

Abstract
We report the development of a tertiary amine-containing β-turn peptide that catalyzes the atroposelective bromination of pharmaceutically relevant 3-arylquinazolin-4(3H)-ones (quinazolinones) with high levels of enantioinduction over a broad substrate scope. The structure of the free catalyst and the peptide-substrate complex were explored using X-ray crystallography and 2D-NOESY experiments. Quinazolinone rotational barriers about the chiral anilide axis were also studied using DFT calculations and are discussed in light of the high enantioselectivities observed. Mechanistic studies also suggest that the initial bromination event is stereo-determining, and the major monobromide intermediate is an atropisomerically stable, mono-ortho-substituted isomer. The observation of stereoisomerically stable monobromides stimulated the conversion of the tribromide products to other, atropisomerically-defined products of interest. For example, (1) a dehalogenation-Suzuki-Miyaura cross-coupling sequence delivers ortho-arylated derivatives, and (2) a regioselective Buchwald-Hartwig amination procedure installs para-amine functionality. Stereochemical information was retained during these subsequent transformations.
Keywords: quinazolinone, atropisomerism, bromination, peptide, catalysis
TOC Graphic

Introduction
It is now becoming well appreciated that atropisomerism1 is an issue of significant importance in medicinal chemistry.2 Chiral compounds that exist as separate atropisomers may present advantages if they have sufficiently low barriers to racemization, such that dynamic kinetic resolution3 is possible during selective binding to a targeted biological receptor. However, if the opposite atropisomer binds to an off-target receptor, alternative, and even deleterious, effects may ensue.4 Perhaps these scenarios are behind an increasing number of reports,5 and even patents,6 that describe the differential biological functions of individual atropisomers. Accordingly, synthetic efforts to prepare individual atropisomers comprise an area of significant innovation in chemistry.7 Among scaffolds that present these stereochemical issues, 3-arylquinazolin-4(3H)-ones8 (1) are an important class of compounds, whose exploding list of characterized biological properties enumerates a large swath of biochemical functions (Figure 1a).9 We report herein our recent studies culminating in catalyst-dependent syntheses of atropisomeric quinazolinone bromides (2) with high levels of enantioinduction. These findings enable efficient access to a range of substituted quinazolinones available through an atroposelective tribromination reaction, followed by either dehalogenation-cross-coupling sequences or regioselective amination reactions.
FIGURE 1.

(a) Some 3-arylquinazolin-4(3H)-one-based bioactive compounds.9 (b) Our strategy for the peptide-catalyzed atroposelective bromination of quinazolinones 1 to access enantioenriched bromides 2.
We recently demonstrated that peptide-based catalysts are effective for the atroposelective bromination of both biaryl10 and tertiary benzamide scaffolds.11 Gratifyingly, a related approach has also been recently reported to access chiral isoquinoline N-oxides using a cinchona alkaloid-derived urea catalyst.12 It is interesting to note that the enantioselective synthesis of axially chiral quinazolinones is a subject with a limited literature,13 and most methods have either involved racemic synthesis followed by resolution or diastereoselective synthesis using chiral auxiliaries. Given the growing interest in these compounds in medicinal chemistry, we targeted a catalytic asymmetric approach (Figure 1b). Notably, unlike many biaryl compounds and benzamides, an opportunity also existed for selective preparation of atropisomerically stable mono-ortho-substituted quinazolinones (e.g. 3, Figure 1b), given their established high barriers to enantiomerization.14 At the same time, these issues could present fundamental challenges to the development of catalysts that might mediate dynamic kinetic resolutions.3 Our assessment of these concepts is presented below.
Results and Discussion
Catalyst Identification
Our exploration of this intriguing system began with a study of peptide-based catalysts for the bromination of quinazolinone 1a using the conditions described in Equation 1.15 Our design principle was guided by previous reports from our group, wherein we were able to embed a tertiary amine-containing β-dimethylaminoalanine (Dmaa) residue within a peptide sequence that was expected to adopt a well-defined β-turn geometry.16 In that way, we hoped to capitalize on an interaction (e.g., hydrogen bonding or even proton transfer) between the phenol of 1a and the tertiary amine moiety of the Dmaa residue, with the potential for additional interactions provided by the functionality and chirality of the peptide (i.e., catalysts such as 4).11 Accordingly, dynamic kinetic resolution would be possible via selective docking and functionalization of one atropisomer of (±)-1a over the other.
 |
(1) |
In the absence of any catalyst, bromination of 1a was sluggish and non-selective under the conditions we examined.17 However, using triethylamine (10 mol%) as a catalyst, bromination of 1a proceeded smoothly, providing 88% yield of racemic tribromide 2a. Thus, we began to assess peptide-based catalysts (4) for this transformation, and the results of our optimization are summarized in Figure 2.18 We were pleased to discover that peptide 4a, an archetypal β-turn-promoting sequence that we have utilized in the past,11,19 provided tribromide 2a in 66:34 er, setting the stage for further optimization. In a number of previous instances, we have observed a marked effect of the C-terminal protecting group on the enantioselectivity of peptide-catalyzed reactions.16 Comparing peptides 4a– f, it is clear that the dimethylamide end cap provides an advantage over the alternatives, which may be attributed to the enhanced hydrogen bond acceptor ability of tertiary amides.20
FIGURE 2.

Assessment of peptide-based catalysts in the bromination of quinazolinone 1a using the conditions described in Equation 1. Data shown in blue are the results of peptide optimization studies, and data shown in green are the results of a truncation study. All results represent the average of two trials. Abbreviations: Boc, tert-butylcarbamate; Dmaa, β-dimethylaminoalanine; Aib, 2-aminoisobutyric acid; Cle, cycloleucine (1-aminocyclopentane carboxylic acid); Acpc, 1-aminocyclopropyl carboxylic acid; Aic, 2-aminoindane carboxylic acid; (R)-α-MBA, (R)-α-methyl benzyl amide; Phe(5F), pentafluorophenylalanine; Nle, norleucine (n-butylglycine); Npg, neopentylglycine; Chg, cyclohexylglycine; Pyrr, pyrrolidinyl.
The enantioselectivity of peptide-catalyzed reactions is often highly sensitive to residue substitutions, especially those at the i+2 residue,16 a position that plays an important role in determining the secondary structural attributes of the peptide.21 Upon examining the i+2 residue (as in 4c and 4g–j), we observed a pronounced increase in enantioselectivity when 1-aminocyclopropyl carboxylic acid (Acpc) was substituted in this position (as in 4h), providing 2a in 88:12 er, an increase in 36% ee compared to 4c. The origin of this remarkable effect is not fully understood, although we have hypothesized that changes in the φ and ψ angles of the β-turn conformation manifest in a more favorable interaction with 1a.16,21 A steric effect is also possible, although the degree of enhancement associated with the cyclopropyl ring of 4h relative to the corresponding gem-dimethyl moiety of peptide 4c may be too pronounced to be explained by sterics alone. Nonetheless, having found a suitable i+2 residue, we elected to reexamine the C-terminal protecting group to ensure that the trends upheld with Acpc at the i+2 position (as in 4h and 4k–l). Indeed, the dimethylamide C-terminal cap (as in 4h) was again found to outperform the other functionalities.
Alterations to the i+3 position of β-turn-containing tetramers allowed for fine-tuning of enantioselectivity. Comparing peptides 4h and 4m–u, a number of trends emerged. First, it is clear that none of these changes produced as dramatic an effect as substitutions to the i+2 residue. Yet, it was also apparent that alkyl-substituted residues (4q–u) provide higher enantioenrichment of 2a than benzyl- (4h, 4m–n) and unsubstituted (4o–p) residues. There was also very little difference among the alkyl-substituted i+3 residues of peptides 4q–u, so we opted to move forward using leucine-containing 4q as the lead catalyst, which provided tribromide 2a in 92:8 er under the conditions of Equation 1. Compared to peptide 4t, which contained a slightly bulkier neopentylglycine (Npg) residue at the i+3 position and provided 2a in 93:7 er, peptide 4q had the added benefit of containing a proteinogenic, inexpensive, and readily available residue at the i+3 position.
Catalyst Structure
Catalyst 4q proved amenable to study with X-ray diffraction (Figure 3). Interestingly, two different conformations of 4q are present in the unit cell, one of which has an extended backbone (as drawn) while the other exhibits a backbone bend of nearly 110° out-of-plane. We have observed this sort of backbone bending previously,22 although it remains unclear if this geometry is representative of active catalytic species. Nonetheless, both structures exhibit intramolecular hydrogen-bonding patterns and φ and ψ dihedral angles consistent with a type II’ β-turn.21 We also studied the structure of 4q in solution using 1H−1H NOESY, and we were able to observe 50 non-sequential nOes consistent with a rigid β-turn geometry (see Supporting Information).
FIGURE 3.

X-ray crystal structure of Boc-Dmaa-d-Pro-Acpc-Leu-NMe2 (4q).24 Two different conformations of 4q are present in the unit cell, both of which show intramolecular hydrogen bonds characteristic of type II’ β-turns.21
Furthermore, we were able to demonstrate the importance of the folded conformation of peptide 4q through a study of truncated peptide catalysts (Figure 2). The effect of sequential residue deletions on the outcome of the bromination reaction was dramatic. For example, we found that the trimer Boc-Dmaa-d-Pro-Acpc-NHMe (4β), which is able to access the intramolecular, tenmembered ring hydrogen bond (NHi+3 to Oi) required for β-turn formation,21 provided 2a in 82:18 er, only modestly reduced compared to tetramer 4q. On the other hand, trimers possessing methyl ester (4z) and dimethylamide (4α) end caps, which cannot form the β-turn hydrogen bond, were significantly less selective. This provides evidence that the β-turn secondary structure, which is accessible in tetramers and secondary amide-terminated trimers, is important for high levels of enantioinduction. Dimers 4w–x were significantly less selective than the corresponding trimers, although we were intrigued to discover that one dimer, Boc-Dmaa-d-Pro-NHMe (4y), was equally selective to trimer 4α, delivering 2a in 72:28 er. It is possible that the enhanced selectivity exhibited by dimer 4y derives from its ability to form a seven-membered ring hydrogen bonded structure (NHi+2 to Oi),23 reinforcing the importance of secondary structure to enantioselectivity. It is also interesting to note that the inherent selectivity of the Dmaa-monomer itself (4v) actually favors the opposite enantiomer of 2a, albeit with very low er.
Optimization of reaction conditions
Having identified catalyst 4q that is able to deliver tribromide 2a in 92:8 er under the conditions of Equation 1, in which N-bromosuccinimide (NBS) was added in one portion at the outset of the reaction, we wondered if we might be able to boost the enantioselectivity by changing the mode of NBS delivery. This speculation was borne of the hypothesis that full and rapid racemization of the starting material is required in order to achieve the best possible results in a dynamic kinetic resolution.3 2-Substituted-3-arylquinazolinones, such as 1a, have been reported to have intrinsic barriers to rotation about the N–CAr bond, even in the absence of ortho-substituents on the phenol ring, that could impede rapid re-racemization as a dynamic kinetic resolution proceeds.13 Therefore, we speculated that, if the barrier to enantiomerization27 of 1a is sufficiently high, the starting material may not be able to re-racemize on the time scale of a very rapid and stereodetermining bromination event. Indeed, DFT calculations25 showed that the barrier to enantiomerization in 1a is 18.8 kcalmol−1 corresponding to a first-order half-life of 6.9 s (Figure 4), which may be slow with respect to the time scale of bromination. After optimization of the relevant parameters, we discovered that slow addition of NBS over 2.5 h at 0 °C, as described in Equation 2 (R = Me), provided 2a in a notably improved 97:3 er.28 It is important to note that this increase in selectivity was observed even in the presence of 5% (by volume) acetone,10a which was added to the delivery solution to facilitate dissolution of the NBS.
 |
(2) |
FIGURE 4.

Computed barriers to rotation about the N–CAr bond in a series of relevant quinazolinones. Barriers were derived from optimization of the stationary points along torsional potential energy profiles using M06-2X/6-311++G(2d,3p)//B3LYP/6-31+G(d,p).25–26
Substrate scope
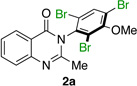
With optimal conditions in hand, we turned our attention to the substrate scope. Catalyst 4q is able to address a wide range of quinazolinones 1 (Table 1). As noted, the bromination of 1a affords tribromide 2a with 97:3 er, and 86% isolated yield (entry 1). In order to establish the absolute configuration of the product, we subjected 2a to standard demethylation conditions using BBr3 to obtain free phenol 2a’,29 which we were able to crystallize and analyze by X-ray diffraction (Equation 3). The crystal structure of 2a’ reveals the (S)-absolute stereochemistry of 2a.
 |
(3) |
Table 1.
| Entry | Quinazolinone | Product | Yieldc | e.r.d |
|---|---|---|---|---|
| 1 | 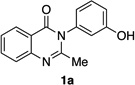 |
 |
86% | 97:3 |
| 2 | 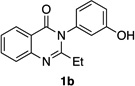 |
 |
79% | 97:3 |
| 3 | 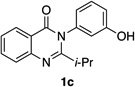 |
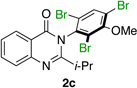 |
78% | 96:4 |
| 4 | 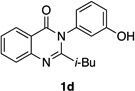 |
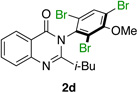 |
79% | 96:4 |
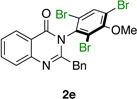
| 5 | 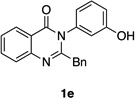 |
 |
75% | 93:7 |
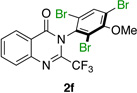
| 6 | 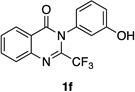 |
 |
63% | 63:37 |
| 7 | 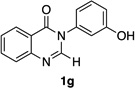 |
 |
80% | 65:35 |
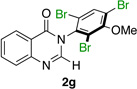
| 8 | 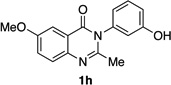 |
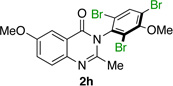 |
85% | 95:5 |
| 9 | 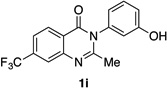 |
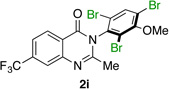 |
85% | 93:7 |
| 10e | 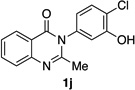 |
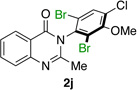 |
93% | 96:4 |
| 11e | 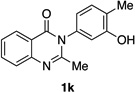 |
 |
92% | 99:1 |
| 12 | 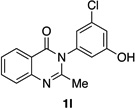 |
 |
89% | 96:4 |
| 13 | 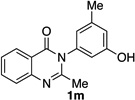 |
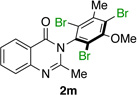 |
77% | 98:2 |
| 14e | 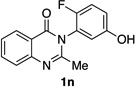 |
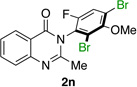 |
84% | 56:44 |
Reaction Conditions: quinazolinone 1 (0.10 mmol, 1 equiv), peptide 4q (0.01 mmol, 10 mol% w.r.t. 1), NBS (0.30 mmol, 3 equiv w.r.t. 1), PhMe/CHCl3 (9:1 v/v) with 5% acetone additive (by volume), slow addition of NBS over 2.5 h.
Data represent the average of two trials.
Isolated yields after chromatography are presented.
Enantiomer ratios were determined by chiral HPLC using OJ-H or AD-H columns.
2.0 equiv NBS was used in the bromination.
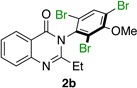
Quinazolinones possessing alkyl substituents at the 2-position (1b–e) were processed by 4q with good yields (75–86%) and high levels of enantioinduction (93:7 to 97:3 er, entries 2–5). However, 2-CF3-substituted 1f did not follow this same trend, as bromination delivered only 63% yield of modestly enriched 2f (63:37 er, entry 6). The result was initially surprising given the high levels of enantioselectivity observed in tribromide 2c (95:5 er, entry 3), as the steric size of a CF3 group is comparable to an i-Pr group on some scales.30 It appears that the electron withdrawing CF3 group of 1f influences the catalyst-substrate complex in a manner we do not understand. Yet, we also observed low levels of enantioenrichment in tribromide 2g (65:35 er, entry 7), in which the 2-position is unsubstituted. The origin of this phenomenon may derive from a low barrier to rotation about the chiral axis in the corresponding ortho-monobromide 3g, which we presume is the immediate product of stereodetermining bromination. Indeed, using DFT calculations,25 we were able to compute a barrier to rotation of 22.6 kcalmol−1 in monobromide 3g, which may be sufficiently low so as to permit racemization on the time scale of the slow addition. By way of comparison, the computed rotational barriers for quinazolinone 1g and tribromide 2g’ were found to be 7.6 and 36.7 kcalmol−1, respectively, using DFT computations (Figure 4).25
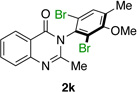
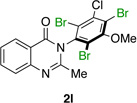
Many other substrates were brominated by 4q with high levels of enantioselectivity. For example, we found that substitution on the benzo moiety of the quinazolinone scaffold is often tolerated (entries 8–9). Tribromide 2h, having an electron donating methoxy group at the 6-position, was isolated in 85% yield and 95:5 er, only slightly lower than the parent compound (entry 8). Likewise, 7-CF3-susbtituted tribromide 2i was isolated in 85% yield and 93:7 er (entry 9). These results suggest a weak effect of distal-substitution on enantioselectivity, though both electron donating and electron withdrawing groups are still largely tolerated by 4q. Substitution on the phenol moiety of the quinazolinone scaffold also revealed interesting data. The p-Cl-substituted dibromide 2j was isolated in excellent yield (93%) and with 96:4 er (entry 10). We also found that tribromide 2l, which was derived from the m-Cl-substituted quinazolinone 1l, was isolated in high yield (89%) and with 96:4 er (entry 12). These results show that peptide 4q is able to process substrates possessing electron withdrawing substituents on the arene with negligible erosion of enantioselectivity relative to 2a. In addition, electron donating Me substituents at the p-(1k) and m-positions (1m) provide excellent substrates, as dibromide 2k was isolated in 92% yield with 99:1 er (entry 11) and tribromide 2m was isolated in 88% yield and 98:2 er (entry 13).31
Lastly, we examined a compound that was pre-functionalized at the o’-position of the phenol moiety. In this case, we anticipated a reduced er value by virtue of a high barrier to racemization in the starting material. Indeed, o’-F-substituted 1n was found to deliver dibromide 2n in only 55:45 er (entry 14). DFT calculations25 showed that 1n has a 26.6 kcalmol−1 barrier to rotation about the chiral axis (Figure 4), corresponding to a half-life to rotation of 3.6 x 106 s. Thus, racemization is presumed to be slow on the time scale of bromination. In this scenario, compound 1n was projected to be a suitable substrate for a traditional kinetic resolution.32 Accordingly, running the bromination of 1n to low conversion using only 0.5 equivalents of NBS under otherwise identical conditions, delivered 2n in 93:7 er (14% isolated yield), showing that quinazolinones with high barriers to racemization may indeed be synthesized enantioselectively employing a classical kinetic resolution.
Mechanism-Driven Experiments
Examination of the substrate scope inspired us to investigate certain mechanistic aspects of this enantioselective bromination reaction. We began with an LC/MS-enabled study of the background and catalyzed bromination of 1a under conditions of reagent-controlled low conversion (Scheme 1). As noted above, in the absence of catalyst, the bromination proceeds slowly, and in fact the major species in the crude reaction mixture is the p-monobromide 5a, although all mono-, di-, and tribromides were observed in minor quantities. When triethylamine is used as a catalyst, the reaction proceeds more efficiently and with significant regioselectivity, also favoring the p-monobromide 5a with no evidence of the other monobromides. The o,p-dibromide was also formed in approximately equal quantities under the triethylamine conditions, and the tribromide was observed as a minor product. Using peptide 4r, which provides enantioselectivity similar to 4q (vide supra), the reaction constrained to reach low conversion exhibits a strikingly different result: the major monobromide is the alternative regioisomer 3a, while only a very small quantity of the p-monobromide 5a is detected, while the o,p-dibromide and tribromide 2a’ are also detected. These results indicate that the peptide-based catalyst is able to overturn the inherent site-selectivity of the bromination reaction, which also implies that the stereodetermining bromination event is the first bromination to give 3a (See Supporting Information for LC/MS data). Furthermore, when the crude reaction mixture of the 4r-catalyzed reaction was subjected to methylation and analyzed by chiral HPLC, it was discovered that monobromide 3a was enantioenriched to 98:2 er. To the best of our knowledge, this is a rare example of an atroposelective reaction in which the chiral axis is set after the first functionalization of an atropisomerically dynamic starting material.33
SCHEME 1.

Study of the major monobromides in the catalyzed and uncatalyzed bromination of 1a
To further our understanding of the basis of stereoselectivity, we studied the peptide-substrate complex using NMR techniques. The results of our findings are summarized in Figure 5. Compared to the 1H-NMR spectra of 1a (Figure 5a) and 4q (Figure 5c), the spectrum of the 1:1 complex (Figure 5b) differs significantly from its isolated constituents under identical conditions.34 Notably, while the other amide signals shift downfield upon complexation, the NHLeu signal barely shifts at all, providing evidence for a strong hydrogen bond between NHLeu and ODmaa. In contrast, the NHAcpc shifts downfield by nearly 1.0 ppm upon complexation, which implicates this proton in an important intermolecular hydrogen bond with 1a. The NHDmaa signal also shifts downfield by about 0.3 ppm, which may suggest a change in the β-hairpin conformation as a result of complexation. It may also implicate the carbamate NH proton in an interaction with the substrate.
FIGURE 5.

(a) 1H-NMR spectrum of quinazolinone 1a. (b) 1H-NMR spectrum of 1a-4q complex (1:1 molar ratio). (c) 1H-NMR spectrum of peptide 4q. (d) NOESY correlation diagram showing the intermolecular nOes observed in the 1a-4q complex. (e) Possible binding model based on NMR data. The model explains the (S)-absolute configuration of the products and also predicts the first site of bromination at the ortho-position of 1a.
Furthermore, we observed a global upfield shifting of all of the i+3 leucine signals, especially the γLeu signal. This suggests that the i+3 residue might be situated beneath an arene, such that electron density in the π-system leads to this anisotropic effect. The αd-Pro signal also shifts upfield, presumably for similar reasons. Similarly, the all of the Dmaa signals tend to shift downfield, in keeping with a decrease in electron density around the side-chain N-atom as would be expected in a Me2NDmaa to HO1a hydrogen bond. It is also evident that the βDmaa signals of 4q become significantly more differentiated in the complex than in the free peptide. With respect to the substrate, all of the 1H–NMR signals of 1a shift downfield in the complex. However, only the ortho-position (Figure 2d) shifts downfield to a significant degree (> 0.30 ppm), which may be consistent with this position disposed most proximally to the peptide. In light of these results, as well as our mechanistic studies that identified this position as the site of the first bromination event, it seems plausible that the peptide delivers Br+ to this position via one of the proximal, Lewis basic carbonyls.35 It should also be noted that many of the 1H-NMR signals corresponding to complexed 1a possess minor shoulder peaks, while this is not evident in any of the signals derived from 4q. This perhaps suggests that 1a might be fluxional in the bound state. We also studied the binding interaction between 1a and 4q using 1H−1H-NOESY, and we were able to observe several intermolecular nOes that supported this model. A summary of these interactions is presented in Figure 5d, where the color-coded distances are derived from integration of the relevant NOESY cross-peaks.36 This experiment highlighted a number of interactions that were not observed in the simple 1H-NMR complexation experiment (vide supra). For instance, strong nOes were observed between (1) Me2NDmaa and the ortho-position of 1a and (2) βAcpc and the 5-position of 1a, suggesting that these groups are very close to one another in space in the complex. Moderate nOes were also observed between βDmaa and the 2-Me group of 1a, as well as between the i-Pr group of leucine and the ortho-position of 1a. Weak nOes were also observed, allowing us to fine-tune our proposed binding orientation.
The culmination of these mechanistically driven studies is a self-consistent binding model from which we were able to rationalize the first site of bromination, as well as the (S)-absolute configuration of products 2 and ortho-mono-bromides 3 (Figure 5e). Our model suggests two intermolecular hydrogen bonds in the catalyst-substrate complex, one between OH1a and Me2NDmaa and the other between NHAcpc and C=O1a.
Cross Coupling
With the atroposelective bromination of quinazolinones established, we turned to the demonstration of further synthetic utility through derivatization of the brominated products.37 Recognizing the prevalence of monosubstituted quinazolinones in the medicinal chemistry literature (Figure 1a),9 we sought to establish that tribromide 2a could be converted to 3a efficiently and without loss of enantioenrichment. As shown in Equation 4, palladium-catalyzed hydrogenation under well-established conditions achieved this goal,38 affording monobrominated compound Me-3a in 80% yield with high levels of regioselectivity presumably governed by sterics. Despite the lack of a second heavy atom ortho’-substituent on the phenol ring, this compound exhibited good atropostablity at ambient temperature. The barrier to rotation about the chiral axis in 3a was calculated to be 35.5 kcalmol−1 using DFT calculations,25 which corresponds to a half-life of 1.13 x 1013 s (Figure 4).
 |
(4) |
Monobromide Me-3a was then examined as a template for diversification. After initial studies of Suzuki-Miyaura cross-coupling39 reactions revealed that some racemization could occur at high temperatures (> 60 °C), we discovered that excellent results could be achieved at lower temperatures (45 °C) when a highly active catalyst was employed. Under the optimized conditions, Pd2(dba)3 and (t-Bu)(Cy)2P•HBF4 enabled the Suzuki coupling of 3a with various arene and heterocycle boronic acids under atropstable conditions, providing good yields of structurally complex products (6) with no loss of enantioenrichment, as shown in Scheme 2a.
SCHEME 2.

Cross-coupling of quinazolinone bromides.
Upon the success of atropstable Suzuki coupling of Me-3a, we sought to expand the scope of our product derivatization beyond the formation of C–C bonds using Buchwald-Hartwig O- and N-arylation.40 Unfortunately, under the conditions we had examined thus far, which required high temperatures of ~80 °C for product formation, products were observed with reduced levels of enantioenrichment, suggesting racemization during the reaction.41 However, the more atropisomerically stable tribromide 2a, having a computed barrier to enantiomerization of over 50 kcalmol−1 (2a’, Figure 4),42 proved a good substrate, wherein regioselective amination could be achieved to form compounds such as 7a–c in good yields and with very little erosion of er (Scheme 2b). The site selectivity of the amination is presumably due to steric differentiation.
Conclusions
Peptide-catalyzed atroposelective bromination has been extended to a highly significant scaffold in medicinal chemistry. Critical to the achievement of this advance were the discovery of an optimized catalyst and an appreciation of the physical organic principles that govern the barriers to rotation in the starting materials and products for these unique reactions. Catalyst 4q was found to be effective for a broad substrate scope, and moreover the unique conformational properties (i.e., barriers to atropisomerization) of the products enable site-selective debromination and cross-coupling reactions of several types to deliver multiple drug-like chemotypes. Mechanism-driven experiments have also revealed a number of interesting features of these asymmetric reactions, including the likely site of the initial and stereo- determining bromination. NMR spectroscopic experiments have unveiled critical features of the catalyst-substrate complex that are consistent with the observation of the absolute configuration of the products that emerge from these reactions. It is our hope that these concepts set the stage for synthetic applications in the context of this biologically relevant scaffold.
Supplementary Material
Acknowledgments
A.J.M. would like to extend his sincerest thanks to the NSF Graduate Research Fellowship Program for financial support. We would like to thank Dr. Eric Paulson for his assistance with our NMR studies and Dr. Brandon Mercado for solving our X-ray crystal structures. We would also like to thank Nadia Abascal, Dr. Guillaume Pelletier, and Dr. Byoungmoo Kim for helpful discussions. All computational work was supported by the facilities and staff of the Yale University Faculty of Arts and Sciences High Performance Computing Center, and by the National Science Foundation under grant #CNS 08-21132 that partially funded acquisition of the facilities.
Funding Sources
We are grateful to the National Institute of General Medical Sciences of the NIH (GM-068649) for support.
Footnotes
ASSOCIATED CONTENT
Additional experimental details, characterization data for all catalysts, substrates, and products, crystallographic information, and computational data are provided in the Supporting Information. This material is available free of charge on the Internet at http://pubs.acs.org.
Notes
Crystallographic data are deposited with the Cambridge Crystallographic Data Centre under the accession number CCDC 1412919 (2a’) and CCDC 1412920 (4q).
REFERENCES
- 1.Ōki M. Recent Advances in Atropisomerism. In: Allinger NL, Eliel EL, Wilen SH, editors. Topics in Stereochemistry. Vol. 14. Hoboken, NJ: Wiley; 1983. pp. 1–81. [Google Scholar]
- 2.For relevant reviews, please see: LaPlante SR, Fader LD, Fandrick KR, Fandrick DR, Hucke I, Kemper R, Miller SPF, Edwards PJ. J. Med Chem. 2011;54:7005–7022. doi: 10.1021/jm200584g. LaPlante SR, Edwards PJ, Fader LD, Jakalian A, Hucke O. ChemMedChem. 2011;6:505–513. doi: 10.1002/cmdc.201000485. Clayden J, Moran WJ, Edwards OJ, LaPlante SR. Angew. Chem. Int. Ed. 2009;48:6398–6401. doi: 10.1002/anie.200901719.
- 3.For reviews on dynamic kinetic resolution, please see: Pellissier H. Tetrahedron. 2011;67:3769–3802. Pellissier H. Tetrahedron. 2008;64:1563–1601. Faber K. Chem.--Eur. J. 2001;7:5004–5010. doi: 10.1002/1521-3765(20011203)7:23<5004::aid-chem5004>3.0.co;2-x. Ward RS. Tetrahedron: Asymmetry. 1995;6:1475–1490.
- 4.For reports describing the differential, and perhaps deleterious, effects of drug enantiomers, please see: Agranat I, Caner H, Caldwell J. Nature Rev. Drug. Discov. 2002;1:753–768. doi: 10.1038/nrd915. Shah RR, Midgley JM, Branch SK. Adv. Drug. React. Toxicol. Rev. 1998;17:145–190. Reist M, Carrupt P-A, Francotte E, Testa B. Chem. Res. Toxicol. 1998;11:1521–1528. doi: 10.1021/tx9801817. Eriksson T, Bjorkman S, Roth B, Fyge A, Hoglund P. Chriality. 1995;7:44–52. doi: 10.1002/chir.530070109. Fabro S, Smith RL, Williams RT. Nature. 1967;215:296. doi: 10.1038/215296a0. Ashrafian H, Horowitz JD, Frenneaux MP. Cardiovasc. Drug Rev. 2007;25:76–97. doi: 10.1111/j.1527-3466.2007.00006.x.
- 5.For examples of differential biological function of individual drug atropisomers, please see: LaPlante SR, Forgione P, Boucher C, Coulombe R, Gillard J, Hucke O, Jakalian A, Joly M-A, Kukolj G, Lemke C, McCollum R, Titolo S, Beaulieu PL, Stammers T. J. Med. Chem. 2014;57:1944–1951. doi: 10.1021/jm401202a. Eveleigh P, Hulme EC, Schudt C, Birdsall NJM. Mol. Pharmacol. 1989;35:477–483.
- 6.Evarts JB, Ulrich RG. (Gilead Galistoga LLC, USA) Atropisomers of 2-purinyl-3-tolyl-quinazolinone derivatives and methods of use. 2013 May 14; US 8,440,677 B2. [Google Scholar]
- 7.For reviews on atroposelective synthesis, please see: Bencivenni G. Synlett. 2015;26:1915–1922. Wencel-Delord J, Panossian A, Leroux FR, Colobert F. Chem. Soc. Rev. 2015;44:3418–3430. doi: 10.1039/c5cs00012b. Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Angew. Chem. Int. Ed. 2005;44:5384–5427. doi: 10.1002/anie.200462661.
- 8.From this point forward, we will refer to 3-arylquinazolin-4(3H)-ones (1) as quinazolinones for simplicity.
- 9.CP-465022: Lazzaro JT, Paternain AV, Lerma J, Chenard BL, Ewing FE, Huang J, Welch WM, Ganong AH, Menniti FS. Neuropharmacology. 2002;42:143–153. doi: 10.1016/s0028-3908(01)00170-8. Benzomalivin A: Sun HH, Barrow CJ, Sedlock DM, Gillum AM, Cooper R. J. Antibiotics. 1994;47:515–522. doi: 10.7164/antibiotics.47.515. Sun HH, Barrow CJ, Cooper R. J. Nat. Prod. 1995;58:1575–1580. Erastin: Gangadhar NM, Stockwell BR. Curr. Opin. Chem. Biol. 2007;11:83–87. doi: 10.1016/j.cbpa.2006.11.033. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR. Nature. 2007;447:864–868. doi: 10.1038/nature05859.
- 10.(a) Gustafson JL, Lim D, Miller SJ. Science. 2010;328:1251–1255. doi: 10.1126/science.1188403. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Garand E, Kamrath MZ, Jordan PA, Wolk AB, McCoy AB, Miller SJ, Johnson MA. Science. 2012;335:694–698. doi: 10.1126/science.1214948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Barrett KT, Miller SJ. J. Am. Chem. Soc. 2013;135:2963–2966. doi: 10.1021/ja400082x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Barrett KT, Metrano AJ, Rablen PR, Miller SJ. Nature. 2014;509:71–75. doi: 10.1038/nature13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyaji R, Asano K, Matsubara S. J. Am. Chem. Soc. 2015;137:6766–6769. doi: 10.1021/jacs.5b04151. [DOI] [PubMed] [Google Scholar]
- 13.For examples of methods providing access to enantioen-riched 3-arylquinazolin-4(3H)-ones, please see: Tokitoh T, Kobayashi T, Nakada E, Inoue T, Yokoshima S, Takahashi H, Natsugari H. Heterocycles. 2006;70:93–99. Penhoat M, Bohn P, Dupas G, Papamicaël C, Marsais F, Levacher V. Tetrahedron: Asymmetry. 2006;17:281–286. Dai X, Wong A, Virgil SC. J. Org. Chem. 1998;63:2597–2600. doi: 10.1021/jo972105z.
- 14.(a) Azanli E, Rothchild R, Sapse A-M. Spectrosc. Lett. 2002;35:257–274. [Google Scholar]; (b) Colebrook LD, Giles HG. Can. J. Chem. 1975;53:3431–3434. [Google Scholar]; (c) Mannschreck A, Kaller H, Stühler G, Davies MA, Traber J. Eur. J. Med. Chem. Chim. Ther. 1984;19:381. [Google Scholar]
- 15.The reaction solvent was rigorously optimized in the early stages of this project. We found that 9:1 v/v PhMe/CHCl3 delivered higher enantioselectivities than pure PhMe, pure CHCl3, 9:1 v/v CHCl3/PhMe, and 9:1 v/v CHCl3/C6F6. Furthermore, the concentration of the reaction (0.01 M) was also optimized at an early stage. Dilution beyond 0.01 M served to decrease yield with no effect on enantioselectivity. Concentrations greater than 0.01 M provided lower er values, presumably due to an acceleration of the background rate.
- 16.For relevant reviews, see: Miller SJ. Acc. Chem. Res. 2004;37:601–610. doi: 10.1021/ar030061c. Jarvo ER, Miller SJ. Tetrahedron. 2002;58:2481–2495. For recent examples of β-turn peptides in enantioselective reactions, see: Metrano AJ, Miller SJ. J. Org. Chem. 2014;79:1542–1554. doi: 10.1021/jo402828f. Mbofana CT, Miller SJ. J Am. Chem. Soc. 2014;136:3285–3292. doi: 10.1021/ja412996f. Romney DK, Miller SJ. Org. Lett. 2012;14:1138–1141. doi: 10.1021/ol3000712. Peris G, Jakobsche CE, Miller SJ. J. Am. Chem. Soc. 2007;129:8710–8711. doi: 10.1021/ja073055a.
- 17.The bromination of 1a in the absence of catalysts provides < 5% yield of tribromide 2a. The majority of 1a is converted to a complex mixture of monobromides and dibromides, with the monobromides being favored. Please consult the “Mechanism-Driven Experiments” section and the Supporting Information for more details. Dissolution of the starting materials is also poor when no catalyst is present.
- 18.Only enantioselectivities were measured in the peptide-optimization studies summarized in Figure 2. Isolated yields were not assessed on small scale. Conversion of 1a was always near quantitative by crude 1H-NMR.
- 19.For examples of peptides taking advantage of a d-Pro-Aib β-turn sequence, see: Fowler BS, Mikochik PJ, Miller SJ. J. A. Chem. Soc. 2010;132:2870–2871. doi: 10.1021/ja9107897. Cowen BJ, Saunders LB, Miller SJ. J. Am. Chem. Soc. 2009;131:6105–6107. doi: 10.1021/ja901279m. See also ref. 11
- 20.Gilli P, Pretto L, Bertolasi V, Gilli G. Acc. Chem. Res. 2009;42:33–44. doi: 10.1021/ar800001k. [DOI] [PubMed] [Google Scholar]
- 21.(a) Haque TS, Little JC, Gellman SH. J. Am. Chem. Soc. 1996;118:6975–6985. [Google Scholar]; (b) Wilmot CM, Thornton JM. J. Mol. Biol. 1988;203:221–232. doi: 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]
- 22.Blank JT, Miller SJ. Biopolymers. 2006;84:38–47. doi: 10.1002/bip.20392. [DOI] [PubMed] [Google Scholar]
- 23.(a) Gellman SH, Dado GP, Liang G-B, Adams BR. J. Am. Chem. Soc. 1991;113:1164–1173. [Google Scholar]; (b) Yang D, Zhang D-W, Hao Y, Wu Y-D, Luo S-W, Zhu N-Y. Angew. Chem. Int. Ed. 2004;116:6887–6890. doi: 10.1002/anie.200454140. [DOI] [PubMed] [Google Scholar]; (c) Trabocchi A, Potenza D, Guarna A. Eur. J. Org. Chem. 2004:4621–4627. [Google Scholar]
- 24.CYLview was used to render the X-ray crystal data: CYL-view, 1.0b; Legault, C. Y., Université de Sherbrooke, 2009 (http://www.cylview.org).
- 25.DFT computations were performed using the Gaussian 09 suite: Gaussian 09, Revision C.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian, Inc. Wallingford CT: 2009. (b) In general, rotational barriers were derived from optimization of the stationary points of a relaxed torsional potential energy scan of the N–CAr anilide axis at the M06-2X/6-311++G(2d,3p)//B3LYP/6-31+G(d,p) level.26 Frequency calculations were performed at the B3LYP/6-31+G(d,p) level, and all transition states were found to have one, single imaginary frequency corresponding to a vibration along the N–CAr torsional reaction coordinate.
- 26. Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241. See also ref 11b
- 27.Reist M, Testa B, Carrupt P-A, Jung M, Schurig V. Chirality. 1995;7:396–400. [Google Scholar]
- 28.Slow addition was accomplished using a syringe pump. We found that using an 18 G needle avoided clogging via NBS precipitation over the 2.5 h slow addition. The rate of addition (1.60 mLmin−1), length of addition (2.5 h), and composition/volumes of delivery (3.5 mL of 9:1 v/v PhMe/CHCl3 with 0.5 mL acetone additive, 4 mL total) and source (6 mL of 9:1 v/v PhMe/CHCl3) solutions were rigorously optimized for the 0.10 mmol-scale reaction. Additional details on the optimization of the reaction conditions may be found in the Supporting Information.
- 29. McOmie JFW, Watts ML, West DE. Tetrahedron. 1968;24:2289–2292. (b) This reaction was performed on small-scale, and isolated yield was not obtained following crystallization.
- 30.Hirsch JA. Table of Conformational Energies. In: Allinger NL, Eliel EL, editors. Topics in Stereochemistry. Vol. 1. Hoboken, NJ: Wiley; 1967. pp. 199–222. [Google Scholar]
- 31.In the case of 2k, we hypothesized that the marked increase in enantioselectivity to 99:1 er (relative to 97:3 er in 2a, corresponding to ΔΔG‡ = 0.61 kcalmol−1 at 0 °C) may be due to (1) the Megroup being electron-releasing and thereby ring activating and (2) the Me-group blocking the position of first bromination in the uncatalyzed background reaction (see Mechanism-Driven Experiments section). In that way, the catalyzed reaction outcompetes the background reaction significantly. In the case of 2m, ring activation of the meta-Me-substituent provides an increase to 98:2 er relative to the parent, but the background pathway is still possible via bromination at the para-position. The results obtained for Cl-substituted 2j and 2l (both 96:4 er) might suggest that ring activation is important to the er of the reaction. Steric effects may also be at play in these instances.
- 32.(a) Keith JM, Larrow JF, Jacobsen EN. Adv. Synth. Catal. 2001;343:5–26. [Google Scholar]; (b) Kagan HB, Fiaud JC. Kinetic Resolution. In: Eliel EL, Wilen SH, editors. Topics in Stereochemistry. Vol. 18. Hoboken, NJ: Wiley; 1988. pp. 249–340. [Google Scholar]
- 33.Based purely on rotational barriers, many previously studied systems, including biaryls and benzamides, require two functionalization events (one at each ortho-position) to lock the asymmetric axis. See refs. 10–11.
- 34.The NMR studies were performed in C6D6 to simulate the bulk PhMe of the reaction conditions (Equation 2) without having two residual solvent lines in the spectrum. Even though the catalytic reaction is run at 0.001 M with respect to peptide 4q, we opted to perform our NMR studies at 0.01 M to obtain better signal-to-noise. We verified that 4q does not aggregate at 0.01 M solution by comparing 1H-NMR spectra of 4q at 0.001 M and 0.01 M under otherwise identical conditions. No change was observed in the proton signals at the higher concentration relative to the more dilute sample.
- 35.For a related example, please see: Denmark SE, Burk MT. Proc. Nat. Acad. Sci. U.S.A. 2010;107:20655–20660. doi: 10.1073/pnas.1005296107.
- 36.(a) Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NG. Protein NMR Spectroscopy: Principles and Practice. 3rd. Boston: Elsevier Academic Press; 2007. [Google Scholar]; (b) Abascal NC, Lichtor PA, Giuliano MW, Miller SJ. Chem. Sci. 2014;5:4504–4511. doi: 10.1039/C4SC01440E. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lichtor PA, Miller SJ. J. Am. Chem. Soc. 2014;136:5301–5308. doi: 10.1021/ja410567a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.For related examples, please see: Barrett KT, Miller SJ. Org. Lett. 2015;17:580–583. doi: 10.1021/ol503593y. Gustafson JL, Lim D, Barrett KT, Miller SJ. Angew. Chem. Int. Ed. 2011;50:5125–5129. doi: 10.1002/anie.201101147.
- 38.For an example of reductive dehalogenation of aryl bromides using Pd/C, please see: Ramanathan A, Jiminez LS. Synthesis. 2010:217–220. For an example of a regioselective dehalogenation of aryl bromides, please see: Chae J, Buchwald SL. J. Org. Chem. 2004;69:3336–3339. doi: 10.1021/jo035819k.
- 39.For examples of Suzuki-Miyaura cross-coupling of aryl halides in the context of quinazolinone scaffolds, please see: Garlapati R, Pottabathini N, Gurram V, Kasani KS, Gundla R, Thulluri C, Machiraju PK, Chaudhary AB, Addepally U, Dayam R, Chunduri VR, Patro B. Org. Biomol. Chem. 2013;11:4778–4791. doi: 10.1039/c3ob40636a. Fang X, Chen YT, Sessions EH, Chowdhurym S, Vojkovsky T, Yin Y, Pocas JR, Grant W, Schröter T, Lin L, Ruiz C, Cameron MD, LoGrasso P, Bannister TD, Feng Y. Bioorg. Med. Chem. Lett. 2011;21:1844–1848. doi: 10.1016/j.bmcl.2011.01.039. Li H-y, Wang Y, McMillen WT, Chaterjee A, Toth JE, Mundla SR, Voss M, Boyer RD, Sawyer JS. Tetrahedron. 2007;63:11763–11770.
- 40.For examples of Buchwald-Hartwig amination reactions of aryl bromides, please see: Driver MS, Hartwig JF. J. Am. Chem. Soc. 1996;118:7217–7218. Wolfe JP, Wagaw S, Buchwald SL. J. Am. Chem. Soc. 1996;118:7215–7216.
- 41.The computed half-life to enantiomerization in monobromide 3a is 27 y at 80 °C,25 which perhaps suggests that the species that undergoes racemization under the attempted amination conditions is not monobromide Me-3a. It is possible that the arylpalladium species formed upon oxidative addition has a lower barrier to enantiomerization by way of a significantly lengthened bond.
- 42.The transitions states to enantiomerization of 2a’ were never found explicitly using the computational methods described in ref. 25. The reported barrier of > 50 kcalmol−1 is derived from the torsional scan computation (see Supporting Information for details).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.