bottom-up approach [67,68], wherein a complex system, for example, a large sunscreen molecule, is broken down into its component parts. These components are then studied in the interaction-free environment offered by in vacuo gas-phase experiments in order of increasing molecular complexity, building up to the large sunscreen molecule of interest. These studies are then extended into a more real-world scenario [69] by the inclusion of solvents through micro-solvation (gas-phase cluster studies) and solvation (solution phase) as well as increasing the number of interacting species. This bottom-up approach has proved very useful in providing insight, for example into the photostability of DNA/RNA nucleobases and their corresponding chromophore (i.e. UV absorbing) subunits [70–72]. This stepwise approach yields a comprehensive understanding of the photochemistry and photophysics of sunscreen molecules following UV radiation absorption.
This section gives a brief overview of the techniques which have been employed as outlined above to study the sunscreen molecules to be discussed in the exemplar case studies presented in §3.
(a). Frequency-resolved spectroscopy
A molecule's photochemistry and photophysics are highly dependent not only on the arrangement of its constituent atoms, but also on its electronic and vibrational energy structure, termed vibronic structure herein [73]. Therefore, an understanding of a molecule's vibronic structure aids the interpretation of that molecule's photoreactivity [73]. Frequency-resolved spectroscopy techniques can provide a picture of the vibronic structure of a given system by photoexciting molecules to specific vibrational levels and then using various methods to probe these excited states.
High-frequency resolution is provided by narrow bandwidth lasers. The spectral bandwidth of a laser pulse, Δν, is related to its temporal full width at half maximum (Δt) by the following [74]:
| 2.1 |
In the above equation, K is a constant which depends on the profile of the pulse. To achieve the sub-wavenumber resolution necessary to resolve vibronic (and in some cases rovibronic) states, nanosecond pulses are required [75–78].
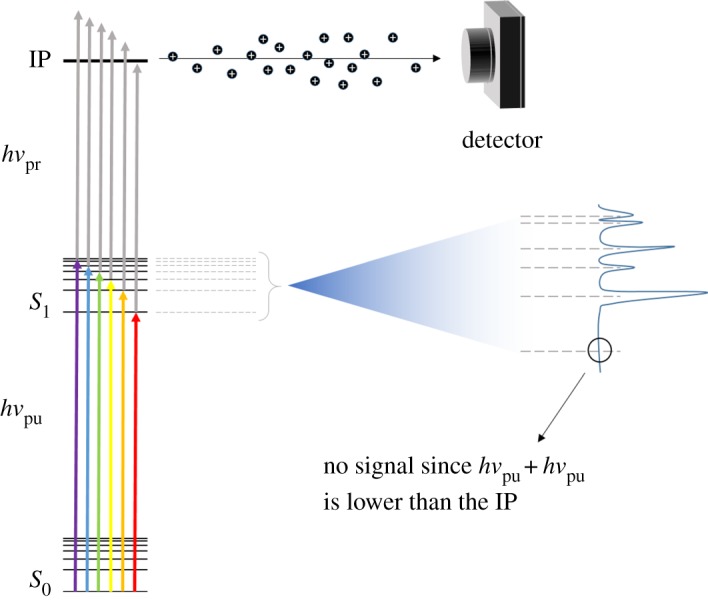
Frequency-resolved techniques can be improved by the use of molecular beam technology, which is the current standard for gas-phase experiments [65,66,79]. By seeding molecules in a buffer gas, such as a noble gas, they are collisionally cooled to a vibrational temperature of just a few kelvins [66]. Rapid expansion of the molecules into vacuum ‘freezes’ the sample at these temperatures and a skimmer removes the outer ‘hot’ region of the beam produced. The skimmer, a cone-shaped orifice (see figure 7 for details), separates the source region (where the gas is introduced into vacuum) from the interaction region (where the gas, in the form of a molecular beam, interacts with the laser pulses). This generates a directed flow of cold molecules with a narrow velocity distribution, effectively reducing the Doppler effect [80], resulting in spectral lines with a small Doppler width and hence improving resolution [66].
Figure 7.

Diagram of a typical gas-phase experimental apparatus, including both a molecular beam and a TOF mass spectrometer. The molecular beam interacts with the laser pulses employed in the experiment. For example, for 1 + 1 REMPI, pump, hνpu, and probe, hνpr, laser pulses are necessary, as represented by the red and blue arrows, respectively. The resulting ions are accelerated towards the detector by a set of ion optics. The last section of this set-up consists of a field-free flight tube (see main text), so that ions reach the detector at different times depending on their m/z ratio. (Online version in colour.)
The main difference between the various frequency-resolved spectroscopic techniques lies in the method used for the detection of the excited states. Some of the possible experimental arrangements are briefly reviewed in the following sub-sections.
(i). Laser-induced fluorescence spectroscopy
In laser-induced fluorescence (LIF) [81,82] spectroscopy, a molecule, initially in its ground vibronic state (S0), is photoexcited by a pump photon, hνpu, of variable wavelength, so that specific vibronic excited states are accessed, e.g. in the first excited electronic state (S1), as represented in figure 5a. The radiative decay of these excited states will then result in a fluorescence signal which is collected by a photomultiplier or similar photodetector. From this, a plot of fluorescence intensity versus excitation wavelength, or the excitation spectrum, is produced. Alternatively, hνpu may be fixed to a wavelength resonant with a specific vibronic excited state. The resulting fluorescence signal is then dispersed by a grating onto a photodetector so that fluorescence from this state onto the several vibrational levels of the ground electronic state is monitored, as depicted in figure 5b [83–85]. This is referred to as dispersed fluorescence (DFL). Fluorescence lifetimes can be measured with either of these set-ups, simply by monitoring the fluorescence signal following photoexcitation over time [86,87]. The temporal resolution of these experiments is then limited by detector response times which, for many common photomultiplier tubes, is approximately 1–10 ns [88].
Figure 5.

Schematic of LIF (a) and DFL (b). In LIF, hνpu is scanned so that when it is resonant with a certain vibronic state, this state may radiatively decay to any vibrational level in the ground electronic state (S0, represented by thick gradient arrow). The fluorescence signal is then collected by a photomultiplier. In DFL, on the other hand, hνpu is set to a particular vibronic state, from which population may fluoresce to any accessible vibrational level of the S0 state. The total fluorescence signal is dispersed by a monochromator in order to deconvolute the information on the vibrational levels of the S0 state, and again collected by a photomultiplier. (Online version in colour.)
The high signal-to-noise levels of this technique, afforded by detecting a bright signal on a dark background, coupled with the high intensity of the lasers utilized, afford a sensitivity not achievable in absorption spectroscopy [81]. As fluorescence intensity is proportional to the population of the emitting state, LIF can provide information on the relative population of excited states [82]. However, as LIF is a one-photon process, it is limited by the efficiency of the transitions involved, in both excitation and fluorescence, which are governed by the same symmetry and spin selection rules of any one-photon vibronic transition [34].
As shall become clear in §3, LIF and DFL can be successfully applied to the study of sunscreen molecules in the gas phase, providing valuable insight into their electronic structure.
(ii). Resonance-enhanced multiphoton ionization spectroscopy
Resonance-enhanced multiphoton ionization (REMPI) is a photoionization technique by which multiple photons are used to first resonantly excite (pump) and then ionize (probe) the molecule of interest [76,89]. REMPI schemes are usually referred to as n + m, where n and m are integers which correspond to the number of photons, of a single colour, used to pump and probe the system of interest, respectively [89–91]. When different colours are used for the pump and probe steps, this is referred to as n + m′ [92,93]. While excited states can usually be accessed and probed simply with a (1 + 1) REMPI scheme, also referred to as resonant two-photon ionization, or R2PI, as shown in figure 6, there are cases in which several photons need to be absorbed simultaneously to reach a certain electronic excited state. These multiphoton absorption processes follow an In power dependence on laser intensity (I), where n corresponds to the number of photons absorbed [94,95], and they therefore require the high photon densities achievable with lasers.
Figure 6.
Schematic showing the basic concept behind R2PI. As hνpu is scanned, each vibronic state is individually accessed. These states are probed by a second photon, hνpr, and resulting ions are directed to an appropriate detector (see main text). (Online version in colour.)
Regardless of the specific ionization scheme, in a REMPI experiment, the ions produced by the ionization step are monitored as a function of excitation wavelength. This produces spectral lines as each new vibronic excited state is accessed, thus revealing the molecule's vibronic structure. The total ion signal, as a function of probe wavelength, can also be monitored in order to calculate the ionization potential (IP, figure 6) of the molecule under study—the cut-off point, where no ion signal is observed, indicates the point at which the total energy provided to the molecule is no longer sufficient to ionize it [96].
Ion detection is usually achieved using a time-of-flight (TOF) spectrometer, as shown in figure 7 [97,98]. In simple terms, a TOF spectrometer consists of a vacuum chamber with an ion source at one end, where a molecular beam interacts with the laser pulses, and an ion detector at the other. Ions are accelerated towards the detector by one or more electric fields (continuous or pulsed). Before they reach the detector, the ions go through a field-free flight path, i.e. a region in which no forces are applied, so that the velocity of the ions in this area is dependent on their mass-to-charge (m/z) ratio. A TOF trace can then be constructed of ion signal against arrival time at the detector. The use of a TOF spectrometer in combination with REMPI spectroscopy thus provides some mass selectivity [89], which allows for contamination from absorbing species other than the molecule of interest that might be present in the molecular beam to be supressed. Like LIF, REMPI spectroscopy benefits from high sensitivity which in this case arises due to the capability to detect all the ions produced by photoionization. In addition, the ionization (probe) step is not as constrained by symmetry selection rules as fluorescence, as photoelectrons will adopt the symmetry required for ionization to take place [99,100].
By careful selection of the temporal pulse width of the lasers used, frequency-resolved (as described above) or time-resolved experiments can be performed in order to gather dynamical information, as shown by the pulse sequences used in figure 7, where the pump and probe are temporally delayed by Δt. This is discussed in greater detail in §2b(i). Some examples of how REMPI spectroscopy, both in a frequency- and a time-resolved context, can be successfully applied to sunscreen molecules will be given in §3.
(iii). Double resonance spectroscopy
Double resonance techniques, such as hole burning (HB) or depletion (ion dip) spectroscopies, are useful when the possibility of contributions to the measured spectra from different conformers needs to be taken into account, as R2PI's mass selectivity cannot help distinguish these [89]. In certain cases, contributions from different conformers in vibrational spectra can be distinguished by the analysis of their rotational structure [101–104]. However, especially for larger molecules, the typical resolution achieved with these techniques (vide supra) is not always enough to resolve closely spaced rotational energy levels [34,79,105]. The ability to distinguish between conformers is particularly relevant when dealing with large molecules, such as the ones discussed herein.
Double resonance spectroscopy requires the use of three pulses: an excitation, hνpu, and ionization, hνpr, laser pulse (as for R2PI) and an extra ‘holeburn’ laser pulse, hνHB [89,106]. The term ‘holeburn’ refers to the creation of a population hole in a certain state (e.g. ground electronic state) by exciting the population to a higher vibronic state [106]. At energies close to the excited-state origin, a wavelength that is resonant with a specific transition in conformer A will likely not be so for a second conformer B. Hence, with reference to figure 8, if the wavelength of hνHB is set to be resonant with a transition in conformer A, thus depopulating its ground state, the R2PI signal will be reduced when hνpu coincides with a resonance in this conformer; however, no analogous signal difference will be observed for conformer B [89]. If the total R2PI spectrum is subtracted from the resulting hole-burning spectrum, the contribution from conformer A can be isolated, with the resulting spectrum consisting of negative peaks.
Figure 8.

Schematic of HB spectroscopy. The HB laser creates a population hole in the ground state of one of the conformers, so that its contribution to the R2PI spectrum is decreased. Subtracting the total R2PI spectrum from the HB spectrum, therefore, yields the isolated contribution from the selected conformer in the form of negative peaks. Multiple headed purple arrows denote the wavelength scanning of the hνpu laser pulse. Various detection techniques (ionization, fluorescence, etc.) can be used. (Online version in colour.)
The same principles apply for depletion spectroscopy, for which the wavelength of hνHB is scanned, with hνpu fixed on a single transition in one conformer [107–109]. In this case, there will be a reduction in the detected signal whenever hνHB is resonant with a vibronic state in this conformer. These techniques, therefore, provide complementary information although one must note that there are differences in the factors that influence ion signal intensities. Depletion spectroscopy monitors changes in ion intensity (as hνHB is scanned) compared with a constant pump–probe signal. As such, the signal intensity depends on the absorption cross section of the transition accessed by hνHB. In HB spectroscopy, on the other hand, as it is the excited state leading to ionization that is being varied, the signal intensity depends on both absorption cross section and also on excited-state dynamics. It is also worth briefly noting that, for both HB and depletion spectroscopy, it is assumed that after photoexcitation by the holeburn laser pulse, population will not return to the lower energy state within the timescale of pump–probe irradiation [110–114].
The HB spectroscopy technique described in figure 8 uses UV radiation in both the hole-burning and pump steps, and is therefore commonly referred to as UV–UV HB spectroscopy. However, as will be demonstrated by the case studies in §3, infrared (IR) radiation can also be used in double resonance techniques, providing conformer selective infrared spectra [112,115].
(b). Time-resolved spectroscopy
Important photochemical and photophysical processes happen very shortly after the absorption of UV radiation, typically on timescales of the order of femtoseconds to picoseconds (fs–ps, where 1 fs = 10−15 s and 1 ps = 10−12 s) [34]. The study of such ultrafast phenomena is usually referred to as femtochemistry, a term largely disseminated by the Nobel laureate Ahmed H. Zewail [116]. These ultrafast dynamics may be used to infer how effectively a sunscreen molecule dissipates excess energy and hence its adequacy for use as a photoprotecting molecule.
In order to be able to follow these dynamical processes, a laser temporal pulse width on the order of femtoseconds is needed and currently the technology to generate laser pulses with less than 50 fs pulse duration is widely available [117–119]. However, a higher time resolution can be achieved only at the expense of reduced frequency resolution and such laser pulses are generally incapable of resolving individual vibronic states. Nevertheless, the obvious strength of time-resolved techniques lies in the ability to follow the molecular dynamics occurring immediately after excitation in real-time, achieved by varying the time delay, Δt, between the pump and probe laser pulses, as will be explored further in the next section.
(i). Time-resolved ion yield spectroscopy
Time-resolved ion yield (TR-IY) is a technique used to study the dynamics following photoexcitation to an excited electronic state, by monitoring the ion signal resulting from ionization of the excited molecule over time. To achieve this, TR-IY follows the pump–probe scheme detailed, for example, by Zewail and co-workers (e.g. [116]), which, in fact, resembles an R2PI scheme (vide supra). Initially, the molecule of interest, seeded into a molecular beam, is photoexcited to an excited electronic state by a pump laser and the system is then allowed to evolve freely over time. After the initial excitation, a probe laser interacts with the photoexcited molecules over a number of time delays, Δt. As the molecule relaxes from its excited electronic state (by any feasible photochemical or photophysical pathway), the population of this state and hence the corresponding parent cation signal will change. Ion detection in TR-IY is carried out as previously described for REMPI experiments, using a TOF mass spectrometer, enabling one to gate on the parent cation (figure 7). By plotting the parent cation signal as a function of time delay, a TR-IY transient is obtained from which the lifetime of the excited electronic state can be extracted. Importantly, by integrating over different mass channels, TR-IY transients of photofragments can also be obtained and the dynamics of their formation extracted. Utilizing such mass selectivity enables one to begin to build up a comprehensive picture of the excited-state decay pathways in operation in a given molecule [68].
The dynamical information obtained from TR-IY thus nicely complements the information gathered from frequency-resolved techniques (and vice versa). It is worth recalling, however, that while frequency-resolved techniques have sufficient resolution to individually access single vibronic states, it is inevitable that the much broader pump pulses used in time-resolved techniques will access several vibrational states within an electronic state. The dynamics and extracted lifetimes correspond, therefore, to a convolution of superposed vibronic states.
Finally, it should be briefly noted that both types of experiment (frequency- and time-resolved) will benefit from insights provided by computational methods [120]. While the specific details of computational studies will not be discussed in the present review, their results will be referred to, where appropriate, in order to inform the discussion of the case studies explored hereafter.
3. Case studies
The interest in the excited-state dynamics of UV absorber molecules in a sunscreen context has only recently gained momentum, but there is already a significant collection of important works reported on this subject. This section will comprise an overview of a sample of the recent work carried out on various sunscreen molecules using the gas-phase methodologies described in §2. The authors acknowledge, however, that there are other works which are also relevant to this field of research but were not discussed in this review for the sake of brevity [121]. Similarly, there is the literature describing work carried out on sunscreen molecules and their precursors using the techniques mentioned in this review, but for which the results were not interpreted in a sunscreen context [122]. These studies were, therefore, not included in this review, but they do provide powerful insight towards an understanding of the photophysics of the artificial and natural sunscreen species we will discuss in §3a and b, respectively.
(a). Cinnamate derivatives
(i). Methoxycinnamates
EHMC/MMC. Many commercial sunscreen lotions use various cinnamate derivatives (figure 2b) as UV absorbing species. In fact, one of the most widely used sunscreen agents, 2-ethylhexyl-4-methoxycinnamate (EHMC, figure 9) [123], falls within this category. However, apart from its wide application as a sunscreen, EHMC has been found to undergo potentially harmful chemistry upon UV radiation absorption, such as photoisomerization and release of reactive oxygen species [124–126].
Figure 9.

Molecular structures of different rotamers of 2-ethylhexyl-4-methoxycinnamate (EHMC) and methyl-4-methoxycinnamate (MMC). The cis/trans isomerization occurs around C8–C9, while the syn/anti isomerization occurs around C4-OCH3. E/Z isomerization occurs around C7=C8.
Tan et al. [127] studied EHMC in the gas phase using frequency-resolved R2PI and UV–UV depletion spectroscopy. In the same paper, the authors report studies on methyl-4-methoxycinnamate (MMC, figure 9), a simplified version of EHMC, in keeping with a bottom-up approach. The R2PI spectra of both MMC and EHMC are presented in figure 10. Computational simulations were also performed in order to identify the vertical excitation energies and characteristics of the relevant electronic excited states.
Figure 10.

(1 + 1′) R2PI excitation spectra for MMC (a, solid black line) and EHMC (b, solid black line), where 1 corresponds to the UV excitation pulse and 1′ to a 193 nm ionization pulse. Solid red and blue lines correspond to the separate UV–UV depletion spectra from the cis and trans rotamers of each molecule, respectively. The arrows indicate the origin bands for each rotamer, to which the molecules were photoexcited when acquiring depletion spectra. Reproduced with permission from reference [127]. (Online version in colour.)
Depletion spectroscopy measurements by Tan et al. shown in figure 10, suggest that, under the conditions of these experiments, two rotamers of both MMC and EHMC were present: cis and trans with respect to rotation around the C8–C9 single bond (figure 9). The syn and anti pairs are also possible for both MMC and EHMC, with respect to rotation around the C4-OCH3 single bond, but these were not considered in this work. Both the cis/trans and syn/anti pairs mentioned are specifically referred to as rotamers, because they interconvert around a single bond and are only distinguishable under conditions of a molecular beam [128–130]. For the benefit of later discussion, it is worthy of note that these molecules can also exist as their corresponding E/Z stereoisomers (cf. rotamers) with respect to rotation around the C7=C8 double bond (figure 9).
The origin bands of the lowest 1ππ* (S1) state of MMC were found to be located at 32 328 and 32 667 cm−1 for the cis and trans rotamers, respectively. In EHMC, these bands are located at 32 258 and 32 562 cm−1 for the cis and trans rotamers, respectively. It was also noted that the R2PI spectrum of MMC is more structured than that of EHMC, which was attributed to the increased molecular complexity of EHMC, due to the addition of an ethylhexyl chain.
Tan et al. were able to extract the excited-state lifetimes of the 1ππ* states for both rotamers of MMC by fitting the R2PI peaks corresponding to the origin bands of each rotamer with a Lorentzian profile. Analysis of these peak-linewidths yielded a reported excited-state lifetime of MMC of 2.9 ps for the cis rotamer and 2.0 ps for the trans rotamer [127]. The authors noted, however, that these values are lower limits owing to rotational broadening in their experiments. For EHMC, the broad onset of absorption and lack of structure in the R2PI spectrum make the analogous measurements more challenging. Nevertheless, rough estimates of the linewidths of the origin bands in the R2PI of EHMC place its excited-state lifetimes in the sub-picosecond range, which is supported by solution-phase fluorescence lifetime measurements of similar systems reported in the literature [131].
In an attempt to corroborate these excited-state lifetimes, the authors monitored the time-resolved R2PI signal from MMC and EHMC by varying the time delay between the UV excitation and 193 nm ionization pulses with a time resolution of nanoseconds (figure 11). The recorded TR-IY transients were fit with a mono-exponential function yielding time constants of 24.0 ± 0.2 ns for MMC, and 17.7 ± 0.7 ns for EHMC; much longer than the lifetimes anticipated from linewidth measurements. The observation of two such different time constants in these experiments suggests that two photophysical processes might be occurring in both MMC and EHMC. While the 1ππ* states of each of these molecules were found to have a picosecond lifetime, an excited state(s) evidently persists for several nanoseconds. Based on the predictions by other authors [132], Tan et al. assigned this nanosecond time constant to a long-lived 1nπ* state. This was also reconciled through computational work by the same authors, which places the 1nπ* state adiabatically lower in energy than the 1ππ* state, in accordance with other cinnamate derivatives [132].
Figure 11.

TR–IY of (a) EHMC photoexcited at 32 526 cm−1 and (b) MMC photoexcited at 32 667 cm−1. Reproduced with permission from reference [127]. (Online version in colour.)
The findings by Tan et al. regarding MMC [127] were expanded on by an independent study by Miyazaki et al. [133], who performed similar R2PI and depletion spectroscopy measurements and complemented their interpretation of these results with theoretical studies. Apart from the cis and trans rotamers previously reported by Tan et al. [127], Miyazaki et al. [133] were also able to identify the syn and anti rotamers of MMC (see figure 9 for structures) with the aid of computational studies. As shown in figure 12, Miyazaki et al. assigned the experimental bands at 32 328 and 32 667 cm−1 to the cis/syn (figure 9a) and cis/anti (figure 9c) rotamers, respectively (cf. assignments by Tan et al. [127]). In addition, while the peak observed at 32 587 cm−1 is assigned to the trans/syn conformer, the peak that might be expected to correspond to the trans/anti conformer was not observed experimentally.
Figure 12.

(a) LIF, (b) R2PI and (c–e) depletion spectra of MMC under molecular beam conditions. Asterisks (*) in (b) indicate hot bands. Calculated electronic transition energies of the different possible conformers (f) were also included for comparison with experimental data. Data collected by Miyazaki et al. Reproduced with permission from reference [133]. (Online version in colour.)
The time-resolved R2PI signal of MMC following photoexcitation to its S1 origin was also measured by Miyazaki et al. [133] with picosecond resolution (cf. nanosecond resolution in Tan et al.'s results [127]), yielding an S1 lifetime of 280 and 80 ps for rotamers A and C, respectively. The TR-IY transients are presented in figure 13. In addition, the authors also reported these lifetimes to be dependent on excess energy, becoming shorter with increasing excitation energy in S1 (for each rotamer). While the authors did not reconcile the discrepancies between these results and those from Tan et al.'s work, namely in terms of much longer initial time constants and the lack of a long-lived state (vide supra), the latter is very likely owing to a difference in probe wavelength. Miyazaki et al. probed for [MMC]+ with 315 nm and measured the IP of MMC to be 63 427 cm−1 (T Ebata 2016, personal communication). Therefore, the total energy provided to the system (approx. 64 000 cm−1) might not be sufficient to ionize from the long-lived, adiabatically lower lying 1nπ* state as observed with the 193 nm probe used by Tan et al. [127]. Nevertheless, the excess energy dependence of the S1 lifetimes measured by Miyazaki et al. suggests the existence of a potential energy barrier for non-radiative decay via 1ππ* → 1nπ* IC. Importantly, Miyazaki et al. have also suggested E/Z photoisomerization to be a viable non-radiative decay pathway for MMC, occurring in competition with IC from 1ππ* → 1nπ*.
Figure 13.

Pump–probe ion signal (circles) and kinetic fits (solid line) of MMC photoexcited to various vibronic levels of the cis/syn, trans/syn and cis/anti conformers and ionized with 315 nm light. Data collected by Miyazaki et al. Reproduced with permission from reference [133].
In summary, both studies (Tan et al. [127] and Miyazaki et al. [133]) are in general agreement with regard to the photophysical processes occurring in MMC in the gas phase, despite understandable discrepancies in the measured time constants; the time-resolution and probe energies of their respective experiments being probable frontline causes. As a final note, it is also important to discuss a most recent study where Yamazaki et al. [134] have presented both experimental and computational data which strongly implicates that triplet states might also be involved in the photophysics of MMC. The authors propose that, upon absorption of UV radiation, and in addition to the aforementioned IC from the bright 1ππ* state to the 1nπ* state, MMC undergoes ISC from both the 1ππ* and 1nπ* states into a manifold of triplet states. The long lifetimes of MMC observed by Tan et al. [127] could then, according to Yamazaki et al. [134], also correspond to the population trapped in these triplet states. These results highlight the role of triplet states in the photophysics of sunscreen molecules, which will be explored later in this review.
MMC-H2O. The lifetimes of the 1nπ* states reported by Tan et al. for both MMC and EHMC in the gas phase [127] are orders of magnitude longer than the excited-state lifetimes of eumelanin, one of the skin's natural photoprotective molecules [135]. This would suggest that neither MMC nor EHMC behaves as an ideal sunscreen molecule. However, as sunscreens are administered as lotions, solvent interactions need to be considered. In order to evaluate how the gas-phase spectroscopy and excited-state lifetimes for these molecules might be affected in the presence of solvents, Tan et al. [127] preformed micro-solvation studies on MMC, using the same methods described previously.
The R2PI excitation spectrum, obtained by Tan et al. resulting from excitation to the origin of the first 1ππ* state of the MMC-H2O cluster, revealed the presence of four different conformers with origins at 32 106, 32 129, 32 390 and 32 529 cm−1, the molecular structures of which were not determined. From the measured linewidths of the bands in the R2PI spectra, there is a factor of two increase from the approximately 2.5 ps 1ππ* state lifetime observed for isolated MMC. However, the nanosecond lifetime attributed to the 1nπ* state was not observed in the TR-IY measurements of the MMC-H2O cluster (cf. isolated MMC), with any dynamics now occurring within the instrument response. From these results, Tan et al. concluded that IC via 1ππ* → 1nπ* is no longer an active relaxation pathway for the MMC-H2O cluster, and that non-radiative decay to the ground state, mediated by E/Z photoisomerization is likely the dominant pathway. Recent theoretical work on MMC by Chang et al. [136], as well as on related systems [132,137], has also shown E/Z isomerization to be the dominant relaxation pathway in MMC-H2O clusters, while MMC undergoes IC via a 1ππ* → 1nπ* transition instead. It is well known that polar solvents will significantly destabilize 1nπ* states while also possibly stabilizing 1ππ* states, resulting in a net increase in the energy difference between these two states [138,139]. Tan et al., therefore, proposed that micro-solvation of MMC is likely to reverse the ordering of its electronic states, effectively inhibiting IC from 1ππ* → 1nπ*. Even though the same studies were not performed for EHMC, it is likely that these observations would still be valid owing to the relatively unperturbing nature of EHMC's extended hydrocarbon chain. If this is the case, then EHMC would undergo fast relaxation to its ground state by dissipating excess energy into its surroundings, justifying its effectiveness as a sunscreen molecule. Miyazaki et al. [133] also studied the MMC-H2O cluster to complement the findings of Tan et al. [127]. Their R2PI spectrum of MMC-H2O revealed that the lowest energy S1 ← S0 transition is approximately 200 cm−1 lower in energy than that of isolated MMC, being located at 32 106 cm−1, labelled b and a′. Another prominent peak, b and b′, was observed in the R2PI spectrum of MMC-H2O at 32 529 cm−1. Comparison between IR–UV depletion spectroscopy measurements (where the hole-burning laser pulse is an IR pulse) and calculated OH vibrational frequencies suggested two conformers of the MMC-H2O cluster were present, the first corresponding to a water molecule bridging the carbonyl oxygen and a vinyl hydrogen (b and a′), the second corresponding to a water molecule bridging the carbonyl oxygen and methyl hydrogen (b and b′).
Miyazaki et al. [133] performed picosecond TR-IY experiments on the MMC-H2O cluster using the same methodology as discussed for the isolated MMC molecule (vide supra). These measurements were taken after excitation to band a′ and b and b′, with both transients modelled with a function consisting of two exponential decays, as shown in figure 14. For band a′, the two time constants extracted were 21 and 150 ps, while for band b′ the time constants were 35 and 150 ps. Importantly, the shorter time components extracted from the TR-IY transients of both conformers of MMC-H2O were shorter than the corresponding time constants for all conformers of the isolated MMC molecule. These observations are in general agreement with the results by Tan et al. [127], which showed that micro-solvation of MMC accelerates its relaxation to the ground state. Also in accordance with the suggestions by Tan et al. [127], computational work carried out by Miyazaki et al. [133] revealed that the micro-solvation of MMC lowers the energy of the 1ππ* state and increases that of the 1nπ* state. This inversion of electronic states causes the observed red shift in the S1 ← S0 transition of the MMC-H2O cluster when compared with isolated MMC, as well as the increase of the potential energy barrier for 1ππ* → 1nπ* IC, which now becomes an unfavourable decay pathway. Potential energy curves also revealed that the energy of the MMC-H2O system decreases with increasing dihedral angle towards E/Z isomerization. E/Z isomerization is, therefore, the likely relaxation mechanism for MMC-H2O, as Tan et al. [127] had previously suggested.
Figure 14.

TR–IY (dotted circles) and kinetic fits (black line—total fit, red and blue lines—components) of the MMC-H2O complex photoexcited at 32 106 cm−1 (a) and 32 529 cm−1 (b), ionized with a 315 nm probe. Data collected by Miyazaki et al. Reproduced with permission from reference [133]. (Online version in colour.)
As a final note, we briefly refer to another conclusion drawn from Miyazaki et al.'s work [133]. While the discussion so far has related specifically to para-MMC, in which the methoxy group is positioned in the para position with respect to the long side chain, Miyazaki et al. [133] also studied ortho- and meta- MMC (which are not used in commercial sunscreens) in order to evaluate the impact of the substituent position in the photodynamics of MMC. These studies revealed that a radiative decay pathway is now preferred for both ortho- and meta- MMC [133]. This demonstrates how substituent position may affect the ultrafast dynamics of UV absorbers and, indeed, how an understanding of the photophysics of these systems may help tailor an efficient sunscreen molecule.
(ii). Ethyl ferulate
Continuing with cinnamate derivatives, ethyl 4-hydroxy-3-methoxycinnamate (ethyl ferulate, EF, figure 15c), a naturally occurring plant product, is another example of a strong UV absorber molecule which is used in commercial sunscreens. Rodrigues et al. [140] studied this sunscreen filter molecule using frequency- and time-resolved techniques as well as computational methods. In keeping with a bottom-up approach, two precursor molecules, 3-methoxy-1-vinylphenol (MVP, figure 15a) and 4-hydroxy-3-methoxycinnamyl alcohol (coniferyl alcohol, ConA, figure 15b), were also studied.
Figure 15.

3-methoxy-1-vinylphenol (MVP, (a)), 4-hydroxy-3-methoxycinnamyl alcohol (coniferyl alcohol, ConA, (b)) and ethyl 4-hydroxy-3-methoxycinnamate (ethyl ferulate, EF, (c)). The blue dashed lines represent the H-bonds between the adjacent hydroxy and methoxy substituent groups. (Online version in colour.)
Frequency-resolved R2PI and UV–UV HB spectroscopy measurements carried out for MVP and ConA by Rodrigo et al. [141] found that both MVP and ConA are present in two different rotamers, labelled A and B. The two rotamers of MVP have S1 origins at 32 802 cm−1 (A) and 33 525 cm−1 (B), while for ConA these origins are located at 32 640 cm−1 (A) and 33 445 cm−1 (B). Comparison of experimental data with computational results suggests that rotamers A and B for both MVP and ConA correspond to their respective syn and anti pairs, with respect to the relative positions of the OH substituent group on C4 and the vinyl group on C1.
The TR-IY studies by Rodrigues et al. [140] showed that photoexcitation to the S1(v = 0) origin band of the syn rotamers of both MVP and ConA directly accesses a long-lived state. This is evidenced by the plateau in the TR-IY transients for MVP+ and ConA+, figure 16a,b respectively, which extend beyond the experimental time window of their measurements (more than 900 ps). The gas-phase studies alone, therefore, suggest that neither of these precursor molecules behave as ideal sunscreens. In EF, however, the addition of the carbonyl moiety was shown to significantly change the behaviour of this molecule when compared with its precursors (figure 16c and below). To investigate this further, R2PI and UV–UV HB spectroscopy were performed on EF by Rodrigues et al. [140], revealing the existence of two conformers with S1(v = 0) origin bands located at 31 491 and 31 507 cm−1. The bandwidth of the laser used for the TR-IY measurements encompasses both conformers (cf. single conformer excitation in MVP and ConA [140,141]), thus conformer-specific dynamics could not be obtained and assignment of the bands was not attempted.
Figure 16.
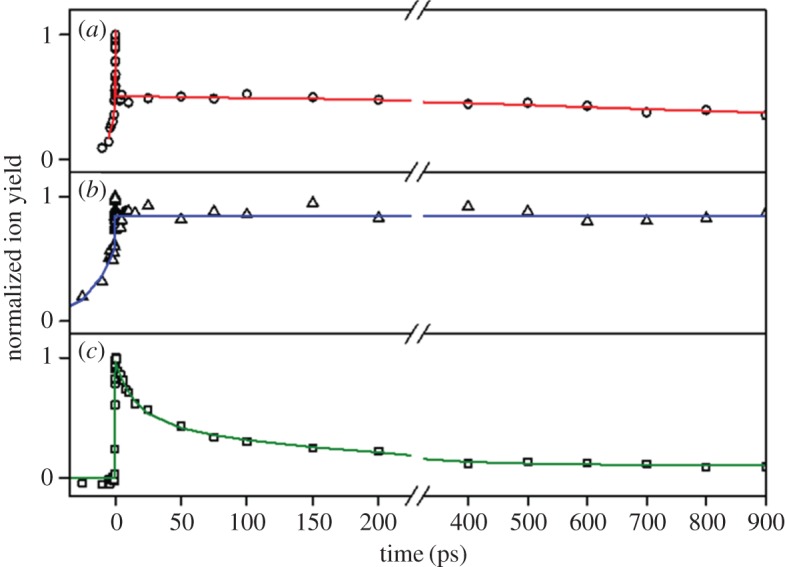
TR–IY (hollow points) and associated kinetic fits of (a) MVP, (b) ConA and (c) EF. Each molecule was excited at the S1 origin and probed with 200 nm light. Data collected by Rodrigues et al. Reproduced with permission from reference [140]. (Online version in colour.)
Using the R2PI and UV–UV HB spectroscopy data, Rodrigues et al. [140] then carried out TR-IY experiments by photoexciting EF with 317.5 nm (approx. 31 500 cm−1) radiation and tracking the excited-state dynamics with a 200 nm probe, the transient of which is shown in figure 16c. The EF+ transient was modelled with a function consisting three exponential decays, returning time constants of τ1 = 15 ± 4 ps, τ2 = 148 ± 47 ps and τ3 > 900 ps. As an accurate lifetime for the long-lived electronic state (τ3) could not be obtained with the TR-IY set-up used in these experiments, fluorescence lifetime measurements were taken, revealing a lifetime of 6.9 ± 0.1 ns. DFL spectra after excitation to the electronic origin of each conformer were consistent with emission from the S1 origin.
In the light of these results, computational methods were employed to evaluate possible relaxation pathways for EF. While results from MMC and EHMC (vide supra) would suggest that an E/Z isomerization pathway is likely involved in the excited-state dynamics of EF, calculations suggest a very large barrier to such isomerization (approx. 8800 cm−1), as shown in figure 17a. Similarly, while calculations suggest that an 1nπ* state is accessible from the S1 origin (figure 17b), transition into this state would likely result in two lifetimes, as seen in MMC and EHMC (vide supra), rather than the three observed in EF.
Figure 17.

Results of the computational work by Rodrigues et al. (a) Potential energy cuts (PECs) of the S0 (black line), S1(11ππ*) (red line), S2(11nπ*) (blue line) and S3(21ππ*) (green line) states of EF along the E/Z isomerization coordinate are shown. Optimized ground state geometries for each isomer and at the S0/S3 conical intersection (CI) are also presented. (b) PECs of the S0 (black line), S1(11ππ*) (red line), S2(11nπ*) (blue line) and S3(21ππ*) (green line) states of EF along linearly interpolated internal coordinates (LIICs). The blue and grey shaded areas represent the S0 to S1 and S1 to S2 (via S1/S2 CI) LIICs, respectively. Optimized geometries and molecular orbitals for the S1 and S2 states are also presented. Reproduced with permission from reference [140]. (Online version in colour.)
Guided by the previous literature, which suggests that the photochemistry of carbonyl compounds is closely linked with the existence of triplet states [54,127,142–149], Rodrigues et al. used computational methods to search for such states in EF. These studies revealed three near isoenergetic triplet states approximately 2000 cm−1 lower in energy than the S2(11nπ*) energy minimum (T2(23ππ*), T3(33ππ*) and T4(13nπ*)) and a fourth, lying approximately 10 500 cm−1 below these (T1(13ππ*)). Apart from being close in energy to both the S1(11ππ*) and S2(11nπ*) energy minima, these triplet states are also the correct orbital type for favourable ISC, according to El-Sayed's rule; ISC is most efficient for transitions where there is a change in orbital type [150].
The complexity of the system under study hindered the unequivocal assignment of time constants to a specific photophysical process. Instead, Rodrigues et al. proposed a scenario where multiple processes occur simultaneously, so that the observed time constants correspond to a convolution of relaxation pathways, as illustrated in figure 18. The authors proposed that photoexcitation of EF at 317.5 nm (approx. S1 origin of both conformers) results in wave packet bifurcation [151,152] to populate both the S1(11ππ*) and S2(11nπ*) states, followed by IVR to the respective energy minima of each state and/or ISC into the triplet state manifold. The convolution of these processes, in competition with one another, would then correspond to τ1 = 15 ± 4 ps. From here, the system evolves via two parallel mechanisms: (i) a convolution of IVR and IC down the ladder of triplet states, happening within τ2 = 148 ± 47 ps, and (ii) radiative decay of population trapped in the energy well of the S1(11ππ*) state, corresponding to the measured fluorescence lifetime of 6.9 ± 0.1 ns. Rodrigues et al. point out, however, that this is not a unique solution to the assignment of the time constants observed, and that contributions from multiple conformers or photofragmentation pathways cannot be ruled out.
Figure 18.

Schematic of the proposed decay mechanisms for excited-state relaxation in EF. Relaxation refers to ISC or phosphorescence back to the ground state; Reaction encompasses such mechanisms as photofragmentation/photocyclization. Note that ‘Reaction’ has been omitted from S1 for clarity, although it is still a valid pathway. Reproduced with permission from reference [140]. (Online version in colour.)
While the authors could not reach a concrete conclusion regarding the relaxation pathways of EF upon UV radiation absorption, this case study serves to demonstrate a number of interesting points. First, these studies suggest that, in the gas phase, EF does not behave as an ideal sunscreen species (nor its precursors MVP and ConA), given the presence of a long-lived excited electronic state. As with the previous case studies involving MMC and EHMC, it is likely that solvent interaction will change this; therefore, solution-phase studies on EF would be highly relevant. Moreover, the case of EF illustrates the complexity of the photophysical processes responsible for the efficiency of sunscreen molecules. When EF is compared with both MVP and ConA, it becomes clear how a simple molecular change can impact the deactivation pathways of a molecule. This also demonstrates how important it is to be able to identify the molecular structures primarily responsible for the dissipation of excess energy, so that future sunscreen species can be tailored for maximum efficiency.
(b). Sinapate derivatives
The term ‘sinapate derivatives’ refers to a group of molecules structurally related to sinapic acid, as shown in figure 19. Sinapate derivatives are ubiquitous in nature, being found in fruits, vegetables, grains and oilseed crops, for example, where they exhibit diverse bioactivity [153]. More importantly for the purpose of this review, these species have an important role in plant photoprotection, as discussed in §1. For example, sinapoyl malate (SM), one of the most complex sinapate derivatives, studied by Dean et al. [142] is found in the epidermis of Arabidopsis plant leaves [154] and it has been shown to be involved in plant photoprotection mechanisms following exposure to UVB radiation [155]. In keeping with a bottom-up approach, Dean et al. [142] studied a series of sinapate derivatives of increasing molecular complexity, as shown in figure 19. These molecules were studied with a combination of spectroscopies, including R2PI (one and two colour), UV–UV HB and DFL, as well as computational methods [142]. The following paragraphs present a summary of the results obtained, focusing mainly on SM owing to its prevalence in plant photoprotection.
Figure 19.

The sinapate series studied by Dean et al. [142]. In the lower right corner, marked with a box, is SM, on which this section will mainly focus owing to its role as a natural sunscreen found in the leaf epidermis of certain plant species. (Online version in colour.)
All of the sinapate species studied show strong UVB absorption, i.e. all have large oscillator strengths for the S1(1ππ*) ← S0 transition. The striking differences between the malate containing species, SM and SDM, and the rest of the sinapate series are immediately evident when comparing their corresponding R2PI spectra, as shown in figure 20. While increased molecular complexity results in increasingly congested spectra, as would be expected, the extensive spectral broadening in both SM and SDM appears to be a unique characteristic in this series. The very broad spectra observed for these two species imply effective absorption of radiation across much of the UVB region, which, at first glance, justifies their sunscreen capabilities. Moreover, such spectral broadness may be linked with short-lived excited-state lifetimes, absent in the simpler derivatives studied. Assuming that SM's dynamics involve a fast relaxation to its ground electronic state, this may also justify SM's efficacy as a sunscreen. Therefore, the origins of the unique spectral broadening in SM are of the utmost interest if its sunscreening capabilities at a molecular level are to be understood. This extensive broadening, however, results in featureless spectra that hinder any reliable vibronic analysis; therefore, extracting information from SM's spectra is not straightforward.
Figure 20.

R2PI spectra for the sinapate derivatives studied by Dean et al. [142]. While simpler molecules show clearly resolved spectra, increasing molecular complexity introduces vibronic congestion. In the case of SM and SDM, the extensive spectral broadening and congestion results in complete loss of vibronic resolution. Reproduced with permission from reference [142].
While many non-dynamical effects can be responsible for spectral broadening, such as increased molecular complexity or multiple stable conformers leading to congested vibronic spectra, upon comparison with the other sinapate derivatives studied, Dean et al. disregarded such reasoning for the featureless spectra in SM and SDM. The less extensive broadening in the structurally similar SML and SMB led Dean et al. to rule out IVR as a potential source of the broadening in SM and SDM. Comparison with theoretical calculations also caused the authors to dismiss Franck–Condon activity and/or molecular complexity arguments as viable explanations for these featureless spectra, while IR ion-dip measurements ruled out absorptions from multiple conformers. As such, an alternative explanation for the observed results was sought.
It is well-documented that locally excited states, such as a 1ππ* state, can couple with nearby charge–transfer states [156,157]. This can lead to complete spectral broadening by virtue of coupling to a manifold of vibronic levels belonging to the adiabatically lower energy charge–transfer state. In essence, the oscillator strength of the optically bright locally excited state is dispersed across the neighbouring charge–transfer levels. Dean et al. [142] carried out theoretical calculations to compute electron density difference maps (EDDMs) of the main singlet excited states of SM. These EDDMs showed that the 1nπ* state in SM has partial charge–transfer character, involving a shift of electron density localized around the first carboxylic acid of the malate group and the antibonding orbital on the ring. The existence of a charge–transfer state in SM is supported by fluorescence measurements carried out by Dean et al. [142]. These measurements, which were performed in aqueous solution for both SA and SM for comparison, showed a considerable Stokes shift in the emission peak of SM when compared with SA. This suggests that the emitting state is stabilized in a polar solvent, which is a general observation for charge–transfer states, as reported in the literature [158,159]. The fluorescence spectra of SM, therefore, support the existence of a charge–transfer state, as proposed by Dean et al. [142].
Despite not having collected any evidence for the existence of long-lived triplet states in SM (owing to experimental constraints), Dean et al. also point out the possibility of ISC being involved in its photodynamics. As discussed in previous sections, triplet states have been reported to be active in the excited-state dynamics of carbonyl-containing species [54], and indeed it has been suggested by Rodrigues et al. [140] that such states are likely to play an important role in the photodynamics of the natural sunscreen molecule, ethyl ferulate (see §3a(ii)). As triplet states have also been linked to the formation of singlet oxygen [160,161], which would be detrimental to SM's performance as a safe UVB sunscreen, this is another aspect of this molecule's spectroscopy which requires further study.
In conclusion, Dean et al. found that, as expected, SM, an important sunscreen molecule in certain plant species, absorbs strongly and broadly across the UVB region. The remarkable broadening of the absorption spectrum of SM was tentatively attributed to a mixing of 1ππ* and 1nπ* electronic states, the latter having some charge-transfer character. The very low fluorescence quantum yields obtained in solution for SA and SM suggest that non-radiative pathways dominate the excited-state dynamics. These authors encourage the importance of future studies aimed at characterizing the intermediate and final states involved in these dynamics, both in the gas- and solution phase.
4. Summary and outlook
While the field of photodynamics of sunscreen molecules is still in its infancy, the work carried out thus far, in both the frequency- and time-domain, has highlighted the wealth of information that may still be gathered, as well as the potential impact of such research to the sunscreen industry. The three case studies reported in this review have revealed a wide range of research opportunities and are likely to encourage further work on the subject. For example, the work by Baker et al. [162], which demonstrates how gas-phase studies may be extended into the solution-phase, offers a tantalizing prospect of a plethora of future studies in environments that more closely mimic the surroundings in which these sunscreen molecules are found. Baker et al. [162] photoexcited and probed the excited-state dynamics of SA, MS and SM (figure 19) using transient electronic absorption spectroscopy (TEAS) [162–164], in both a non-polar and polar solvent (cf. the gas-phase studies by Dean et al. [142], §3b). While frequency-resolved gas-phase studies [142] revealed marked differences in the spectra of these molecules, with SM yielding an exceptionally broadened R2PI spectrum when compared with simpler analogues, the TEAS studies showed remarkably similar data for SA, MS and SM. Importantly, it had been concluded from gas-phase studies that an effective non-radiative process was likely to be responsible for SM's sunscreen properties, but a photoprotection mechanism could not be drawn from the data available. The TEAS studies by Baker et al. [162] (guided by the theoretical work of Karsili et al. [137] for similar systems) were able to expand on this prediction and provide a detailed picture of the excited-state dynamics occurring in SA, MS and SM. For all three molecules, Baker et al. suggest that, after initial photoexcitation to the 11ππ* electronic state, IC to a second 21ππ* state occurs via a 11ππ*/21ππ* conical intersection (CI). This, in turn, is followed by the formation of either the E or Z conformer of the molecule in the ground state via a second 21ππ*/S0 CI. Importantly, for all three molecules and in both non-polar and polar solvents, the molecules return to their ground electronic states in tens of picoseconds, making all three good candidates for photoprotection. However, only SM is used in nature as a photoprotective agent, which highlights that there may be other factors that govern the selection of SM as a natural sunscreen agent over the biological precursor SA.
Although the information on the photodynamics of individual sunscreen molecules is valuable (as discussed), commercially available sunscreen lotions are a complex mixture of components (absorbers, scatterers, photostabilizers, fragrances to name but a few). Therefore, the next step within the bottom-up approach would be to evaluate how interactions within such mixtures affect the photodynamics of, say, a sunscreen filter molecule. This would be particularly important for sunscreen filter molecules which populate long-lived states upon absorption of UV radiation when studied in isolation. In a mixture, such long-lived states could be reactive. Indeed, it is well known that some sunscreen blends incorporate the use of photostabilizers [165,166] to counter this. It is, therefore, crucial to explore these types of interactions.
In conclusion, we have seen how frequency- and time-resolved studies in both the gas- and solution phase, as well as computational work, all have the potential to provide crucial insight into the photodynamics of sunscreen molecules. Indeed, as mentioned in the introduction to this review, these techniques (and in particular the bottom-up approach) are also useful in the study of any such photoabsorbing species such as DNA and other biomolecules [70–72]. This information, in turn, can aid in the development of a new generation of sunscreens, tailor-made in order to maximize their efficiency and safety, to make them capable of more effectively tackling the increasing risks of UV radiation exposure in today's society.
Acknowledgements
The authors are grateful to Prof. Ebata and his group for providing further details regarding their experiments. The authors thank Neil-Cole Filipiak, Michael Horbury (University of Warwick) and Yoann Peperstraete (ENS de Cachan) for their helpful comments when preparing the manuscript.
Authors' contributions
The writing of the manuscript was shared between authors.
Competing interests
We declare we have no competing interests.
Funding
N.D.N.R. and M.S. thank the Engineering and Physical Sciences Research Council (EPSRC) for doctoral and postdoctoral funding, respectively. V.G.S. thanks the Royal Society for a University Research Fellowship.
References
- 1.World Healh Organization 2003. Intersun: The global UV project (A guide and compendium). Nairobi, Kenya: United Nations Environment Programme. [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. 2015. Cancer statistics, 2015. CA Cancer J. Clin. 65, 5–29. (doi:10.3322/caac.21254) [DOI] [PubMed] [Google Scholar]
- 3.Cancer Research UK. 2013. Statistics by cancer type. http://www.cancerresearchuk.org/ health-professional/cancer-statistics/statistics-by-cancer-type/skin-cancer (accessed 15 November 2016). [Google Scholar]
- 4.Vallejo-Torres L, Morris S, Kinge JM, Poirier V, Verne J. 2014. Measuring current and future cost of skin cancer in England. J. Public Health (Oxf.) 36, 140–148. (doi:10.1093/pubmed/fdt032) [DOI] [PubMed] [Google Scholar]
- 5.Gasparro FP, Mitchnick M, Nash JF. 1998. A review of sunscreen safety and efficacy. Photochem. Photobiol. 68, 243–256. (doi:10.1111/j.1751-1097.1998.tb09677.x) [PubMed] [Google Scholar]
- 6.Ananthaswamy HN, Pierceall WE. 1990. Molecular mechanisms of ultraviolet radiation carcinogenesis. Photochem. Photobiol. 52, 1119–1136. (doi:10.1111/j.1751-1097.1990.tb08452.x) [DOI] [PubMed] [Google Scholar]
- 7.D'Orazio J, Jarrett S, Amaro-Ortiz A, Scott T. 2013. UV Radiation and the skin. Int. J. Mol. Sci. 14, 12222–12248. (doi:10.3390/ijms140612222). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elwood JM, Jopson J. 1997. Melanoma and sun exposure: an overview of published studies. Int. J. Cancer 73, 198–203. (doi:10.1002/(SICI)1097-0215(19971009)73:2<198::AID-IJC6>3.0.CO;2-R) [DOI] [PubMed] [Google Scholar]
- 9.Lo JA, Fisher DE. 2014. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science 346, 945–949. (doi:10.1126/science.1253735) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowe NJ. 2006. An overview of ultraviolet radiation, sunscreens, and photo-induced dermatoses. Dermatol. Clin. 24, 9–17. (doi:10.1016/j.det.2005.08.001) [DOI] [PubMed] [Google Scholar]
- 11.Paul ND, Gwynn-Jones D. 2003. Ecological roles of solar UV radiation: towards an integrated approach. Trends Ecol. Evol. 18, 48–55. (doi:10.1016/s0169-5347(02)00014-9) [Google Scholar]
- 12.National Renewable Energy Laboratory (NREL). See http://www.nrel.gov/rredc/ (accessed June 2016).
- 13.World Health Organization 2002. Global solar UV index: a practical guide, Meteorological Organization, UN Environment Programme, International Commission on Non-Ionizing Radiation Protection. Geneva, Switzerland: WHO. [Google Scholar]
- 14.Frederick JE, Snell HE, Haywood EK. 1989. Solar ultraviolet radiation at the earth's surface. Photochem. Photobiol. 50, 443–450. (doi:10.1111/j.1751-1097.1989.tb05548.x) [Google Scholar]
- 15.Webb AR, Steven MD. 2007. Solar ultraviolet-B radiation under cloudless skies. Q. J. R. Meteor. Soc. 113, 393–400. (doi:10.1002/qj.49711347521) [Google Scholar]
- 16.Coulson K. 1975. Solar and terrestrial radiation: methods and measurements. New York, NY: Academic Press Inc. [Google Scholar]
- 17.Brenner M, Hearing VJ. 2008. The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 84, 539–549. (doi:10.1111/j.1751-1097.2007.00226.x). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorshelev V, Serdyuchenko A, Weber M, Chehade W, Burrows JP. 2014. High spectral resolution ozone absorption cross-sections; part 1: measurements, data analysis and comparison with previous measurements around 293 K. Atmos. Meas. Tech. 7, 609–624. (doi:10.5194/amt-7-609-2014). [Google Scholar]
- 19.Seckmeyer G, et al. . 2008. Variability of UV irradiance in Europe. Photochem. Photobiol. 84, 172–179. (doi:10.1111/j.1751-1097.2007.00216.x). [DOI] [PubMed] [Google Scholar]
- 20.Sinha RP, Häder DP. 2002. UV-induced DNA damage and repair: a review. Photochem. Photobiol. Sci. 1, 225–236. (doi:10.1039/b201230h) [DOI] [PubMed] [Google Scholar]
- 21.de Gruijl FR. 2000. Photocarcinogenesis: UVA vs UVB. Methods Enzymol. 319, 359–366. (doi:10.1016/s0076-6879(00)19035-4) [DOI] [PubMed] [Google Scholar]
- 22.Schulz I, Mahler H-C, Boiteux S, Epe B. 2000. Oxidative DNA base damage induced by singlet oxygen and photosensitization: recognition by repair endonucleases and mutagenicity. Mutat. Res. DNA Repair 461, 145–156. (doi:10.1016/s0921-8777(00)00049-5) [DOI] [PubMed] [Google Scholar]
- 23.Meyskens FL Jr, Farmer P, Fruehauf JP. 2001. Redox regulation in human melanocytes and melanoma. Pigm. Cell. Res. 14, 148–154. (doi:10.1034/j.1600-0749.2001.140303.x) [DOI] [PubMed] [Google Scholar]
- 24.Rössle S, Friedrichs J, Frank I. 2010. The formation of DNA photodamage: the role of exciton localization. Chem. Phys. Chem. 11, 2011–2015. (doi:10.1002/cphc.201000081) [DOI] [PubMed] [Google Scholar]
- 25.Brash DE. 2015. UV signature mutations. Photochem. Photobiol. 91, 15–26. (doi:10.1111/php.12377). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Gruijl FR, Rebel H. 2008. Early events in UV carcinogenesis—DNA damage, target cells and mutant p53 foci. Photochem. Photobiol. 84, 382–387. (doi:10.1111/j.1751-1097.2007.00275.x) [DOI] [PubMed] [Google Scholar]
- 27.Narayanan DL, Saladi RN, Fox JL. 2010. Ultraviolet radiation and skin cancer. Int. J. Dermatol. 49, 978–986. (doi:10.1111/j.1365-4632.2010.04474.x) [DOI] [PubMed] [Google Scholar]
- 28.Rigel DS. 2008. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J. Am. Acad. Dermatol. 58, S129–S132. (doi:10.1016/j.jaad.2007.04.034) [DOI] [PubMed] [Google Scholar]
- 29.Gasparro FP. 1997. Sunscreen photobiology. New York, NY: Springer. [Google Scholar]
- 30.Cabrera MI, Alfano OM, Cassano AE. 1996. Absorption and scattering coefficients of titanium dioxide particulate suspensions in water. J. Phys. Chem. 100, 20 043–20 050. (doi:10.1021/jp962095q) [Google Scholar]
- 31.Newman MD, Stotland M, Ellis JI. 2009. The safety of nanosized particles in titanium dioxide- and zinc oxide-based sunscreens. J. Am. Acad. Dermatol. 61, 685–692. (doi:10.1016/j.jaad.2009.02.051) [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Arjona D, Lopez-Perez G, Dominguez MM, Cuesta Van Looken S. 2015. Study of sunscreen lotions, a modular chemistry project. J. Lab. Chem. Ed. 3, 9 (doi:10.5923/j.jlce.20150303.02) [Google Scholar]
- 33.Barel AO. 2005. Handbook of cosmetic science and technology. New York, NY: Basel. [Google Scholar]
- 34.Atkins P, De Paula J. 2014. Atkins’ physical chemistry. Oxford, UK: OUP Oxford. [Google Scholar]
- 35.Pavia DL, Lampman GM, Kriz GS, Vyvyan JR. 2009. Introduction to spectroscopy. Belmont, CA: Brooks/Cole. [Google Scholar]
- 36.Kalsi PS. 2004. Spectroscopy of organic compounds. New Delhi, India: New Age International. [Google Scholar]
- 37.Tanner PR. 2006. Sunscreen product formulation. Dermatol. Clin. 24, 53–62. (doi:10.1016/j.det.2005.09.002) [DOI] [PubMed] [Google Scholar]
- 38.Verheugen G. 2006. Commission recommendation on the efficacy of sunscreen products and the claims made relating thereto. Brussels, Belgium: European Commission. [Google Scholar]
- 39.Fuehring S. 2006. Standardisation mandate assigned to CEN concerning methods for testing efficacy of sunscreen products. Brussels, Belgium: European Commission. [Google Scholar]
- 40.Cole C. 2014. Sunscreens—what is the ideal testing model? Photodermatol. Photoimmunol. Photomed. 30, 81–87. (doi:10.1111/phpp.12095) [DOI] [PubMed] [Google Scholar]
- 41.Vergou T, Patzelt A, Schanzer S, Meinke MC, Weigmann HJ, Thiede G, Sterry W, Lademann J, Darvin ME. 2013. Methods for the evaluation of the protective efficacy of sunscreen products. Skin Pharmacol. Physiol. 26, 30–35. (doi:10.1159/000343576) [DOI] [PubMed] [Google Scholar]
- 42.Wong T, Orton D. 2011. Sunscreen allergy and its investigation. Clin. Dermatol. 29, 306–310. (doi:10.1016/j.clindermatol.2010.11.002) [DOI] [PubMed] [Google Scholar]
- 43.Kerr A, Ferguson J. 2010. Photoallergic contact dermatitis. Photodermatol. Photoimmunol. Photomed. 26, 56–65. (doi:10.1111/j.1600-0781.2010.00494.x) [DOI] [PubMed] [Google Scholar]
- 44.Ortonne JP. 2002. Photoprotective properties of skin melanin. Br. J. Dermatol. 146, 7–10. (doi:10.1046/j.1365-2133.146.s61.3.x) [DOI] [PubMed] [Google Scholar]
- 45.Gröniger A, Sinha RP, Klisch M, Häder DP. 2000. Photoprotective compounds in cyanobacteria, phytoplankton and macroalgae—a database. J. Photochem. Photobiol. B 58, 115–122. (doi:10.1016/s1011-1344(00)00112-3) [DOI] [PubMed] [Google Scholar]
- 46.Niyogi KK. 1999. Photoprotection revisited: genetic and molecular approaches. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 333–359. (doi:10.1146/annurev.arplant.50.1.333) [DOI] [PubMed] [Google Scholar]
- 47.Jansen MAK, Gaba V, Greenberg BM. 1998. Higher plants and UV-B radiation: balancing damage, repair and acclimation. Trends Plant Sci. 3, 131–135. (doi:10.1016/s1360-1385(98)01215-1) [Google Scholar]
- 48.Ballare CL, Scopel AL, Stapleton AE, Yanovsky MJ. 1996. Solar ultraviolet-B radiation affects seedling emergence, DNA integrity, plant morphology, growth rate, and attractiveness to herbivore insects in datura ferox. Plant Physiol. 112, 161–170. (doi:10.1104/pp.112.1.161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landry LG, Chapple C, Last RL. 1995. Arabidopsis mutants lacking phenolic sunscreens exhibit enhanced ultraviolet-B injury and oxidative damage. Plant Physiol. 109, 1159–1166. (doi:10.1104/pp.109.4.1159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shirley BW. 1996. Flavonoid biosynthesis: ‘new’ functions for an ‘old’ pathway. Trends Plant. Sci. 1, 377–382. (doi:10.1016/s1360-1385(96)80312-8) [Google Scholar]
- 51.Bieza K, Lois R. 2001. An arabidopsis mutant tolerant to lethal ultraviolet-B levels shows constitutively elevated accumulation of flavonoids and other phenolics. Plant Physiol. 126, 1105–1115. (doi:10.1104/pp.126.3.1105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheahan JJ. 1996. Sinapate esters provide greater UV-B attenuation than flavonoids in arabidopsis thaliana (brassicaceae). Am. J. Bot. 83, 679–686. (doi:10.2307/2445845) [Google Scholar]
- 53.Pathak MA. 1982. Molecular aspects of drug photosensitivity with special emphasis on psoralen photosensitization reaction. J. Natl Cancer Inst. 69, 163–170. (doi:10.1093/jnci/69.1.163). [DOI] [PubMed] [Google Scholar]
- 54.Turro NJ, Ramamurthy V, Scaiano JC. 2010. Modern molecular photochemistry of organic molecules. Sausalito, CA: University Science Books. [Google Scholar]
- 55.Klán P, Wirz J. 2009. Photochemistry of organic compounds: from concepts to practice. New York, NY: John Wiley & Sons Ltd. [Google Scholar]
- 56.Jaffe HH, Miller AL. 1966. The fates of electronic excitation energy. J. Chem. Ed. 43, 469–473. (doi:10.1021/ed043p469) [Google Scholar]
- 57.Tramer A, Jungen C, Lahmani F. 2005. Energy dissipation in molecular systems. Berlin, Germany: Springer. [Google Scholar]
- 58.Bixon M, Jortner J. 1968. Intramolecular radiationless transitions. J. Chem. Phys. 48, 715–726. (doi:10.1063/1.1668703) [Google Scholar]
- 59.Lakowicz JR. 2006. Principles of fluorescence spectroscopy. Berlin, Germany: Springer. [Google Scholar]
- 60.Lewis GN, Kasha M. 1944. Phosphorescence and the triplet state. J. Am. Chem. Soc. 66, 2100–2116. (doi:10.1021/ja01240a030) [Google Scholar]
- 61.Forestier S. 2008. Rationale for sunscreen development. J. Am. Acad. Dermatol. 58, S133–S138. (doi:10.1016/j.jaad.2007.05.047) [DOI] [PubMed] [Google Scholar]
- 62.Harris DC. 1978. Symmetry and spectroscopy: an introduction to vibrational and electronic spectroscopy. New York, NY: Oxford University Press. [Google Scholar]
- 63.Chergui M. 1996. Femtochemistry, Lausanne, Switzerland: World Scientific Publishing. [Google Scholar]
- 64.Berezin MY, Achilefu S. 2010. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 110, 2641–2684. (doi:10.1021/cr900343z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Demtröder W. 2015. Laser spectroscopy 1: basic principles. Berlin, Germany: Springer; (10.1007/978-3-540-73418-5) [Google Scholar]
- 66.Demtröder W. 2015. Laser spectroscopy 2: experimental techniques. Berlin, Germany: Springer. [Google Scholar]
- 67.Stavros VG. 2014. Photochemistry: a bright future for sunscreens. Nat. Chem. 6, 955–956. (doi:10.1038/nchem.2084) [DOI] [PubMed] [Google Scholar]
- 68.Staniforth M, Stavros VG. 2013. Recent advances in experimental techniques to probe fast excited-state dynamics in biological molecules in the gas phase: dynamics in nucleotides, amino acids and beyond. Proc. R. Soc. A 469, 20130458 (doi:10.1098/rspa.2013.0458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ariens EJ. 1973. Drug design. London, UK: Academic Press, Inc. (London) Ltd. [Google Scholar]
- 70.Roberts GM, Stavros VG. 2014. The role of πσ* states in the photochemistry of heteroaromatic biomolecules and their subunits: insights from gas-phase femtosecond spectroscopy. Chem. Sci. 5, 1698–1722. (doi:10.1039/c3sc53175a) [Google Scholar]
- 71.Stavros VG, Verlet JR. 2016. Gas-phase femtosecond particle spectroscopy: a bottom-up approach to nucleotide dynamics. Annu. Rev. Phys. Chem. 67, 211–232. (doi:10.1146/annurev-physchem-040215-112428) [DOI] [PubMed] [Google Scholar]
- 72.de Vries MS, Hobza P. 2007. Gas-phase spectroscopy of biomolecular building blocks. Annu. Rev. Phys. Chem. 58, 585–612. (doi:10.1146/annurev.physchem.57.032905.104722) [DOI] [PubMed] [Google Scholar]
- 73.Pratt DW. 1998. High resolution spectroscopy in the gas phase: even large molecules have well-defined shapes. Annu. Rev. Phys. Chem. 49, 481–530. (doi:10.1146/annurev.physchem.49.1.481) [DOI] [PubMed] [Google Scholar]
- 74.Rulliere C. 2003. Femtosecond laser pulses. Paris, France: Springer. [Google Scholar]
- 75.Hollas MJ. 1982. High resolution spectroscopy. London, UK: Butterworths. [Google Scholar]
- 76.Ashfold MNR, Howe JD. 1994. Multiphoton spectroscopy of molecular species. Annu. Rev. Phys. Chem. 45, 57–82. (doi:10.1146/annurev.pc.45.100194.000421) [Google Scholar]
- 77.Velarde L, Zhang XY, Lu Z, Joly AG, Wang Z, Wang HF. 2011. Communication: Spectroscopic phase and lineshapes in high-resolution broadband sum frequency vibrational spectroscopy: Resolving interfacial inhomogeneities of ‘identical’ molecular groups. J. Chem. Phys. 135, 241102 (doi:10.1063/1.3675629) [DOI] [PubMed] [Google Scholar]
- 78.Muller-Dethlefs K, Schlag EW. 1991. High-resolution zero kinetic energy (ZEKE) photoelectron spectroscopy of molecular systems. Annu. Rev. Phys. Chem. 42, 109–136. (doi:10.1146/annurev.pc.42.100191.000545) [Google Scholar]
- 79.Levy DH. 1980. Laser spectroscopy of cold gas-phase molecules. Annu. Rev. Phys. Chem. 31, 197–225. (doi:10.1146/annurev.pc.31.100180.001213) [Google Scholar]
- 80.Dicke RH. 1953. The effect of collisions upon the doppler width of spectral lines. Phys. Rev. 89, 472–473. (doi:10.1103/PhysRev.89.472) [Google Scholar]
- 81.Zare RN. 2012. My life with LIF: a personal account of developing laser-induced fluorescence. Annu. Rev. Anal. Chem. 5, 1–14. (doi:10.1146/annurev-anchem-062011-143148) [DOI] [PubMed] [Google Scholar]
- 82.Kinsey JL. 1977. Laser-induced fluorescence. Annu. Rev. Phys. Chem. 28, 349–372. (doi:10.1146/annurev.pc.28.100177.002025) [Google Scholar]
- 83.Bouwens RJ, Hammerschmidt JA, Grzeskowiak MM, Stegink TA, Yorba PM, Polik WF. 1996. Pure vibrational spectroscopy of S0 formaldehyde by dispersed fluorescence. J. Chem. Phys. 104, 460–479. (doi:10.1063/1.470844) [Google Scholar]
- 84.Abe H, Mlkaml N, Ito M, Udagawa Y. 1982. Dispersed fluorescence spectra of hydrogen-bonded phenols in a supersonic free jet. J. Phys. Chem. 86, 2567–2569. (doi:10.1021/j100211a005) [Google Scholar]
- 85.Fabbi JC, Langenberg JD, Costello QD, Morse MD. 2001. Dispersed fluorescence spectroscopy of jet-cooled AgAu and Pt2. J. Chem. Phys. 115, 7542–7549. (doi:10.1063/1.1407273) [Google Scholar]
- 86.O'Brian TR, Lawler JE. 1991. Radiative lifetimes in Si I from laser-induced fluorescence in the visible, ultraviolet, and vacuum ultraviolet. Phys. Rev. A. 44, 7134–7143. (doi:10.1103/PhysRevA.44.7134) [DOI] [PubMed] [Google Scholar]
- 87.Chin W, Mons M, Dimicoli I, Piuzzi F, Tardivel B, Elhanine M. 2002. Tautomer contributions to the near UV spectrum of guanine: towards a refined picture for the spectroscopy of purine molecules. Eur. Phys. J. D 20, 347–355. (doi:10.1140/epjd/e2002-00171-6) [Google Scholar]
- 88.Hakamata T, et al. 2006. Photomultiplier tubes: basics and applications. Tokyo, Japan: Hamamatsu Photonics K. K. [Google Scholar]
- 89.Dessent CEH, Müller-Dethlefs K. 2000. Hydrogen-bonding and van der Waals complexes studied by ZEKE and REMPI spectroscopy. Chem. Rev. 100, 3999–4022. (doi:10.1021/cr990060r) [DOI] [PubMed] [Google Scholar]
- 90.Jacobs DC, Zare RN. 1986. Reduction of 1+1 resonance enhanced MPI spectra to populations and alignment factors. J. Chem. Phys. 85, 5457–5468. (doi:10.1063/1.451556) [Google Scholar]
- 91.Bominaar J, Schoemaecker C, Dam N, Ter Meulen JJ, Groenenboom GC. 2007. (2+1)REMPI on molecular nitrogen through the (II)-state. Chem. Phys. Lett. 435, 242–246. (doi:10.1016/j.cplett.2006.12.100) [Google Scholar]
- 92.Takahashi M. 1992. Two-color (2+1′) multiphoton ionization threshold photoelectron study of the Ar–NO van der Waals complex: observation of intermolecular vibrational progressions of the Ar–NO+ cation. J. Chem. Phys. 96, 2594–2599. (doi:10.1063/1.462010) [Google Scholar]
- 93.Müller-Dethlefs K, Sander M, Schlag EW. 1984. Two-colour photoionization resonance spectroscopy of NO: complete separation of rotational levels of NO+ at the ionization threshold. Chem. Phys. Lett. 112, 291–294. (doi:10.1016/0009-2614(84)85743-7) [Google Scholar]
- 94.Ropers C, Solli DR, Schulz CP, Lienau C, Elsaesser T. 2007. Localized multiphoton emission of femtosecond electron pulses from metal nanotips. Phys. Rev. Lett. 98, 043907 (doi:10.1103/PhysRevLett.98.043907) [DOI] [PubMed] [Google Scholar]
- 95.Lin SH, Fujimura Y, Neusser HJ, Schlag EW. 1984. Multiphoton spectrosocpy of molecules. London, UK: Academic Press, Inc. [Google Scholar]
- 96.Johnson PM, Otis CE. 1981. Molecular multiphoton spectroscopy with ionization detection. Annu. Rev. Phys. Chem. 32, 139–157. (doi:10.1146/annurev.pc.32.100181.001035) [Google Scholar]
- 97.Kruit P, Read FH. 1983. Magnetic field paralleliser for 2π electron-spectrometer and electron-image magnifier. J. Phys. E: Sci. Inst. 16, 313–324. (doi: 10.1088/0022-3735/16/4/016) [Google Scholar]
- 98.Wiley WC, McLaren IH. 1955. Time-of-flight mass spectrometer with improved resolution. Rev. Sci. Inst. 26, 1150–1157. (doi:10.1063/1.1715212) [Google Scholar]
- 99.Boesl U. 1991. Multiphoton excitation and mass-selective ion detection for neutral and ion spectroscopy. J. Phys. Chem. 95, 2949–2962. (doi:10.1021/j100161a005) [Google Scholar]
- 100.Letokhov VS. 1987. Laser photoionization spectroscopy. London, UK: Academic Press, Inc. (London) Ltd. [Google Scholar]
- 101.Philips LA, Levy DH. 1988. Rotationally resolved electronic spectroscopy of tryptamine conformers in a supersonic jet. J. Chem. Phys. 89, 85–90. (doi:10.1063/1.455464) [Google Scholar]
- 102.Zwier TS. 2001. Laser spectroscopy of jet-cooled biomolecules and their water-containing clusters: water bridges and molecular conformation. J. Phys. Chem. A 105, 8827–8839. (doi:10.1021/jp011659) [Google Scholar]
- 103.Martinez SJ, Alfano JC, Levy DH. 1992. Rotationally resolved fluorescence excitation spectra of phenol and 4-ethylphenol in a supersonic jet. J. Mol. Spec. 152, 80–88. (doi:10.1016/0022-2852(92)90118-8) [Google Scholar]
- 104.Luo X, Rizzo TR. 1990. Rotationally resolved vibrational overtone spectroscopy of hydrogen peroxide at chemically significant energies. J. Chem. Phys. 93, 8620–8633. (doi:10.1063/1.459249) [Google Scholar]
- 105.Brumfield BE, Stewart JT, McCall BJ. 2012. Extending the limits of rotationally resolved absorption spectroscopy: Pyrene. J. Phys. Chem. Lett. 3, 1985–1988. (doi:10.1021/jz300769k) [Google Scholar]
- 106.Demtrӧder W. 2003. Laser spectroscopy: basic concepts and instrumentation. New York, NY: Springer. [Google Scholar]
- 107.Mehta-Hurt DN, Korn JA, Navotnaya P, Parobek AP, Clayton RM, Zwier TS. 2015. The spectroscopy and photochemistry of quinioline structural isomers: (E)- and (Z)-phenylvinylnitrile. J. Chem. Phys. 143, 074304 (doi:10.1063/1.4928191) [DOI] [PubMed] [Google Scholar]
- 108.Rijs AM, Oomens J. 2015. Gas-phase IR spectroscopy and structure of biological molecules. Cham, Switzerland: Springer. [Google Scholar]
- 109.Tan EM, Amirjalayer S, Smolarek S, Vdovin A, Rijs AM, Buma WJ. 2013. Conformational heterogeneity of methyl 4-hydroxycinnamate: a gas-phase UV-IR spectroscopic study. J. Phys. Chem. B 117, 4798–4805. (doi:10.1021/jp312624e) [DOI] [PubMed] [Google Scholar]
- 110.Gerhards M, Perl W, Kleinermanns K. 1995. Rotamers and vibrations of resorcinol obtained by spectral hole burning. Chem. Phys. Lett. 240, 506–512. (doi:10.1016/0009-2614(95)00567-n) [Google Scholar]
- 111.Patwari GN, Doraiswamy S, Wategaonkar S. 1998. Hole-burning spectroscopy of jet-cooled hydroquinone. Chem. Phys. Lett. 289, 8–12. (doi:10.1016/s0009-2614(98)00398-4) [Google Scholar]
- 112.Inokuchi Y, Kobayashi Y, Ito T, Ebata T. 2007. Conformation of L-tyrosine studied by fluorescence-detected UV-UV and IR-UV double-resonance spectroscopy. J. Phys. Chem. A 111, 3209–3215. (doi:10.1021/jp070163a) [DOI] [PubMed] [Google Scholar]
- 113.Isozaki T, Iga H, Suzuki T, Ichimura T. 2007. Low-frequency vibrations specific for conformers of 1-aminoindan studied by UV-UV hole-burning spectroscopy. J. Chem. Phys. 126, 214304 (doi:10.1063/1.2736687) [DOI] [PubMed] [Google Scholar]
- 114.Snoek LC, Robertson EG, Kroemer RT, Simons JP. 2000. Conformational landscapes in amino acids: infrared and ultraviolet ion-dip spectroscopy of phenylalanine in the gas phase. Chem. Phys. Lett. 321, 49–56. (doi:10.1016/s0009-2614(00)00320-1) [Google Scholar]
- 115.Sakota K, Yamamoto N, Ohashi K, Saeki M, Ishiuchi S-I, Sakai M, Fujii M, Sekiya H. 2002. IR-dip and IR–UV hole-burning spectra of jet-cooled 4-aminobenzonitrile–(H2O)1. Observation of π-type and σ-type hydrogen-bonded conformers in the CN site. Chem. Phys. 283, 209–219. (doi:10.1016/s0301-0104(02)00501-3) [Google Scholar]
- 116.Zewail AH. 1988. Laser femtochemistry. Science 242, 1645–1653. (doi:10.1126/science.242.4886.1645) [DOI] [PubMed] [Google Scholar]
- 117.Fermann ME, Galvanauskas A, Sucha G. 2005. Ultrafast lasers: technology and applications. New York, NY: Marcel Dekker, Inc. [Google Scholar]
- 118.Keller U. 2003. Recent developments in compact ultrafast lasers. Nature 424, 831–838. (doi:10.1038/nature01938) [DOI] [PubMed] [Google Scholar]
- 119.Backus S, Durfee CG, Murnane MM, Kapteyn HC. 1998. High power ultrafast lasers. Rev. Sci. Inst. 69, 1207–1223. (doi:10.1063/1.1148795) [Google Scholar]
- 120.Seel M, Domcke W. 1991. Femtosecond time-resolved ionization spectroscopy of ultrafast internal-conversion dynamics in polyatomic molecules: theory and computational studies. J. Chem. Phys. 95, 7806–7822. (doi:10.1063/1.461816) [Google Scholar]
- 121.Shimada D, Kusaka R, Inokuchi Y, Ehara M, Ebata T. 2012. Nonradiative decay dynamics of methyl-4-hydroxycinnamate and its hydrated complex revealed by picosecond pump-probe spectroscopy. Phys. Chem. Chem. Phys. 14, 8999–9005. (doi:10.1039/c2cp24056d) [DOI] [PubMed] [Google Scholar]
- 122.Ryan WL, Gordon DJ, Levy DH. 2002. Gas-phase photochemistry of the photoactive yellow protein chromophore trans-p-coumaric acid. J. Am. Chem. Soc. 124, 6194–6201. (doi:10.1021/ja017505p) [DOI] [PubMed] [Google Scholar]
- 123.Broadbent KK, Martincigh BS, Raynor MW, Salter LF, Moulder R, Sjöberg P, Markides KE. 1996. Capillary supercritical fluid chromatography combined with atmospheric pressure chemical ionisation mass spectrometry for the investigation of photoproduct formation in the sunscreen absorber 2-ethylhexyl-p-methoxycinnamate. J. Chromatogr. A 732, 101–110. (doi:10.1016/0021-9673(95)01199-4) [Google Scholar]
- 124.Kockler J, Oelgemöller M, Robertson S, Glass BD. 2012. Photostability of sunscreens. J. Photochem. Photobiol. C 13, 91–110. (doi:10.1016/j.jphotochemrev.2011.12.001) [Google Scholar]
- 125.Allen JM, Gossett CJ, Allen SK. 1996. Photochemical formation of singlet molecular oxygen in illuminated aqueous solutions of several commercially available sunscreen active ingredients. Chem. Res. Toxicol. 9, 605–609. (doi:10.1021/tx950197m) [DOI] [PubMed] [Google Scholar]
- 126.Hanson KM, Gratton E, Bardeen CJ. 2006. Sunscreen enhancement of UV-induced reactive oxygen species in the skin. Free Radic. Biol. Med. 41, 1205–1212. (doi:10.1016/j.freeradbiomed.2006.06.011) [DOI] [PubMed] [Google Scholar]
- 127.Tan EM, Hilbers M, Buma WJ. 2014. Excited-state dynamics of isolated and microsolvated cinnamate-based UV-B sunscreens. J. Phys. Chem. Lett. 5, 2464–2468. (doi:10.1021/jz501140b) [DOI] [PubMed] [Google Scholar]
- 128.Felder P, Günthard HH. 1982. Conformational interconversions in supersonic jets: Matrix IR spectroscopy and model calculations. Chem. Phys. 71, 9–25. (doi:10.1016/0301-0104(82)87002-x) [Google Scholar]
- 129.Johnson JR, Jordan KD, Plusquellic DF, Pratt DW. 1990. High resolution S1←S0 fluorescence excitation spectra of the 1- and 2-hydroxynaphthalenes. Distinguishing the cis and trans rotamers. J. Chem. Phys. 93, 2258–2273. (doi:10.1063/1.459059) [Google Scholar]
- 130.Nesselrodt DR, Potts AR, Baer T. 1995. Observations of ethyl-substituted cyclohexanone and cyclopentanone rotamers using resonance-enhanced multiphoton ionization spectroscopy. J. Phys. Chem. 99, 4458–4465. (doi:10.1021/j100013a015) [Google Scholar]
- 131.Lewis FD, Quillen SL, Elbert JE, Schneider S, Geiselhart P. 1989. The singlet states of methyl cinnamate and methyl indenoate. J. Photochem. Photobiol. A 47, 173–179. (doi:10.1016/1010-6030(89)87063-7) [Google Scholar]
- 132.Gromov EV, Burghardt I, Hynes JT, Köppel H, Cederbaum LS. 2007. Electronic structure of the photoactive yellow protein chromophore: ab initio study of the low-lying excited singlet states. J. Photochem. Photobiol. A 190, 241–257. (doi:10.1016/j.jphotochem.2007.04.033) [Google Scholar]
- 133.Miyazaki Y, Yamamoto K, Aoki J, Ikeda T, Inokuchi Y, Ehara M, Ebata T. 2014. Experimental and theoretical study on the excited-state dynamics of ortho-, meta-, and para-methoxy methylcinnamate. J. Chem. Phys. 141, 244313 (doi:10.1063/1.4904268) [DOI] [PubMed] [Google Scholar]
- 134.Yamazaki K, et al. . 2016. Multistep intersystem crossing pathways in cinnamate-based UV-B sunscreens. J. Phys. Chem. Lett. 7, 4001–4007. (doi:10.1021/acs.jpclett.6b01643) [DOI] [PubMed] [Google Scholar]
- 135.Nofsinger JB, Ye T, Simon JD. 2001. Ultrafast nonradiative relaxation dynamics of eumelanin. J. Phys. Chem. B 105, 2864–2866. (doi:10.1021/jp004045y) [Google Scholar]
- 136.Chang XP, Li CX, Xie BB, Cui G. 2015. Photoprotection mechanism of p-methoxy methylcinnamate: a CASPT2 study. J. Phys. Chem. A 119, 11 488–11 497. (doi:10.1021/acs.jpca.5b08434) [DOI] [PubMed] [Google Scholar]
- 137.Karsili TNV, Marchetti B, Ashfold MNR, Domcke W. 2014. Ab initio study of potential ultrafast internal conversion routes in oxybenzone, caffeic acid, and ferulic acid: Implications for sunscreens. J. Phys. Chem. A 118, 11999–12010. (doi:10.1021/jp507282d) [DOI] [PubMed] [Google Scholar]
- 138.Kasha M. 1950. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 9, 14–19. (doi:10.1039/df9500900014) [Google Scholar]
- 139.McConnell H. 1952. Effect of polar solvents on the absorption frequency of n→π electronic transitions. J. Chem. Phys. 20, 700–704. (doi:10.1063/1.1700519) [Google Scholar]
- 140.Das Neves Rodrigues N, et al. In press FDDYNAM16 Towards elucidating the photochemistry of the sunscreen filter ethyl ferulate using time-resolved gas-phase spectroscopy. Faraday Discuss. (doi:10.1039/c6fd00079g). [DOI] [PubMed] [Google Scholar]
- 141.Rodrigo CP, James WH 3rd, Zwier TS. 2011. Single-conformation ultraviolet and infrared spectra of jet-cooled monolignols: p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol. J. Am. Chem. Soc. 133, 2632–2641. (doi:10.1021/ja109218j) [DOI] [PubMed] [Google Scholar]
- 142.Dean JC, Kusaka R, Walsh PS, Allais F, Zwier TS. 2014. Plant sunscreens in the UV-B: ultraviolet spectroscopy of jet-cooled sinapoyl malate, sinapic acid, and sinapate ester derivatives. J. Am. Chem. Soc. 136, 14 780–14 795. (doi:10.1021/ja5059026) [DOI] [PubMed] [Google Scholar]
- 143.Dym S. 1968. Assignment of the lowest triplet state of the carbonyl group. J. Chem. Phys. 48, 646–652. (doi:10.1063/1.1668695) [Google Scholar]
- 144.Ledger MB, Porter G. 1972. Primary photochemical processes in aromatic molecules. Part 15—the photochemistry of aromatic carbonyl compounds in aqueous solution. J. Chem. Soc. Faraday Trans. 1 68, 539–553. (doi:10.1039/f19726800539) [Google Scholar]
- 145.Satzger H, Schmidt B, Root C, Zinth W, Fierz B, Krieger K, Kiefhaber T, Gilch P. 2004. Ultrafast quenching of the xanthone triplet by energy transfer: new insight into the intersystem crossing kinetics. J. Phys. Chem. A 108, 10 072–10 079. (doi:10.1021/jp047583) [Google Scholar]
- 146.Peters KS, Lee J. 1993. Picosecond dynamics of the photoreduction of benzophenone by DABCO. J. Phys. Chem. 97, 3761–3764. (doi:10.1021/j100117a022) [Google Scholar]
- 147.Yabumoto S, Shigeto S, Lee YP, Hamaguchi HO. 2010. Ordering, interaction, and reactivity of the low-lying nπ* and ππ* excited triplet states of acetophenone derivatives. Angew. Chem. Int. Ed. Engl. 49, 9201–9205. (doi:10.1002/anie.201004571) [DOI] [PubMed] [Google Scholar]
- 148.Tomas F, Olba A, Medina P, Zabala I. 1986. Vibronic interaction between nπ and ππ states and phosphorescence of Norharmane. J. Mol. Struct. 142, 143–146. (doi:10.1016/0022-2860(86)85082-7) [Google Scholar]
- 149.Joseph J, Cavaleri KP, Bowman RM. 1996. An investigation of the solvent dependence on the ultrafast intersystem crossing kinetics of xanthone. Chem. Phys. Lett. 259, 495–502 (10.1016/0009-2614(96)00782-8) [Google Scholar]
- 150.El-Sayed MA. 1968. Triplet state. Its radiative and nonradiative properties. Acc. Chem. Res. 1, 8–16. (doi:10.1021/ar50001a002) [Google Scholar]
- 151.Takatsuka K, Arasaki Y, Wang K, McKoy V. 2000. Introductory lecture. Probing wavepacket dynamics with femtosecond energy- and angle-resolved photoelectron spectroscopy. Faraday Discuss. 115, 1–15. (doi:10.1039/B002739L) [PubMed] [Google Scholar]
- 152.Arasaki Y, Takatsuka K, Wang K, McKoy V. 2003. Pump-probe photoionization study of the passage and bifurcation of a quantum wave packet across an avoided crossing. Phys. Rev. Lett. 90, 248303 (doi:10.1103/PhysRevLett.90.248303) [DOI] [PubMed] [Google Scholar]
- 153.Nićiforović N, Abramovič H. 2014. Sinapic acid and its derivatives: natural sources and bioactivity. Compr. Rev. Food Sci. Food Saf. 13, 34–51. (doi:10.1111/1541-4337.12041) [DOI] [PubMed] [Google Scholar]
- 154.Ruegger M, Chapple C. 2001. Mutations that reduce sinapoylmalate accumulation in arabidopsis thaliana define loci with diverse roles in phenylpropanoid metabolism. Genetics 159, 1741–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chapple CC, Vogt T, Ellis BE, Somerville CR. 1992. An arabidopsis mutant defective in the general phenylpropanoid pathway. Plant Cell. 4, 1413–1424. (doi:10.1105/tpc.4.11.1413) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.van Dantzig NA, Shou H, Alfano JC, Yang NCC, Levy DH. 1994. Photoinduced charge transfer in bichromophoric molecules in the gas phase. J. Chem. Phys. 100, 7068–7078. (doi:10.1063/1.466907) [Google Scholar]
- 157.Jiang S, Levy DH. 2002. Supersonic jet studies on the photophysics of substituted benzenes and naphthalenes. J. Phys. Chem. A 106, 8590–8598. (doi:10.1021/jp025764a) [Google Scholar]
- 158.Grabowski ZR, Rotkiewicz K, Rettig W. 2003. Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures. Chem. Rev. 103, 3899–4032. (doi:10.1021/cr940745l) [DOI] [PubMed] [Google Scholar]
- 159.Gorse A-D, Pesquer M. 1995. Intramolecular charge transfer excited state relaxation processes in para-substituted N,N-dimethylaniline: a theoretical study including solvent effects. J. Phys. Chem. 99, 4039–4049. (doi:10.1021/j100012a026) [Google Scholar]
- 160.DeRosa M. 2002. Photosensitized singlet oxygen and its applications. Coordin. Chem. Rev. 233–234, 351–371. (doi:10.1016/s0010-8545(02)00034-6) [Google Scholar]
- 161.Grewer C, Brauer HD. 1993. Temperature dependence of the oxygen quenching of ππ* singlet and ππ* triplet states of singlet oxygen sensitizers. J. Phys. Chem. 97, 5001–5006. (doi:10.1021/j100121a024) [Google Scholar]
- 162.Baker LA, Horbury MD, Greenough SE, Allais F, Walsh PS, Habershon S, Stavros VG. 2016. Ultrafast photoprotecting sunscreens in natural plants. J. Phys. Chem. Lett. 7, 56–61. (doi:10.1021/acs.jpclett.5b02474) [DOI] [PubMed] [Google Scholar]
- 163.Berera R, van Grondelle R, Kennis JT. 2009. Ultrafast transient absorption spectroscopy: principles and application to photosynthetic systems. Photosynth. Res. 101, 105–118. (doi:10.1007/s11120-009-9454-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Greenough SE, Horbury MD, Thompson JOF, Roberts GM, Karsili TNV, Marchetti B, Townsend D, Stavros VG. 2014. Solvent induced conformer specific photochemistry of guaiacol. Phys. Chem. Chem. Phys. 16, 16 187–16 195. (doi:10.1039/c4cp02424a) [DOI] [PubMed] [Google Scholar]
- 165.Chatelain E, Gabard B. 2001. Photostabilization of butyl methoxydibenzoylmethane (Avobenzone) and ethylhexyl methoxycinnamate by bis-ethylhexyloxyphenol methoxyphenyl triazine (Tinosorb S), a new UV broadband filter. Photochem. Photobiol. 74, 401–406. (doi:10.1562/0031-8655(2001)0740401POBMAA2.0.CO2) [DOI] [PubMed] [Google Scholar]
- 166.Kikuchi A, Hata Y, Kumasaka R, Nanbu Y, Yagi M. 2013. Photoexcited singlet and triplet states of a UV absorber ethylhexyl methoxycrylene. Photochem. Photobiol. 89, 523–528. (doi:10.1111/php.12017) [DOI] [PubMed] [Google Scholar]