

Abstract
Key points
Candesartan, an inverse agonist of the type 1 angiotensin II receptor (AT1R), causes a concentration‐dependent inhibition of pressure‐dependent myogenic tone consistent with previous reports of mechanosensitivity of this G protein‐coupled receptor.
Mechanoactivation of the AT1R occurs independently of local angiotensin II production and the type 2 angiotensin receptor.
Mechanoactivation of the AT1R stimulates actin polymerization by a protein kinase C‐dependent mechanism, but independently of a change in intracellular Ca2+.
Using atomic force microscopy, changes in single vascular smooth muscle cell cortical actin are observed to remodel following mechanoactivation of the AT1R.
Abstract
The Gq/11 protein‐coupled angiotensin II type 1 receptor (AT1R) has been shown to be activated by mechanical stimuli. In the vascular system, evidence supports the AT1R being a mechanosensor that contributes to arteriolar myogenic constriction. The aim of this study was to determine if AT1R mechanoactivation affects myogenic constriction in skeletal muscle arterioles and to determine underlying cellular mechanisms. Using pressure myography to study rat isolated first‐order cremaster muscle arterioles the AT1R inhibitor candesartan (10−7–10−5 m) showed partial but concentration‐dependent inhibition of myogenic reactivity. Inhibition was demonstrated by a rightward shift in the pressure–diameter relationship over the intraluminal pressure range, 30–110 mmHg. Pressure‐induced changes in global vascular smooth muscle intracellular Ca2+ (using Fura‐2) were similar in the absence or presence of candesartan, indicating that AT1R‐mediated myogenic constriction relies on Ca2+‐independent downstream signalling. The diacylglycerol analogue 1‐oleoyl‐2‐acetyl‐sn‐glycerol (OAG) reversed the inhibitory effect of candesartan, while this rescue effect was prevented by the protein kinase C (PKC) inhibitor GF 109203X. Both candesartan and PKC inhibition caused increased G‐actin levels, as determined by Western blotting of vessel lysates, supporting involvement of cytoskeletal remodelling. At the single vascular smooth muscle cell level, atomic force microscopy showed that cell swelling (stretch) with hypotonic buffer also caused thickening of cortical actin fibres and this was blocked by candesartan. Collectively, the present studies support growing evidence for novel modes of activation of the AT1R in arterioles and suggest that mechanically activated AT1R generates diacylglycerol, which in turn activates PKC which induces the actin cytoskeleton reorganization that is required for pressure‐induced vasoconstriction.
Keywords: actin stress fibres, cytoskeleton, mechanotransduction, myogenic response, protein kinase C, type 1 angiotensin II receptor, vascular smooth muscle cell signalling
Key points
Candesartan, an inverse agonist of the type 1 angiotensin II receptor (AT1R), causes a concentration‐dependent inhibition of pressure‐dependent myogenic tone consistent with previous reports of mechanosensitivity of this G protein‐coupled receptor.
Mechanoactivation of the AT1R occurs independently of local angiotensin II production and the type 2 angiotensin receptor.
Mechanoactivation of the AT1R stimulates actin polymerization by a protein kinase C‐dependent mechanism, but independently of a change in intracellular Ca2+.
Using atomic force microscopy, changes in single vascular smooth muscle cell cortical actin are observed to remodel following mechanoactivation of the AT1R.
Abbreviations
- AFM
atomic force microscopy
- Ang II
angiotensin II
- AT1R
angiotensin II type 1 receptor
- [Ca2+]i
intracellular Ca2+
- DAG
diacylglycerol
- DMSO
dimethyl sulfoxide
- GPCRs
G protein‐coupled receptors
- HSP‐27
heat shock protein‐27
- IP3
inositol trisphosphate
- MLCK
myosin light chain kinase
- OAG
1‐oleoyl‐2‐acetyl‐sn‐glycerol
- PIP2
phosphatidylinositol 4,5‐bisphosphate
- PLC
phospholipase C
- PKC
protein kinase C
- SR
sarcoplasmic reticulum
- TRP
transient receptor potential
- VDCC
voltage‐dependent Ca2+ channel
- VSMCs
vascular smooth muscle cells
Introduction
Resistance arteries and arterioles exhibit mechanosensitivity by vasoconstricting and dilating in response to increases and decreases in intravascular pressure (myogenic response), respectively. The myogenic response is an inherent property of vascular smooth muscle cells (VSMCs). It occurs independently of endothelial cells or innervation and functions to regulate peripheral resistance and blood pressure while protecting capillaries from damage due to high intraluminal pressure (Davis & Hill, 1999; Hill et al. 2003). Importantly, as abnormalities in pressure‐induced vasoconstriction are often found in pathophysiological conditions including systemic hypertension, vasospasm, stroke, or diabetes (Hill et al. 2009), considerable attention has been given to understanding mechanisms by which VSMCs detect changes in intraluminal pressure and how this subsequently leads to an appropriate vasomotor response.
Signalling events including VSMC membrane depolarization, Ca2+ mobilization via voltage‐dependent Ca2+ channels, inhibition of an outward K+ conductance, and phosphorylation of myosin light chain in VSMCs have been shown to contribute to pressure‐induced vasoconstriction (Davis et al. 1992; Hill & Meininger, 1994; Knot & Nelson, 1998; Davis & Hill, 1999; Hill et al. 2001, 2010; Kotecha & Hill, 2005). However, less is known regarding exactly how VSMCs detect mechanical stresses and in turn initiate critical steps for myogenic constriction such as depolarization and Ca2+ entry. A number of VSMC elements/proteins have been proposed as mechanosensors including ion channels, VSMC‐extracellular matrix adhesion through integrins, cell–cell adhesion through cadherins, cytoskeletal components, and membrane‐bound proteins (Martinez‐Lemus et al. 2003; Sun et al. 2008, 2012; Jackson et al. 2010; Schwartz, 2010; Mederos y Schnitzler et al. 2011; Hill & Meininger, 2012). Considerable support has been given to members of the superfamily of transient receptor potential canonical (TRPC) channels (for example TRPC6) acting as mechanosensors in VSMCs (Spassova et al. 2006; Kim et al. 2013; Gonzales et al. 2014). However, studies have demonstrated that TRPC6 may not be the primary mechanotransduction event, as pressure‐induced vasoconstriction is observed in TRPC6 channel‐deficient mesenteric arteries (Schleifenbaum et al. 2014). As a result of such observations attention has turned to the role of additional mechanosensing mechanisms underlying myogenic constriction.
G protein‐coupled receptors (GPCRs) are the largest class of integral membrane proteins and are involved in signal transduction by hormones, neurotransmitters, or mechanical stimuli to intracellular signalling pathways. Of the characterized GPCRs, the angiotensin II type 1 receptor (AT1R), in particular, has been suggested to be mechanosensitive in both cardiac myocytes (Zou et al. 2004) and arteriolar VSMCs (Mederos y Schnitzler et al. 2008; Blodow et al. 2014; Schleifenbaum et al. 2014). Furthermore, overexpression of the AT1R in aortic VSMCs (generally considered mechano‐insensitive) confers mechanosensitivity to those cells (Mederos y Schnitzler et al. 2008). Notably, mechanical activation of the AT1R occurs independently of its classical agonist, angiotensin II (Ang II), consistent with a direct effect on receptor conformation (Zou et al. 2004).
Mechanosensitivity of GPCRs in small arteries does not appear to be limited to the AT1R. Thus, P2Y4 and P2Y6 purinergic receptors have been shown to mediate myogenic constriction in cerebral parenchymal arterioles (Brayden et al. 2013) while cysteinyl leukotriene 1 receptors have been reported to be mechanoactivated in mesenteric vessels from wild‐type and mice lacking both AT1a and AT1b receptors (Storch et al. 2015). However, as other studies have reported that knockout of the AT1R in mouse mesenteric vessels abolishes myogenic reactivity (Schleifenbaum et al. 2014), it appears that the contribution of particular GPCRs may differ between tissues or that methodological differences impact the studies.
Consistent with a role for GPCRs as vascular mechanosensors, myogenic tone and reactivity were previously reported to couple to phospholipase C (PLC) and G protein activation in small cerebral arteries (Osol et al. 1993). More recently, GPCR‐mediated activation of PLCγ1 has been suggested to lead to Ca2+ influx via TRPC6 channels, subsequently causing inositol trisphosphate (IP3)‐dependent Ca2+ release from the sarcoplasmic reticulum, at a local level, that activates transient receptor potential melastatin 4 (TRPM4) channels to initiate membrane depolarization and myogenic constriction (Gonzales et al. 2014). However, studies have reported that TRPM4−/− mice show myogenic responses despite deletion of this channel (Mathar et al. 2010). Further, myogenic constriction has been suggested to also involve mechanisms independent of membrane potential‐driven changes in intracellular Ca2+ and myosin light chain phosphorylation. For example, disruption or stabilization of actin cytoskeleton proteins results in decreased or increased myogenic constriction, respectively (Cipolla & Osol, 1998; Cipolla et al. 2002). From a biochemical perspective, remodelling of the actin cytoskeleton during myogenic constriction appears to be regulated by protein kinase C (PKC) and the heat shock protein‐27 (HSP‐27) (Moreno‐Dominguez et al. 2013, 2014; El‐Yazbi et al. 2015). As ligand‐dependent activation of the AT1R results in diacylglycerol (DAG) and PKC‐dependent vasoconstriction (Wynne et al. 2009), it is likely that mechanoactivation of the AT1R activates this pathway during myogenic constriction. However, whether mechanoactivation of the AT1R and subsequent myogenic constriction involve these intracellular signalling mechanisms is yet to be defined.
The aim of the present studies was to first confirm that the AT1R can be activated independently of Ang II and that this contributes to myogenic constriction in skeletal muscle arterioles. Secondly, the present study also sought to define mechanisms underlying how AT1R‐mechanoactivation by intraluminal pressure leads to myogenic constriction. It was found that pharmacological blockade of the AT1R caused a partial and concentration‐dependent inhibition of myogenic reactivity as shown by a rightward shift in the pressure–diameter relationship. Further, inhibition of myogenic constriction by candesartan was (1) reversed by membrane permeable DAG, which in turn was abrogated by PKC inhibition, and (2) associated with alterations to the actin cytoskeleton. Thus, mechanoactivation of the AT1R appears to induce generation of DAG and subsequently arteriolar myogenic constriction that results, in part, from PKC‐dependent actin cytoskeleton rearrangement.
Methods
Ethical approval and animals
Studies used male Sprague‐Dawley rats aged 6–8 weeks, with body weight between 150 and 250g. This age of animals was chosen to be consistent with our earlier studies and as these vessels show robust in vitro myogenic responses (Zou et al. 1995; Hill et al. 2000; Kotecha & Hill, 2005). Animals were housed in a temperature, humidity and light controlled animal facility and were allowed free access to a standard rat chow and water. Rats were anaesthetized with sodium pentobarbital (Nembutal, 100 mg (kg body weight)−1) given by intraperitoneal injection. All experiments were terminal in nature with killing occurring by cardiac arrest and exsanguination while under anaesthesia. Protocols were approved by the Animal Care and Use Committee of the University of Missouri‐Columbia (USA).
Vessel isolation
The left or right cremaster muscle was excised from anaesthetized rats and placed in a cooled chamber (4°C) containing dissection solution (in mm): NaCl 140; KCl 5.6; MgSO4 1; NaH2PO4 1.2; EDTA 0.02; sodium pyruvate 2; glucose 5; Mops 3; albumin 0.1mg ml−1 (USB Corporation, Cleveland, OH, USA). Segments of the first order arteriole (1A) were dissected as previously described (Kotecha & Hill, 2005) and subsequently used for vessel myography or biochemical studies.
Vessel myography
Segments of 1A arteriole (approximately 1–2 mm in length) were cannulated and secured onto glass micropipettes with 11‐0 monofilament suture and positioned in a custom‐designed superfusion chamber. The cannulation stage was placed on an inverted microscope and the arterioles were continually superfused (4 ml min−1) with Krebs buffer (in mm: NaCl 111; NaHCO3 25.7; KCl 4.9; CaCl2 2.5; MgSO4 1.2; KH2PO4 1.2; glucose 11.5; Hepes 10; pH of the buffer was adjusted to 7.3 with NaOH at room temperature). Arterioles were initially pressurized to 70 mmHg (in the absence of luminal flow) by connecting the inflow pipettes to a height‐adjustable fluid reservoir (the outflow pipette was closed using a 3‐way stopcock). Length of the cannulated arterioles was set, as previously described (Kotecha & Hill, 2005), by stretching the vessels until lateral bowing did not occur during changes in intraluminal pressure (30–110 mmHg). The temperature of the superfusate was adjusted to 33–34°C and the vessels allowed to develop spontaneous myogenic tone at 70 mmHg (90 ± 2 μm, n = 77; approximately 40–50% of maximal passive diameter (170 ± 2 μm, n = 77)) during a 60‐min equilibration period. Changes in intraluminal diameter in response to alteration of intraluminal pressures were monitored using video microscopy and a video‐based calliper system. Intraluminal pressures were increased from 10 to 110 mmHg in a stepwise manner (at 5 min intervals) to determine pressure–diameter relationships. For the various experimental protocols, Krebs buffer (control), AT1R blockers (losartan, 10−6 m–10−5 m; candesartan, 10−7 m–10−5 m; EXP3174, 10−6 m–10−5 m; olmesartan, 10−6 m–10−5 m), the angiotensin II type 2 receptor (AT2R) antagonist (PD‐123,319, 10−6 m), the angiotensin‐converting enzyme inhibitor (captopril, 10−6 m), and the PKC inhibitor (GF 109203X, 3×10−6 m) were superfused into the vessel chamber. The membrane permeable DAG analogue OAG (10−7 m–10−5 m) was directly applied into the bath containing the pressurized arterioles. At the end of all pressure myography experiments, vessels were superfused with 0 mm Ca2+ Krebs buffer containing 2 mm EGTA and maximal passive diameters at each pressure step were recorded. Vessel diameters were normalized to the passive diameter at 70 mmHg (%d 70passive). Myogenic constriction was calculated as the decrease in diameter from passive diameter at each pressure: myogenic constriction (%) = [(passive diameter − constricted diameter)/passive diameter] × 100.
Measurement of intracellular global [Ca2+]i in pressurized rat skeletal muscle arterioles
Cannulated and pressurized cremaster arterioles were incubated abluminally with Fura 2‐acetoxymethyl ester (Fura 2‐AM, 2×10−6 m, Molecular Probes, Eugene, OR, USA) for 60 min at room temperature in a buffer solution containing 0.5% dimethyl sulfoxide (DMSO) and 0.01% pluronic F‐127 as previously described (Hill et al. 2000). At completion of dye loading, vessels were washed by superfusion with fresh Krebs buffer (34°C) for 30 min. Fura‐2‐loaded arterioles were exposed to alternating excitation wavelengths (340 and 380 nm) using a computer‐controlled, motorized filter wheel and a 75 W xenon light source. Fluorescence emission (510 nm) was detected and quantified using a photomultiplier‐based system (IonOptix LLC, Milton, MA, USA). The photometry system was set at a sampling rate of 10 Hz for collection of Ca2+ data. Fluorescent intensity following changes in intraluminal pressures was measured in the absence or presence of candesartan (10−5 m) and was expressed as the 340:380 nm ratio for quantification of changes in [Ca2+]i in arteriolar wall VSMCs. To measure vessel diameter simultaneously with Ca2+ ratios, the vessel was transilluminated with near‐infrared light and a dichroic mirror directed the image to a video‐based calliper system connected to a charge‐coupled device camera. Ca2+ ratio and diameter measurements were normalized to passive Ca2+ ratio and diameter at 70 mmHg, respectively.
Isolation of cremaster arteriolar myocytes and whole‐cell patch clamp
VSMCs were freshly isolated from cremaster muscle arterioles using a previously enzymatic described enzymatic technique (Yang et al. 2009). Whole‐cell L‐type voltage‐dependent Ca2+ channel (VDCC) currents were determined using standard whole‐cell voltage clamp techniques as described previously (Mueller et al. 2015). Data acquisition was performed using an EPC‐10 USB amplifier and Patchmaster software (HEKA, Lambrecht, Germany). Cells were superfused with a physiological salt solution (PSS; in mm, 1 CaCl2, 138 NaCl, 1 MgCl2, 5 KCl, 10 Hepes, 10 glucose, pH 7.4) during gigaseal formation and then switched to PSS with tetraethylammonium chloride (TEA‐Cl) substituted for NaCl and 1 mm Ca2+, 20 mm Ba2+ as the charge carrier. Heat‐polished glass pipettes (2–5 MΩ) were filled with a solution containing (mm): 120 CsCl, 10 TEA‐Cl, 1 MgCl2, 20 Hepes, 2 MgATP, 5 EGTA and 0·5 Na.GTP. Whole‐cell currents were digitized at 500 μs intervals. Current densities (pA pF−1) were obtained for each cell by normalization of whole‐cell current to cell capacitance. Voltage stimuli (duration = 400 ms) were applied across the range, −60 to +50 mV, from a holding potential of −70 mV. Leak subtraction was not performed. Cells were continuously perfused using Automate Scientific perfusion system (1ml min−1 flow) with Bay K8644 (2×10−6 m), candesartan (10−5 m) and nifedipine (10−6 m) added as required. All experiments were conducted at room temperature (22–25°C). I–V curves were plotted and presented as group data.
Western blot measurement of G‐actin content in pressurized rat skeletal muscle arterioles
Methods for G‐actin measurement were modified from those used previously (Moreno‐Dominguez et al. 2013, 2014). Briefly 1A arterioles were pressurized to 20 or 70 mmHg in the absence or presence of candesartan (10−5 m) and then frozen with acetone (–70°C) cooled by a dry ice–ethanol bath. Cold acetone was directly applied to the cannulation chamber containing cremaster arterioles. The frozen vessels were removed from glass pipettes, and the cannulated ends cut from the central portion of the vessel and discarded. Each sample was stored in a 1.5 ml tube at –80°C prior to SDS‐PAGE and Western blotting. For actin measurements LAS2 (lysis and stabilization) buffer including 1 ml lysis and F‐actin stabilization buffer, 10 μl ATP (100 mm), 10 μl 100× protease inhibitor cocktail stock (actin assay kit, Cytoskeleton Inc., Denver, CO, USA) was prepared in advance. Frozen arterioles were homogenized in 5 μl LAS2 buffer and incubated with 90 μl LAS2 buffer for 20 min at room temperature; 80 μl of the homogenate was then transferred to an ultracentrifuge tube and centrifuged at 100,000 × g for 1 h at 22°C to separate G‐ and F‐actin and cellular debris. The supernatant containing G‐actin was transferred to another tube and stored at –80°C. For electrophoresis, G‐actin samples were mixed with an equivalent volume of Laemmli sample buffer (Bio‐Rad, Hercules, CA, USA) and heated at 95°C for 10 min. The protein samples were kept at room temperature for 10–15 min and loaded onto 10% SDS‐PAGE gels for separation by electrophoresis at 80 V for 2 h using a Mini Protean 3 Cell (Bio‐Rad). After electrophoresis, the protein samples were transferred to a polyvinylidene difluoride (PVDF) membrane (Bio‐Rad) at 0.15–0.2A for 2 h using a Mini Trans‐Blot Electrophoretic Transfer Cell (Bio‐Rad). Membranes were washed with Tris‐buffered saline buffer and 0.05% Tween 20 (TBST) twice for 5 min. Membranes were then blocked with 5% non‐fat dry milk in 0.05% TBST buffer at room temperature for 1 h and then cut at a molecular weight of 35 kDa. Proteins on the upper or lower membrane were detected with mouse monoclonal anti‐actin (1:5000; Sigma‐Aldrich, St Louis, MO, USA) or goat polyclonal anti‐SM22α (1:2000; Novus Biologicals, Littleton, CO, USA) antibodies, respectively. After overnight blotting at 4°C in 1% dry milk in 0.05% TBST, the membranes were washed in 0.05% TBST three times for 15 min and incubated with anti‐mouse IgG‐horseradish peroxidase‐conjugated secondary antibody (1:10,000; Sigma‐Aldrich) or anti‐goat IgG‐horseradish peroxidase‐conjugated secondary antibody (1:5000; R&D Systems, Minneapolis, MN, USA), respectively, in 1% dry milk and 0.05% TBST at room temperature for 1 h. The membranes were washed and Dura Chemiluminescence Detection Reagent (Thermo Fisher Scientific, Waltham, MA, USA) incubated with the upper or lower membranes for 2 and 5 min, respectively. A Bio‐Rad Chemi‐DOC XRS digital system and ImageLab software (Bio‐Rad) were used to quantify chemiluminescence signals. G‐actin content was normalized to expression of SM‐22α.
Isolation of cremaster arteriolar myocytes and cell culture
Freshly isolated arteriolar myocytes were prepared as previously described (Yang et al. 2009). Segments of cremaster arterioles were incubated with low‐Ca2+ PSS containing 26 U ml−1 papain and 1 mg ml−1 dithiothreitol at 37°C for 30 min along with intermittent agitation and transferred to low‐Ca2+ PSS containing 1.95 U ml−1 type H collagenase (Sigma‐Aldrich), 1 mg ml−1 soybean trypsin inhibitor (Sigma‐Aldrich), and 75 U ml−1 elastase (Elastin Products Company, Owensville, MO, USA) for incubation (8–10 min at 37°C.). After further digestion, the remaining segments of cremaster arterioles were washed 2–3 times with low‐Ca2+ PSS and triturated gently using a fire‐polished Pasteur pipette to release single cells. Isolated arteriolar myocytes were cultured in conditional medium (DMEM/F‐12) supplemented with 20% fetal bovine serum (FBS, Atlanta Biologicals, Lawrenceville, GA, USA), 1% Hepes, 1% l‐glutamine, 1% sodium pyruvate, 1% penicillin, and 1% streptomycin for 3–5 days without passage prior to cell scanning experiments using an atomic force microscopy (AFM). Arteriolar myocytes were placed in 60 mm tissue culture dishes and kept in a humidified and water‐jacked incubator with 5 % CO2 at 37°C. Cells were serum‐starved for 12 h prior to the AFM experiments to eliminate any effects of Ang II in the 20% serum medium.
Visualization and quantification of cortical actin fibres using atomic force microscopy (AFM)
An Asylum AFM System (Model MFP‐3D‐BIO, Asylum Research, Santa Barbara, CA, USA) was used in contact mode to obtain the live cell topography images (i.e. deflection images) of randomly selected arteriolar myocytes as described previously (Hong et al. 2014). The AFM system was positioned on an inverted microscope (Model IX81, Olympus America Inc.). AFM measurements were performed at room temperature (approximately 25°C) in serum‐free medium and cells were incubated in the absence or presence of hypotonic buffer (200 mosmol l−1, 30 min) and/or candesartan (10−5 m, 1 h). The size of the cell surface area involved in cell scanning experiments was 60 × 60 μm and the digital density of the scanned area was 512 × 512 pixels. A stylus type AFM probe (Model: MLCT‐C, k = 15 pN nm−1, Bruker, Santa Barbara, CA, USA) was employed to conduct the AFM imaging at 0.2 Hz frequency with a 300—500 pN tracking force at room temperature. Cultured cremaster arteriolar VSMCs were used at passage zero and treated with serum‐free medium or hypotonic solutions in the absence or presence of 10−5 m candesartan.
AFM images were analysed with custom‐developed software using previously described methods (Hong et al. 2014). Briefly, the stress fibre area fraction was evaluated by computing the ratio of pixel number of the stress fibre to the total pixel number of the cell mask. To determine stress fibre orientation the AFM deflection image was subdivided into 2 × 2 pixel size sub‐regions and the orientation of each sub‐region was evaluated by computing the gradient magnitude using the horizontal and vertical spatial gradient of each pixel (Karlon et al. 1999). The dominant orientation of stress fibre was the fibre angles that fall in the range of ±20 deg in the histogram of the normalized orientations of the sub‐regions in the entire image.
Phalloidin staining of actin fibres
A subset of VSMCs used for AFM studies were fixed with 4% paraformaldehyde for 20 min at room temperature, followed by 2× incubation in glycine buffer (0.1 m glycine) for 10 min. The cells was then incubated with Alexa 488–phalloidin (1:200 dilution in antibody buffer (NaCl 0.1 m, sodium citrate 7.5 mm, Triton X‐100 0.1%, BSA 1%) for 1 h at room temperature, followed by washing (4×) with antibody dilution buffer. The cells were imaged using an Olympus FV1000 confocal microscope, through a ×60 oil objective at 0.2 μm step size. The z‐stack images were then summated to provide an image of the actin cytoskeleton.
RNA purification from intact skeletal muscle arterioles and real‐time quantitative polymerase chain reaction (PCR)
As described previously (Nourian et al. 2014; Lee et al. 2015), total RNA was extracted from 1A arterioles using the Arcturus PicoPure RNA isolation kit (Applied Biosystems, Carlsbad, CA, USA) and on‐column DNase I (Qiagen, Valencia, CA, USA) to minimize contamination from genomic DNA. Prepared RNA was then used for cDNA synthesis via reverse transcription using random hexamer and oligod (T) primers, and SuperScript III First‐Strand Synthesis System (Life Technologies, Grand Island, NY, USA). Real‐time quantitative PCR was conducted in duplicate using KAPA SYBER FAST qPCR Kit Master mix (KAPA Biosystems, Woburn, MA, USA). PCR was performed using a Mastercycler EP Realplex 2 thermocycler (Eppendorf‐North America, Westbury, NY, USA) to detect mRNA expression of angiotensin II type 1a receptor (AT1aR), type 1b receptor (AT1bR), and type 2 receptor (AT2R) in the 1A arterioles. PCR reactions (20 μl) contained 10 μl of the KAPA SYBR Mix, 1 μl of forward and reverse primers, 7 μl of DNase‐free water, and 1 μl of first‐strand cDNA as a template. For each real‐time PCR determination, a negative control without template cDNA was included. The forward and reverse primers used for AT1aR, AT1bR, and AT2R were pre‐designed and purchased from Integrated DNA Technologies (Coralville, IA, USA). The pre‐designed primers were intron‐spanning and covered multiple exons (2–3 exons) to prevent amplification of genomic DNA. As an internal control, β‐actin was selected as a representative housekeeping gene. Primers used for real‐time PCR and amplification efficiencies between target and housekeeping genes are shown in Table 1. The PCR thermal programme consisted of polymerase activation at 95°C for 2 min, 42 cycles of denaturation at 95°C for 3 s, annealing, and extension at 56°C for 30 s. A melting curve was performed to confirm specificity of the amplified products. The comparative threshold (C t) method (2−∆∆Ct; Livak & Schmittgen, 2001) was used to evaluate relative expression levels. Expression levels were then normalized to AT1aR expression.
Table 1.
Properties of primers used for real‐time quantitative PCR
| Target gene | Accession number | Primer sequence | Amplicon length | Amplification efficiency |
|---|---|---|---|---|
| AT1aR | NM_030985 | F: 5′‐CACTATTCGAAATCCACTTGACC‐3′ | 129 bp | 1.97 |
| R: 5′‐CTCTCAGCTCTGCCACATTC‐3′ | ||||
| AT1bR | NM_031009 | F: 5′‐TGCTCTCTGACACTATTTAAAATGC‐3′ | 114 bp | 1.99 |
| R: 5′‐GACACACACAGCCTTTCCA‐3′ | ||||
| AT2R | NM_012494 | F: 5′‐GCAGAAACATCACCAGCAGTC‐3′ | 97 bp | 1.99 |
| R: 5′‐GCAGAAACATCACCAGCAGTC‐3′ | ||||
| β‐actin | NM_031144 | F: 5′‐CCTCTATGCCAACACAGTGCTGTCT‐3′ | 128 bp | 1.99 |
| R: 5′‐GCTCAGGAGGAGCAATGATCTTGA‐3′ |
Chemicals
Candesartan and GF 109203X were purchased from Selleckchem (Houston, TX, USA) and Enzo Life Science (Farmingdale, NY, USA), respectively. Olmesartan and EXP3174 were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Losartan, PD‐123,319, and captopril were purchased from Sigma‐Aldrich. OAG was obtained from Cayman (Ann Arbor, MI, USA). Unless stated general chemicals and reagents were purchased from Sigma‐Aldrich. Chemical agents were dissolved in physiological salt solutions or DMSO on the basis of their solubility.
Statistical analysis
All data are presented in graphical form using Prism GraphPad software (La Jolla, CA, USA) and expressed as means ± SEM. Statistical analyses were conducted using either IBM SPSS Statistics 22 or Prism GraphPad. Student's t test was used for comparison of data obtained from two different treatments at constant intraluminal pressures. One‐way ANOVA, with or without repeated measures, was performed to analyse statistical differences between repetitively measured data or where there were more than three different treatments, respectively. If significant differences were shown by ANOVA, a Bonferroni correction was used for post hoc testing. A P value of <0.05 was considered as statistically significant.
Results
Expression of mRNA for AT1aR, AT1bR and AT2R in 1A cremaster muscle arterioles
qPCR was performed to confirm expression of ATR subtypes in cremaster 1A. As shown Fig. 1, while all three subtypes (AT1aR, AT1bR and AT2R) were evident at the mRNA level, the predominant species was the AT1aR.
Figure 1. Expression of the AT1R subtypes in cremaster muscle arterioles .
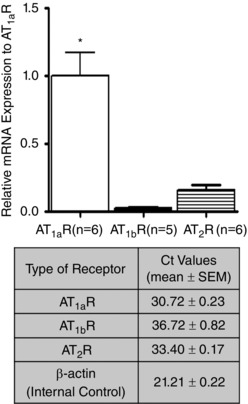
Measurements were performed on RNA extracted from whole vessel segment homogenates. Quantitative PCR was performed according to procedures described under Methods and results normalized relative to mRNA expression levels for the AT1aR. * P < 0.05 vs. AT1bR or AT2R. Results are presented as means ± SEM.
Inhibition of AT1R mechanoactivation attenuates pressure‐induced vasoconstriction in skeletal muscle arterioles
Initial pharmacological studies were performed using the AT1R antagonist losartan. Ang II concentration–response curves and pressure–diameter relationships were studied in rat cremaster arterioles in the absence or presence of losartan to determine its inhibitory effects on vasoconstriction following both ligand‐dependent and ‐independent activation of the AT1R (Fig. 2 A–D). At a concentration of 10−6 m, losartan significantly blunted Ang II‐dependent vasoconstriction (Fig. 2 A and B), whereas the antagonist had only a modest effect on steady‐state myogenic tone causing dilatation at only 50 mmHg at a concentration of 10−5 m (Fig. 2 C and D).
Figure 2. Inhibitory effects of losartan on Ang II‐mediated vasoconstriction .
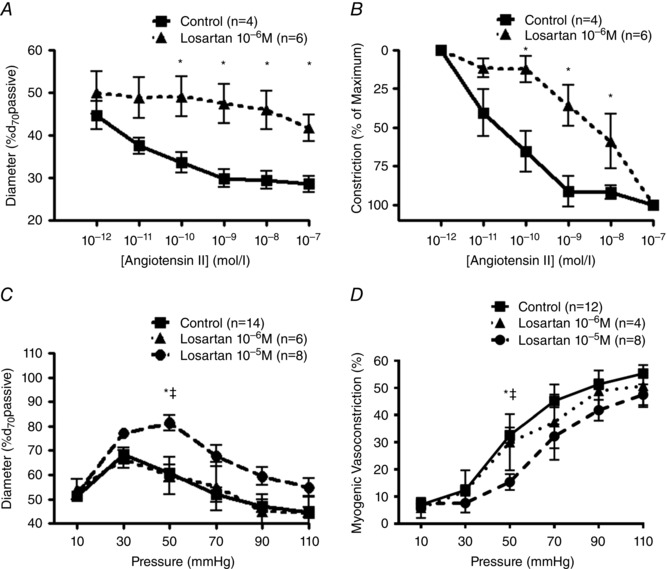
A, Ang II–dose response curves of rat cremaster arterioles pressurized to 70 mmHg were measured in the absence (n = 4) or presence of losartan 10−6 m (n = 6). Diameters observed at each concentration were normalized to passive diameter at 70 mmHg (%d 70passive). * P < 0.05 vs. control. B, data from panel A presented as percentage maximal constriction. C and D, pressure–diameter relationships and calculated myogenic constriction (%) in the absence or presence of losartan (10−6 m–10−5 m). Intraluminal pressure was increased from 10 to 110 mmHg in 20 mmHg steps. Diameters observed at each pressure were normalized to passive diameter at 70 mmHg (%d 70passive; C) and myogenic constriction calculated (Panel D) as myogenic constriction (%) = [(passive diameter − constricted diameter)/passive diameter] × 100. * P < 0.05 vs. 10−6 m; ‡ P < 0.05 vs. 10−5 m. Group data are presented as means ± SEM.
As it has been reported that losartan inhibits Ang II‐induced, but not mechanical stress‐mediated, secretion of vascular endothelial growth factor protein (Gruden et al. 1999) due to the antagonist not possessing carboxyl or hydroxyl functional groups that are requisite for inverse agonist function (Milligan, 2003; Akazawa et al. 2009), additional studies were performed in the presence of candesartan. Candesartan (10−7–10−5 m), with a carboxyl group in the benzimidazole ring, showed a partial and concentration‐dependent inhibition of myogenic reactivity, as indicated by a significant rightward shift in the pressure–diameter relationship and myogenic constriction (Fig. 3 A and B). Alternative AT1R inverse agonists olmesartan (including both hydroxyl and carboxyl groups) and EXP3174 (containing a carboxyl group, but structurally similar to losartan) both significantly diminished pressure‐induced vasoconstriction (Fig. 3 C and D). Appropriate time controls showed that myogenic responses were stable over the time period of the study (not shown). Based on these results candesartan was chosen as the AT1R blocker for the following studies.
Figure 3. Inverse agonists of the AT1R inhibit AT1R mechanoactivation‐dependent vasoconstriction .
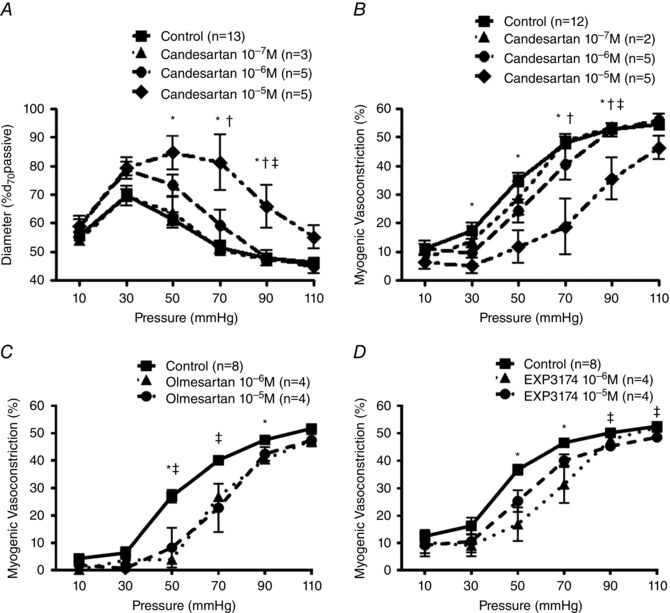
A and B, pressure–diameter relationships were measured in rat cremaster arterioles (A) and myogenic constriction (%) was calculated (B) in the absence or presence of candesartan (10−7 m–10−5 m). Myogenic reactivity was first measured in the absence of candesartan. After 1 h incubation of the inverse agonist via a superfusion system, myogenic responsiveness for the same arterioles was repeated (control, n = 13; 10−7 m, n = 3; 10−6 m, n = 5; 10−5 m, n = 5). Diameters observed at each pressure were normalized to passive diameter at 70 mmHg (%d 70passive; A). Myogenic constriction (%) was calculated as follows (B; control, n = 12; 10−7 m, n = 2; 10−6 m, n = 5; 10−5 m, n = 5): myogenic constriction (%) = [(passive diameter − constricted diameter)/passive diameter] × 100. * P < 0.05 control vs. 10−5 m; † P < 0.05 10−7 m vs. 10−5 m; ‡ P < 0.05 10−6 m vs. 10−5 m. C and D, pressure–diameter relationships in the absence or presence of olmesartan (10−6 m‒10−5 m; C) or EXP3174 (10−6 m‒10−5 m; D). Myogenic constriction was calculated as described above. * P < 0.05 control vs. 10−6 m; ‡ P < 0.05 control vs. 10−5 m. Group data are presented as means ± SEM.
Reduced myogenic reactivity is not a result of AT2R‐mediated vasodilatation or inhibition of locally generated Ang II
Mechanical stretch leads to local production of Ang II from cardiac myocytes, with this endogenous Ang II contributing to stretch‐induced cardiac hypertrophy (Sadoshima et al. 1993). To rule out locally generated Ang II acting in an autocrine or paracrine manner, pressure–diameter relationships were evaluated in the absence or presence of an angiotensin converting enzyme inhibitor (captopril, 10−6 m) and/or candesartan (10−5 m). Pre‐treatment with captopril alone had no effect on myogenic reactivity and inhibition of myogenic reactivity was still observed following treatment with captopril and candesartan (Fig. 4 A). Thus, pressure‐mediated Ang II generation does not appear to occur in skeletal muscle resistance arterioles and the inhibitory effect of candesartan on pressure‐induced constriction is not a result of locally produced Ang II interacting with AT1R.
Figure 4. Decreased pressure‐induced vasoconstriction by candesartan is not a result of AT2R‐dependent vasodilatation and/or suppression of local Ang II production .
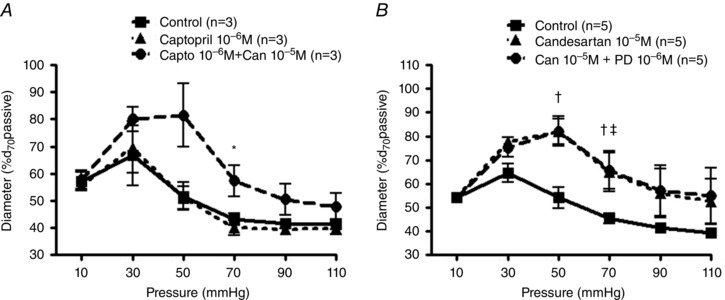
A, myogenic reactivity was examined in pressurized rat cremaster arterioles with or without captopril alone (angiotensin converting enzyme inhibitor; 10−6 m; n = 3) or in the presence of a combination of captopril (10−6 m) and candesartan (10−5 m; n = 3). Experimental protocols and calculation of myogenic constriction were performed as described previously. ‘Capto’ and ‘Can’ indicate captopril and candesartan, respectively. * P < 0.05 captopril 10−6 m vs. captopril 10−6 m + candesartan 10−5 m. B, myogenic reactivity was examined in pressurized cremaster arterioles with or without candesartan alone (10−5 m; n = 5) or combination of candesartan (10−5 m) and PD‐123,319 (AT2R inhibitor; 10−5 m; n = 5). Experimental protocols and calculation of myogenic constriction were performed as described previously. ‘Can’ and ‘PD’ represent candesartan and PD‐123,319, respectively. † P < 0.05 control vs. candesartan 10−5 m, ‡ P < 0.05 control vs. candesartan 10−5 m + PD‐123,319 10−5 m. Group data are presented as means ± SEM.
As activation of the AT2R has protective effects on the cardiovascular system by preventing vascular remodelling, oxidative stress, or atherosclerosis (Horiuchi & Mogi, 2011) and promotes vasodilatation (Maron & Leopold, 2014), it is conceivable that the AT2R may be activated and subsequently involved in vasodilatation once the AT1R is suppressed by candesartan. To examine whether the candesartan‐mediated inhibition of myogenic response is linked to activation of the AT2R, pressure–diameter relationships were monitored in the absence or presence of candesartan and/or the specific AT2R inhibitor (PD‐123,319, 10−6 m). Pressure–diameter relationships in the presence of candesartan alone or in response to the combination of candesartan + PD‐123,319 were similar (Fig. 4 B), implying that the AT2R activation is not involved in the candesartan‐mediated reduction in myogenic reactivity. Collectively, these studies in arterioles from skeletal muscle provide additional support for a role for the AT1R in mechanosensory events underlying myogenic constriction.
Candesartan inhibition of myogenic constriction is not associated with reduced global intracellular Ca2+ in VSMCs
Ca2+ influx (i.e. primarily through voltage‐dependent Ca2+ channels/TRP ion channels) and Ca2+ release from sarcoplasmic reticulum (SR) are important determinants for pressure‐induced vasoconstriction. We therefore addressed the question of whether the candesartan‐induced reduction in myogenic constriction is related to a reduction in global intracellular Ca2+ concentration ([Ca2+]i) in arteriolar myocytes. Using the Ca2+‐sensitive ratiometric indicator Fura‐2, increased intraluminal pressure caused a similar steady‐state increase in Ca2+ ratio both in the absence and presence of 10−5 m candesartan, suggesting that mechanoactivation of the AT1R may not directly affect Ca2+ dynamics in pressurized rat skeletal muscle arterioles (Fig. 5 A and B).
Figure 5. Candesartan‐dependent inhibition of myogenic constriction does not require a decrease in arteriolar VSMC global [Ca2+]i .
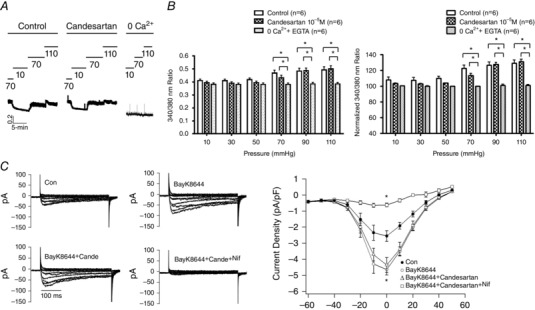
Ca2+ fluorescence ratios were determined in pressurized rat cremaster arterioles loaded with Fura‐2 (2×10−6 m) in the absence or presence of candesartan 10−5 m (n = 6). A, example tracing showing that candesartan does not prevent pressure‐induced changes in intracellular Ca2+ levels. B, intraluminal pressures were elevated from 10 to 110 mmHg in 20 mmHg steps. Values are shown as raw 340:380 nm fluorescence ratios and as a percentage change in the ratio. Results are normalized to the Ca2+ ratio under passive conditions (0 Ca2++EGTA) at 70 mmHg. * P < 0.05 control or candesartan 10−5 m vs. zero Ca2+ + EGTA. Group data are presented as means ± SEM. C, patch clamp data showing that candesartan (10−5 m) does not have an effect on VSMC VDCC currents. Cells (12 cells from n = 3 independent cell preparations) were stimulated with the agonist Bay K8644 to provide a level of basal activation (2 × 10−6 m). Example tracings are shown on the left and group data on the right. Compared with control (Con), Bay K8644 caused a significant (* P < 0.05) increase in Ca2+ current while nifedipine caused significant (* P < 0.05) inhibition. Group data are presented as means ± SEM.
As candesartan was used at a high concentration (10−5 m), whole‐cell patch clamp of freshly isolated cremaster muscle arteriolar VSMCs was employed to confirm that the blocker did not exert direct effects on L‐type VDCCs. Cells were first stimulated with the VDCC agonist Bay K8644 (2 × 10−6 m) to simulate the basal activity expected in myogenically active vessels, and then treated with candesartan. As shown in Fig. 5 C, candesartan did not significantly affect VDCCs consistent with a lack of a direct action of the blocker on Ca2+ dynamics.
Membrane permeable DAG restores decreased arteriolar myogenic constriction following candesartan
Ligation of GqPCRs results in heterotrimeric G proteins activating PLC‐β or PLC‐γ that cleaves phosphatidylinositol 4,5‐bisphosphate (PIP2) to IP3 and DAG. IP3 and DAG function as second messengers that induce vasoconstriction through Ca2+ release from SR (McCarron et al. 2004) and activation of TRPC channels (Large et al. 2009), respectively. To indirectly address whether pressure‐induced activation of the AT1R requires DAG to elicit myogenic constriction (supported by previous studies showing intraluminal pressure‐induced DAG and IP3 production; Narayanan et al. 1994), increasing concentrations of the membrane permeable DAG analogue OAG were applied to pressurized cremaster arterioles showing spontaneous tone at 70 mmHg in the absence or presence of candesartan (Fig. 6 A). Membrane permeable DAG, alone, potentiated steady‐state myogenic tone in the absence of candesartan (Fig. 6 A, squares). Further, the loss of myogenic tone at 70 mmHg, following candesartan treatment, was partially restored by membrane permeable DAG (Fig. 6 A, triangles). This was assumed to be consistent with AT1R mechanoactivation‐induced DAG production contributing to myogenic constriction in rat skeletal muscle arterioles.
Figure 6. Decreased myogenic constriction due to candesartan is reversed by membrane permeable DAG .
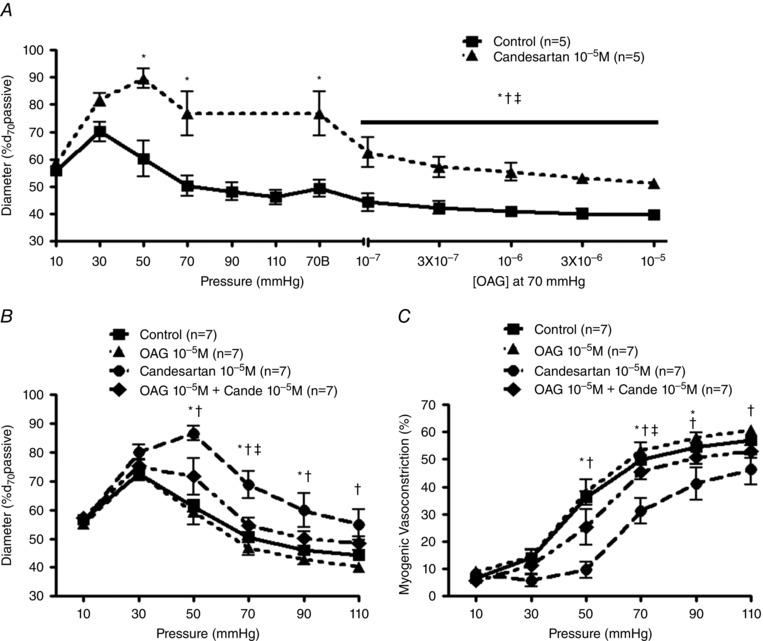
A, myogenic reactivity was observed in pressurized rat cremaster arterioles with or without candesartan pre‐treatment 10−5 m (left panel; n = 5). Subsequently OAG–concentration response curves (10−7 m–10−5 m; right panel; n = 5) were studied in the same arterioles pressurized to 70 mmHg in the absence or presence of candesartan (10−5 m). ‘70B’ indicates baseline of each dose–response curve at 70 mmHg in the absence or presence of candesartan. * P < 0.05 control vs. candesartan 10−5 m, † P < 0.05 70B without candesartan 10−5 m vs. each concentration of OAG without candesartan 10−5 m, ‡ P < 0.05 70B with candesartan 10−5 m vs. each concentration of OAG with candesartan 10−5 m. B and C, myogenic reactivity and vasoconstriction were evaluated in the absence or presence of OAG (10−5 m), candesartan (10−5 m), or combination of OAG and candesartan (n = 7). Experimental protocols and calculation of myogenic vasoconstriction were performed as described previously. ‘Cande’ indicates candesartan. * P < 0.05 control vs. candesartan 10−5 m, † P < 0.05 OAG 10−5 m vs. candesartan 10−5 m, ‡ P < 0.05 candesartan 10−5 m vs. OAG 10−5 m + candesartan 10−5 m. Group data are presented as means ± SEM.
Subsequently, the effect of a single OAG concentration (10−5 m) on arteriolar pressure–diameter relationships was determined (Fig. 6 B and C). In the absence of candesartan, membrane permeable DAG had little effect on pressure‐induced vasoconstriction (Fig. 6 B and C, triangles). Candesartan, alone, inhibited steady‐state myogenic tone and myogenic reactivity as previously observed (Fig. 6 B and C, circles). In contrast, the inhibitory effect of candesartan on myogenic constriction (50–110 mmHg) was significantly attenuated by application of membrane permeable DAG (10−5 m; Fig. 6 B and C, diamonds). Similar to above, these data support AT1R mechanoactivation‐mediated generation of DAG playing an important role in skeletal muscle arteriolar myogenic constriction.
Mechanoactivation of the AT1R elicits PKC‐dependent myogenic vasoconstriction of skeletal muscle resistance arterioles
Pharmacological inhibition of PKC has been shown to abolish myogenic constriction in a number of tissues including rat cremaster and cerebral arteries (Hill et al. 1990; Osol et al. 1991). More recently, PKC‐mediated mechanisms underlying Ca2+ sensitization and/or cytoskeleton remodelling have been reported to contribute to force generation during myogenic constriction (Moreno‐Dominguez et al. 2013, 2014). We therefore examined whether PKC inhibition (GF 109203X, 3×10−6 m) blunted the ‘rescue effects’ of membrane permeable DAG (Fig. 6).
Candesartan significantly diminished myogenic reactivity in the presence of GF 109203X (Fig. 7). To assess the possible contribution of downstream DAG–PKC pathways to AT1R‐mediated myogenic constriction, membrane permeable DAG was applied to arterioles pretreated with both GF 103209X and candesartan. Whereas membrane permeable DAG rescued myogenic reactivity following candesartan alone (Fig. 7), OAG application did not restore myogenic reactivity in the presence of the PKC inhibitor (Fig. 7). Thus these data are consistent with mechanoactivation of the AT1R resulting in activation of DAG–PKC‐dependent pathways and subsequently eliciting myogenic constriction in skeletal muscle arterioles.
Figure 7. DAG/PKC‐mediated intracellular signalling is associated with myogenic constriction in response to mechanoactivation of the AT1R .
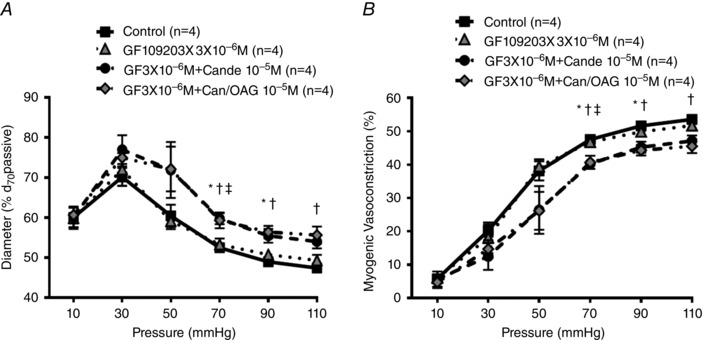
A and B, myogenic reactivity and vasoconstriction of cremaster arterioles were determined in the absence or presence of GF 109203X (a PKC inhibitor; 3 × 10−6 m) alone and in combination with candesartan (10−5 m), and/or OAG (10−5 m) (n = 4). Experimental protocols and calculation of myogenic vasoconstriction were conducted as described previously. ‘GF’ and ‘Cande/Can’ represent GF 109203X and candesartan, respectively. * P < 0.05 control vs. GF109203X 3 × 10−6 m + candesartan 10−5 m, † P < 0.05 control vs. GF109203X 3 × 10−6 m + candesartan 10−5 m + OAG 10−5 m, ‡ P < 0.05 GF109203X 3 × 10−6 m vs. GF109203X 3 × 10−6 m + candesartan 10−5 m. Group data are presented as means ± SEM.
Actin cytoskeleton remodelling contributes to AT1R‐mediated mechanoactivation and myogenic constriction
Current evidence suggests that a mechanosensitive increase in [Ca2+]i alone leading to activation of myosin light chain kinase (MLCK) and phosphorylation at serine 19 of 20 kDa regulatory light chain is not sufficient to fully explain the molecular mechanisms underlying arteriolar myogenic constriction (Cole & Welsh, 2011; Hill & Meininger, 2012; Walsh & Cole, 2013; Turner & Macdonald, 2014; Abd‐Elrahman et al. 2015). Consistent with these findings, we found that candesartan inhibited myogenic constriction without a requirement for a reduction in [Ca2+]i (Fig. 5) and mechanoactivation of the AT1R appeared coupled to PKC‐related signalling pathways (Fig. 7). Thus, we hypothesized that mechanical activation of the AT1R initiates actin cytoskeleton remodelling consistent with the work of Cole and colleagues (Moreno‐Dominguez et al. 2013, 2014). In agreement with these studies, a significant decrease in G‐actin content was observed in cremaster arterioles exposed to an acute increase in intraluminal pressure (20–70 mmHg; Fig. 8 A). Subsequently, increased intraluminal pressure‐dependent actin cytoskeleton rearrangement was evaluated in the absence or presence of candesartan to determine if myogenic constriction initiated by mechanoactivation of the AT1R is accompanied by increased actin polymerization. Increased G‐actin levels (indicative of decreased F‐actin and actin polymerization) were observed in the presence of 10−5 m candesartan (Fig. 8 B). G‐actin levels were normalized to the density of SM‐22α as a loading control as used in previous studies (Moreno‐Dominguez et al. 2013, 2014). Collectively, the data suggest that mechanoactivation of the AT1R activates PKC and in turn causes actin cytoskeleton remodelling contributing to myogenic constriction.
Figure 8. Blockade of the AT1R or inhibition of PKC causes a reduction in mechanically activated actin polymerization .
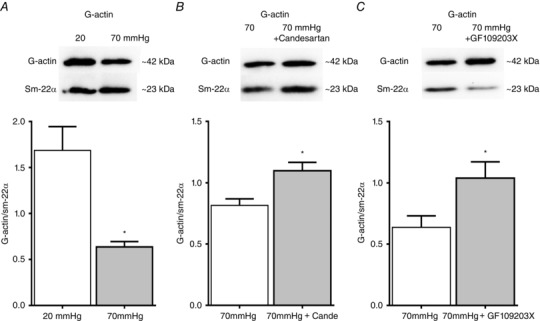
A, alteration in G‐actin levels in response to increased intraluminal pressure (i.e. from 20 to 70 mmHg) was evaluated in rat cremaster arterioles using Western blotting analysis (each pressure, n = 4). G‐actin content was normalized to expression of SM‐22α, an indicator of total VSMC protein. * P < 0.05 vs. 20 mmHg. B and C, G‐actin levels of pressurized (70 mmHg) cremaster arterioles in the absence or presence of candesartan (B, n = 7 per group) or GF 109203X (C, n = 6 per group) were measured. G‐actin content was normalized to expression of SM‐22α, an indicator of total VSMC protein. * P < 0.05 vs. 70 mmHg. Group data are presented as means ± SEM.
To further confirm the involvement of PKC in pressure‐induced actin remodelling, G‐actin levels were determined (at an intravascular pressure of 70 mmHg) in the absence and presence of the PKC inhibitor GF 109203X. Similarly to candesartan, GF 109203X caused an increase in G‐actin levels (Fig. 8 C) consistent with PKC inhibition blocking pressure‐induced G‐F actin transition.
Candesartan inhibits actin polymerization as assessed by atomic force microscopy (AFM) at the single cell level
To provide further support for a role of the AT1R receptor in mechanoactivation of actin polymerization, studies were undertaken using cremaster muscle VSMCs exposed to hypotonic buffer. This approach was used to elicit membrane stretch and mechanoactivation (Welsh et al. 2000; Bulley et al. 2012). Experiments were performed in the presence and absence of candesartan and imaging of the cortical cytoskeleton was undertaken using AFM. As shown in Fig. 9 A, exposing cells to hypotonic buffer resulted in an increase in stress fibre thickness. Pre‐treatment of VSMCs with candesartan (10−5 m) attenuated both the thickening and reorganization of the fibres. Figure 9 B is a histogram plot of total actin stress fibre orientation for the entire deflection image for control, hypotonic buffer, and hypotonic buffer plus candesartan. The results show that hypotonic solution significantly enhances the alignment of stress fibres, while this was prevented by candesartan. The summarized data from 10 cells for each group showed that the area fraction of the dominant stress fibre in the range of ±20 deg fibre angle significantly increased on treatment with hypotonic buffer but decreased following the addition of candesartan (Fig. 9 C). The dominant area fraction was defined as the ratio of the area covered by stress fibres to the area devoid of stress fibres, thus providing a measurement of density (Hong et al. 2014).
Figure 9. Mechanoactivation at the single VSMC level causes actin remodelling which is prevented by candesartan .
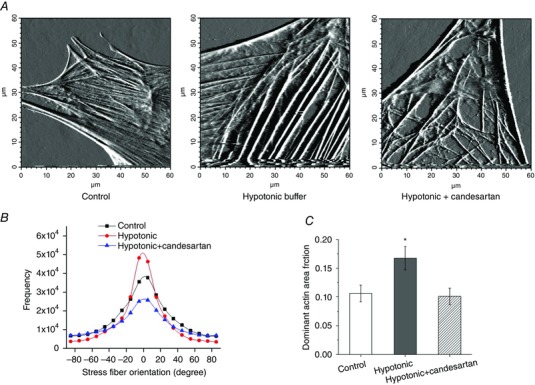
A, example AFM deflection images under control (left), hypotonic buffer (200 mosmol l−1) (centre) and hypotonic + candesartan (10−5 m) (right) conditions. B, hypotonic conditions caused greater alignment of stress fibre fragments around a defined axis while candesartan decreased apparent organization. C, images were quantified with respect to the ‘dominant actin area fraction’ (Hong et al. 2014) supporting a significant increase in actin polymerization that was prevented by the AT1R inhibition. * P < 0.05 hypotonic vs. control or hypotonic + candesartan. Group data are presented as means ± SEM. [Colour figure can be viewed at wileyonlinelibrary.com]
As the above data were collected using differing populations of cells for control and hypotonic conditions, two additional approaches were undertaken to confirm these observations. Firstly, a cell was scanned using the AFM under control conditions, after which the cell was gently perfused with hypotonic buffer before repeating the AFM scan. As shown in Fig. 10 A hypotonic buffer again caused an increase in prominence of the actin fibres. As a second approach to confirm these observations, cells subjected to AFM scanning were subsequently stained with phalloidin, again illustrating that hypotonic buffer causes an increased density of stress fibres (Fig. 10 B).
Figure 10. Additional examples of mechanoactivation at the single VSMC level causing actin remodelling .

A, AFM performed on a single VSMC before and after exposure to hypotonic buffer (200 mosmol l−1). A greater density of fibres is apparent after exposure to the hypotonic buffer. B, immediately after AFM studies (left column), phalloidin staining was performed on cells exposed to isosmotic or hypotonic buffer (right column) to provide additional evidence of increased reorganization of actin stress fibres. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
Arteriolar myogenic responsiveness to changes in intraluminal pressure plays a major role in regulating local haemodynamics in line with metabolic needs while also protecting tissues/organs from fluctuations in blood pressure and flow (Davis & Hill, 1999; Hill et al. 2001; Schubert et al. 2008; Kauffenstein et al. 2012; Lidington et al. 2013; Tan et al. 2013). Impaired blood flow resulting from an exaggerated myogenic response as described in hypertension and heart failure (Hughes & Bund, 2002; Gschwend et al. 2003) or excessive flow caused by impaired myogenic responsiveness is likely to contribute to ischaemic stroke, vascular rupture, and blood–brain barrier disruption (Walsh & Cole, 2013). However, despite its physiological and clinical significance, the precise cellular mechanisms underlying exactly how VSMCs sense changes in intraluminal pressure remain uncertain.
Recently, GPCRs have emerged as potential mechanosensors for myogenic constriction. In particular, growing evidence supports the AT1R acting as a novel mechanosensor and contributing to myogenic autoregulation in mouse mesenteric, cerebral, and renal arteries (Mederos y Schnitzler et al. 2008; Brayden et al. 2013; Blodow et al. 2014; Schleifenbaum et al. 2014). An augmented myogenic response in congestive heart failure was attenuated by losartan or candesartan, implicating AT1R involvement in myogenic reactivity during pathophysiological situations (Gschwend et al. 2003). Although studies have shown a total loss of myogenic reactivity in vessels from AT1R knockout mice, histamine, endothelin, vasopressin, muscarinic (Mederos y Schnitzler et al. 2008), and α‐adrenergic (Blodow et al. 2014) receptors have also been reported to be mechanically activated. Similarly, P2Y4 and P2Y6 receptors (purinergic receptors) have been reported to play a role in contraction of cerebral arterioles following an increase in intraluminal pressure (Brayden et al. 2013; Li et al. 2014). In contrast, β‐adrenergic receptors, despite being a class of GPCR, do not appear to be mechanosensitive (Mederos y Schnitzler et al. 2008; Blodow et al. 2014). This points towards Gq/11‐coupled receptors, not Gs‐coupled receptors, being mechanosensors and relevant to the myogenic response (Mederos y Schnitzler et al. 2008).
Consistent with a role for the AT1R in mesenteric, cerebral and renal arteries, the present studies show that in rat cremaster muscle arterioles, pharmacological inhibition of the receptor attenuates myogenic reactivity. In addition, these data add support to AT1R mechanosensitivity existing across species. Despite this, the exact contribution of the AT1R to arteriolar myogenic constriction is uncertain, with doubt relating to its absolute requirement in the myogenic signalling pathway(s). While this, in part, may reflect differences in vascular beds it may also relate to differences in expression of subtypes of the AT1R. For example, in contrast to humans, rodents express two isoforms of AT1R, AT1aR and AT1bR, in arteriolar VSMCs, with both isoforms signalling through the same downstream pathways (Zhou et al. 1993; Tian et al. 1996). Both isoforms appear to be mechanosensitive in mouse mesenteric and renal arterioles (Blodow et al. 2014; Schleifenbaum et al. 2014). However, the relative expression level of either subtype of AT1R may dictate the pathway used by a given vasculature. Thus Blodow et al. (2014) have shown that the AT1bR exhibits greater apparent mechanosensitivity compared to the AT1aR and that the relative mRNA expression of the AT1bR predominates in mouse mesenteric arterioles (i.e. 96.1% (AT1bR) vs. 3.9% (AT1aR)). In the present studies we found that rat cremaster muscle arterioles expressed mRNA for the AT1aR at considerably higher levels than that for the AT1bR (Fig. 1). Thus, it appears that the AT1aR is the major AT1R‐mediated mechanosensor in cremaster muscle arterioles and that the inverse agonists used in this study (e.g. candesartan, olmesartan, and EXP3174; Fig. 3) are likely to block its contribution to myogenic responsiveness.
In the present studies several control experiments were performed to confirm that the apparent mechanosensitivity of the AT1R is related to activation of the receptor, per se. Previous studies have shown that Ang II is locally produced in response to stretch and causes cardiac hypertrophy in an autocrine or paracrine manner (Parker et al. 1990; Sadoshima et al. 1993). Similarly, a number of studies have demonstrated that the vascular wall contains the components necessary for local Ang II production (Nguyen Dinh & Touyz, 2011). Further, it has recently been reported in isolated carotid arteries that increased intraluminal pressure (80–150 mmHg; 3 h) causes local production of Ang II and reactive oxygen species, which impairs dilatation in response to acetylcholine (Zhao et al. 2015). It is unlikely that such a mechanism contributes to myogenic constriction in the cremaster muscle arterioles, at least in the context of conditions used in the present study (5 min changes in pressure). Supporting this, ACE inhibition with captopril did not inhibit myogenic reactivity (Fig. 4 A), consistent with similar studies of Mederos y Schnitzler and colleagues (2008) and the finding of normal myogenic responses in mesenteric arteries isolated from angiotensinogen gene knockout mice (Schleifenbaum et al. 2014). Further supporting differences in signalling events underlying acute (5 min) and short term (3 h) pressure changes are previous studies showing that vascular remodelling is initiated in less than 4 h of Ang II exposure, but not after 5 min exposure (Martinez‐Lemus et al. 2011). In addition, the present studies showed that attenuated myogenic reactivity following candesartan treatment does not result from an unopposed AT2R activation‐mediated vasodilatation (Fig. 4 B). Collectively, these studies add support to the hypothesis that the AT1R per se is mechanically activated by increased intraluminal pressure (Mederos y Schnitzler et al. 2008; Blodow et al. 2014; Schleifenbaum et al. 2014) and contributes to the cellular mechanisms underlying myogenic constriction.
Myogenic constriction is considered to involve the following sequence of signals: (1) membrane depolarization in response to stretch of, or increased tension experienced by, arteriolar VSMCs; (2) extracellular Ca2+ influx via VDCC and intracellular Ca2+ release from sarcoplasmic reticulum; (3) increased global intracellular Ca2+ levels activating calmodulin; and (4) phosphorylation of myosin light chain via MLCK (Davis et al. 1992; Zou et al. 1995, 2000; Davis & Hill, 1999; Knot & Nelson, 1998; Kotecha & Hill, 2005). While these events lead to contractile force generation, it remains less certain how these steps are coupled to the initial mechanosensory events. It has been suggested that mechanical activation of GPCRs gives rise to PLC‐dependent generation of 2nd messengers including IP3 and DAG which promote the opening of cation channels, including TRPC6 or TRPM4 channel (Osol et al. 1993; Slish et al. 2002; Welsh et al. 2002; Earley et al. 2004, 2007; Large et al. 2009), which in turn result in membrane depolarization and opening of voltage‐gated Ca2+ channels. Interestingly DAG has also been implicated, via activation of PKC, in modulating VSMC cytoskeletal dynamics and contractile protein Ca2+ sensitivity (Hill et al. 2001; Hill & Meininger, 2012; Moreno‐Dominguez et al. 2013, 2014; Walsh & Cole, 2013).
Direct application of a membrane permeable DAG analogue to intact pressurized rat cremaster arterioles without candesartan did not significantly potentiate myogenic reactivity, perhaps as a result of a tonic production of endogenous DAG in the pressurized vessels (Fig. 6 B and C). In contrast, membrane permeable OAG treatment was able to cause constriction and overcome the reduction in steady‐state myogenic tone (Fig. 6 A) and reactivity (Fig. 6 B and C) caused by candesartan. Thus these data are consistent with pressure‐induced AT1R activation being associated with DAG‐dependent vasoconstriction.
DAG is known to directly activate cation channels including TRPC6 channels giving rise to membrane depolarization‐mediated Ca2+ influx and vasoconstriction (Slish et al. 2002; Ju et al. 2010; Large et al. 2009). Based on this, it was initially hypothesized that mechanoactivation of the AT1R in skeletal muscle arterioles would utilize these pathways. As a result, we examined intracellular global Ca2+ levels in pressurized cremaster arterioles in the absence or presence of candesartan (Fig. 5). Somewhat surprisingly, increased pressure‐induced changes in Ca2+ i were not markedly affected by candesartan (10−5 m; Fig. 5 A and B). Further, candesartan (10−5 m) did not exert direct actions on VDCC currents as measured by whole‐cell patch clamp. (Fig. 5 C). These data suggest that mechanoactivation of the AT1R may involve additional cellular mechanisms such as Ca2+ sensitization and/or cytoskeletal remodelling for myogenic constriction (Hill et al. 2001; Muranyi et al. 2005; Eto, 2009; Hill & Meininger, 2012; Moreno‐Dominguez et al. 2013, 2014; Walsh & Cole, 2013), rather than only direct effects on VSMC [Ca2+]i. As DAG stimulates several PKC isoforms and PKC has been implicated in myogenic constriction (Hill et al. 1990; Osol et al. 1991; Moreno‐Dominguez et al. 2013, 2014), we then used a pharmacological approach to investigate its role in the effects of membrane permeable DAG. In the presence of a PKC inhibitor (GF 109203X), membrane permeable DAG was not able to attenuate the inhibitory effect of candesartan on myogenic reactivity (Fig. 7). Collectively these data suggested that a signalling sequence involving AT1R–DAG–PKC plays an important role in myogenic constriction in skeletal muscle arterioles.
Increasing evidence supports the contribution of dynamic remodelling of the VSMC actin cytoskeleton to contraction and maintenance of contraction. While the actin cytoskeleton is actively rearranged in a number of physiological circumstances actin polymerization has been shown to specifically contribute to arteriolar myogenic constriction (Cipolla et al. 2002; Flavahan et al. 2005; Moreno‐Dominguez et al. 2013). Consistent with this effect, decreased levels of globular α‐actin (G‐actin; presumably correlated to increased levels of filamentous α‐actin (F‐actin)) following an acute increase in arteriolar intraluminal pressure were initially shown by Cipolla and colleagues (2002). It was further shown that myogenic tone or reactivity could be modulated using activators (e.g. jasplakinolide) or inhibitors (e.g. cytochalasin D and latrunculin B) of actin polymerization (Cipolla et al. 2002). More recently, and using a biochemical approach, changes in intravascular pressure (from 10 to 80 or 120 mmHg) have been demonstrated to cause reduced G‐actin content up to by 66% or 87%, respectively (Moreno‐Dominguez et al. 2014). The present investigation similarly showed a 62% reduction in G‐actin levels when skeletal muscle arterioles were exposed to an increase in intravascular pressure (20–70 mmHg; Fig. 8 A).
In regard to the Western blot methodology used in the current study we chose to normalize G actin to measurements of the protein SM‐22α as used in a number of previous studies (Luykenaar et al. 2009; Walsh et al. 2011; Moreno‐Dominguez et al. 2013, 2014; Colinas et al. 2015). The underlying reason for this related to concerns in the reproducibility of direct F‐actin measurements in the small samples we used. This has previously been discussed by Walsh et al. (2011), who suggested that during ultracentrifugation, F‐actin pellets may be contaminated by incompletely lysed material that would contain small/variable amounts of G‐actin. To initially validate our measurements, we showed in small arteries that we could detect that jasplakinolide concentration‐dependently decreased levels of G‐actin (data not shown). To study actin dynamics in situ, a quantitative fluorescence‐based technique would be ideal. While progress has been made in following actin polymerization in small blood vessels (Foote et al. 2016), this is not yet suitable for measurement of depolymerization which would have been required for the present study.
On the basis of this and to add further support for VSMC mechanoactivation causing acute actin remodelling, studies were also undertaken with primary (non‐passaged) cultured arteriolar VSMCs. Cells were exposed to hypotonic buffer to induce membrane stretch as used in a number of previous studies (Welsh et al. 2000; Bulley et al. 2012). AFM was then used to image cellular cortical actin fibres, allowing quantification of actin density and orientation as indices of remodelling. Consistent with the Western blotting studies described above, mechanoactivation resulted in remodelling of the cytoskeleton by increasing cortical filament size, density and orientation and these events were prevented by pre‐treatment with candesartan (Fig. 9). In addition, an alternate approach (i.e. phalloidin staining) supported mechanical stretch of VSMC membranes causing remodelling of actin stress fibres (Fig. 10). Further, it has been recently shown that Ang II can cause rapid increases in VSMC stiffness and adhesion in isolated cremaster VSMCs (Hong et al. 2014). The increase in cell stiffness correlated strongly with reorganization of the cortical actin stress fibres (i.e. increased stress fibre size, density and orientation; Hong et al. 2014). This further supports a direct link between activation of AT1R and cytoskeletal remodelling as observed with mechanoactivation of the receptor.
The present studies demonstrate that the AT1R can be activated by mechanical stimuli and play an important role in myogenic constriction of skeletal muscle arterioles. The findings are therefore also consistent with a growing literature that mechanical forces can directly activate certain GPCRs (Mederos y Schnitzler et al. 2008, 2011; Storch et al. 2012, 2015; Brayden et al. 2013; Blodow et al. 2014; Gonzales et al. 2014; Li et al. 2014; Schleifenbaum et al. 2014). An additional significant finding of the present study is that mechanoactivation of the AT1R appears to result in the generation of second messengers (e.g. DAG) and vasoconstriction that may be related to PKC‐dependent actin cytoskeleton reorganization.
Additional information
Competing interests
The authors have no conflicts of interest relevant to this manuscript.
Author contributions
K.H.: conception, design; data acquisition and analysis/interpretation; drafting and revision; G.Z.: data acquisition and analysis; Z.H.: design; data acquisition and analysis; Z. S.: design; data acquisition and analysis; Y.Y.: data acquisition and analysis; P.S.C.: design; interpretation; drafting and revision; M.J.D. design; interpretation; drafting and revision; G.A.M. design; analysis and interpretation; drafting and revision; M.A.H. conception, design; analysis and interpretation; drafting and revision. All authors have approved submission of the final version of the manuscript. All authors agree to be accountable for all aspects of the work. All authors qualify for authorship and all those who qualify for authorship are listed.
Funding
Studies were supported by grants from the National Institutes of Health, USA (HL092241, M.A.H.; HL095486, G.A.M.) and American Heart Association (15SDG25420001, Z.H.).
Translational perspective
Small arteries exhibit (myogenic) vasoconstriction in response to an acute increase in intraluminal pressure. Similarly, a decrease in pressures causes vasodilatation. Under physiological conditions, these mechanically induced vasomotor responses underlie the autoregulatory control of local haemodynamics and protect capillaries from the damaging effects of excessive intraluminal pressure. Abnormalities in myogenic constriction, however, are likely to contribute to the pathology of a number of important vascular diseases including hypertension and heart failure. As a consequence, it is important to have an exact understanding of the events that sense and transduce pressure changes to the contractile proteins. The present studies add support to the hypothesis that type 1 angiotensin receptor is a mechanosensor and its activation leads to activation of events leading to cytoskeletal remodelling and contraction. The studies thus provide candidate signalling mechanisms that can be targeted for modulating myogenic reactivity.
Acknowledgements
We thank Rong Li M.D. for assisting with functional vascular studies.
References
- Abd‐Elrahman KS, Walsh MP & Cole WC (2015). Abnormal Rho‐associated kinase activity contributes to the dysfunctional myogenic response of cerebral arteries in type 2 diabetes. Can J Physiol Pharmacol 93, 177–184. [DOI] [PubMed] [Google Scholar]
- Akazawa H, Yasuda N & Komuro I (2009). Mechanisms and functions of agonist‐independent activation in the angiotensin II type 1 receptor. Mol Cell Endocrinol 302, 140–147. [DOI] [PubMed] [Google Scholar]
- Blodow S, Schneider H, Storch U, Wizemann R, Forst AL, Gudermann T & Schnitzler M (2014). Novel role of mechanosensitive AT1B receptors in myogenic vasoconstriction. Pflugers Arch 466, 1343–1353. [DOI] [PubMed] [Google Scholar]
- Brayden JE, Li Y & Tavares MJ (2013). Purinergic receptors regulate myogenic tone in cerebral parenchymal arterioles. J Cereb Blood Flow Metab 33, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulley S, Neeb ZP, Burris SK, Bannister JP, Thomas‐Gatewood CM, Jangsangthong W & Jaggar JH (2012). TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res 111, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolla MJ, Gokina NI & Osol G (2002). Pressure‐induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J 16, 72–76. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ & Osol G (1998). Vascular smooth muscle actin cytoskeleton in cerebral artery forced dilatation. Stroke 29, 1223–1228. [DOI] [PubMed] [Google Scholar]
- Cole WC & Welsh DG (2011). Role of myosin light chain kinase and myosin light chain phosphatase in the resistance arterial myogenic response to intravascular pressure. Arch Biochem Biophys 510, 160–173. [DOI] [PubMed] [Google Scholar]
- Colinas O, Moreno‐Dominguez A, Zhu HL, Walsh EJ, Perez‐Garcia MT, Walsh MP, Cole WC (2015). α5‐Integrin‐mediated cellular signaling contributes to the myogenic response of cerebral resistance arteries. Biochem Pharmacol 97, 281–291. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Donovitz JA & Hood JD (1992). Stretch‐activated single‐channel and whole cell currents in vascular smooth muscle cells. Am J Physiol Cell Physiol 262, C1083–C1088. [DOI] [PubMed] [Google Scholar]
- Davis MJ & Hill MA (1999). Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 79, 387–423. [DOI] [PubMed] [Google Scholar]
- Earley S, Straub SV & Brayden JE (2007). Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circ Physiol 292, H2613–H2622. [DOI] [PubMed] [Google Scholar]
- Earley S, Waldron BJ & Brayden JE (2004). Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res 95, 922–929. [DOI] [PubMed] [Google Scholar]
- El‐Yazbi AF, Abd‐Elrahman KS & Moreno‐Dominguez A (2015). PKC‐mediated cerebral vasoconstriction: Role of myosin light chain phosphorylation versus actin cytoskeleton reorganization. Biochem Pharmacol 95, 263–278. [DOI] [PubMed] [Google Scholar]
- Eto M (2009). Regulation of cellular protein phosphatase‐1 (PP1) by phosphorylation of the CPI‐17 family, C‐kinase‐activated PP1 inhibitors. J Biol Chem 284, 35273–35277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan NA, Bailey SR, Flavahan WA, Mitra S & Flavahan S (2005). Imaging remodeling of the actin cytoskeleton in vascular smooth muscle cells after mechanosensitive arteriolar constriction. Am J Physiol Heart Circ Physiol 288, H660–H669. [DOI] [PubMed] [Google Scholar]
- Foote CA, Castorena‐Gonzalez JA, Staiculescu MC, Clifford PS, Hill MA, Meininger GA & Martinez‐Lemus LA (2016). Brief serotonin exposure initiates arteriolar inward remodeling processes in vivo that involve transglutaminase activation and actin cytoskeleton reorganization. Am J Physiol Heart Circ Physiol 310, H188–H198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales AL, Yang Y, Sullivan MN, Sanders L, Dabertrand F, Hill‐Eubanks DC, Nelson MT & Earley S (2014). A PLCgamma1‐dependent, force‐sensitive signaling network in the myogenic constriction of cerebral arteries. Sci Signal 7, ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruden G, Thomas S, Burt D, Zhou W, Chusney G, Gnudi L & Viberti G (1999). Interaction of angiotensin II and mechanical stretch on vascular endothelial growth factor production by human mesangial cells. J Am Soc Nephrol 10, 730–737. [DOI] [PubMed] [Google Scholar]
- Gschwend S, Henning RH, Pinto YM, de ZD, van Gilst WH & Buikema H (2003). Myogenic constriction is increased in mesenteric resistance arteries from rats with chronic heart failure: instantaneous counteraction by acute AT1 receptor blockade. Br J Pharmacol 139, 1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Falcone JC & Meininger GA (1990). Evidence for protein kinase C involvement in arteriolar myogenic reactivity. Am J Physiol Heart Circ Physiol 259, H1586–H1594. [DOI] [PubMed] [Google Scholar]
- Hill MA & Meininger GA (1994). Calcium entry and myogenic phenomena in skeletal muscle arterioles. Am J Physiol Heart Circ Physiol 267, H1085–H1092. [DOI] [PubMed] [Google Scholar]
- Hill MA & Meininger GA (2012). Arteriolar vascular smooth muscle cells: mechanotransducers in a complex environment. Int J Biochem Cell Biol 44, 1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Meininger GA, Davis MJ & Laher I (2009). Therapeutic potential of pharmacologically targeting arteriolar myogenic tone. Trends Pharmacol Sci 30, 363–374. [DOI] [PubMed] [Google Scholar]
- Hill MA, Potocnik SJ, Martinez‐Lemus LA & Meininger GA (2003). Delayed arteriolar relaxation after prolonged agonist exposure: functional remodeling involving tyrosine phosphorylation. Am J Physiol Heart Circ Physiol 285, H849–H856. [DOI] [PubMed] [Google Scholar]
- Hill MA, Yang Y, Ella SR, Davis MJ & Braun AP (2010). Large conductance, Ca2+‐activated K+ channels (BKCa) and arteriolar myogenic signaling. FEBS Lett 584, 2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Zou H, Davis MJ, Potocnik SJ & Price S (2000). Transient increases in diameter and [Ca2+]i are not obligatory for myogenic constriction. Am J Physiol Heart Circ Physiol 278, H345–H352. [DOI] [PubMed] [Google Scholar]
- Hill MA, Zou H, Potocnik SJ, Meininger GA & Davis MJ (2001). Invited review: arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J Appl Physiol (1985 ) 91, 973–983. [DOI] [PubMed] [Google Scholar]
- Hong Z, Sun Z, Li M, Li Z, Bunyak F, Ersoy I, Trzeciakowski JP, Staiculescu MC, Jin M, Martinez‐Lemus L, Hill MA, Palaniappan K & Meininger GA (2014). Vasoactive agonists exert dynamic and coordinated effects on vascular smooth muscle cell elasticity, cytoskeletal remodelling and adhesion. J Physiol 592, 1249–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M & Mogi M (2011). Role of angiotensin II receptor subtype activation in cognitive function and ischaemic brain damage. Br J Pharmacol 163, 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JM & Bund SJ (2002). Arterial myogenic properties of the spontaneously hypertensive rat. Exp Physiol 87, 527–534. [DOI] [PubMed] [Google Scholar]
- Jackson TY, Sun Z, Martinez‐Lemus LA, Hill MA & Meininger GA (2010). N‐cadherin and integrin blockade inhibit arteriolar myogenic reactivity but not pressure‐induced increases in intracellular Ca. Front Physiol 1, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju M, Shi J, Saleh SN, Albert AP & Large WA (2010). Ins(1,4,5)P3 interacts with PIP2 to regulate activation of TRPC6/C7 channels by diacylglycerol in native vascular myocytes. J Physiol 588, 1419–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlon WJ, Hsu PP, Li S, Chien S, McCulloch AD & Omens JH (1999). Measurement of orientation and distribution of cellular alignment and cytoskeletal organization. Ann Biomed Eng 27, 712–720. [DOI] [PubMed] [Google Scholar]
- Kauffenstein G, Laher I, Matrougui K, Guerineau NC & Henrion D (2012). Emerging role of G protein‐coupled receptors in microvascular myogenic tone. Cardiovasc Res 95, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EC, Choi SK, Lim M, Yeon SI & Lee YH (2013). Role of endogenous ENaC and TRP channels in the myogenic response of rat posterior cerebral arteries. PLoS One 8, e84194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ & Nelson MT (1998). Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N & Hill MA (2005). Myogenic contraction in rat skeletal muscle arterioles: smooth muscle membrane potential and Ca2+ signaling. Am J Physiol Heart Circ Physiol 289, H1326–H1334. [DOI] [PubMed] [Google Scholar]
- Large WA, Saleh SN & Albert AP (2009). Role of phosphoinositol 4,5‐bisphosphate and diacylglycerol in regulating native TRPC channel proteins in vascular smooth muscle. Cell Calcium 45, 574–582. [DOI] [PubMed] [Google Scholar]
- Lee S, Yang Y, Tanner MA, Li M & Hill MA (2015). Heterogeneity in Kv7 channel function in the cerebral and coronary circulation. Microcirculation 22, 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Baylie RL, Tavares MJ & Brayden JE (2014). TRPM4 channels couple purinergic receptor mechanoactivation and myogenic tone development in cerebral parenchymal arterioles. J Cereb Blood Flow Metab 34, 1706–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidington D, Schubert R & Bolz SS (2013). Capitalizing on diversity: an integrative approach towards the multiplicity of cellular mechanisms underlying myogenic responsiveness. Cardiovasc Res 97, 404–412. [DOI] [PubMed] [Google Scholar]
- Livak KJ & Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Luykenaar KD, El‐Rahman RA, Walsh MP & Welsh DG (2009). Rho‐kinase‐mediated suppression of KDR current in cerebral arteries requires an intact actin cytoskeleton. Am J Physiol Heart Circ Physiol 296, H917–H926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarron JG, MacMillan D, Bradley KN, Chalmers S & Muir TC (2004). Origin and mechanisms of Ca2+ waves in smooth muscle as revealed by localized photolysis of caged inositol 1,4,5‐trisphosphate. J Biol Chem 279, 8417–8427. [DOI] [PubMed] [Google Scholar]
- Maron BA & Leopold JA (2014). The role of the renin—angiotensin–aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series). Pulm Circ 4, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Lemus LA, Wu X, Wilson E, Hill MA, Davis GE, Davis MJ & Meininger GA (2003). Integrins as unique receptors for vascular control. J Vasc Res 40, 211–233. [DOI] [PubMed] [Google Scholar]
- Martinez‐Lemus LA, Zhao G, Galinanes EL & Boone M (2011). Inward remodeling of resistance arteries requires reactive oxygen species‐dependent activation of matrix metalloproteinases. Am J Physiol Heart Circ Physiol 300, H2005–H2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathar I, Vennekens R, Meissner M, Kees F, Van der Mieren G, Camacho Londono JE, Uhl S, Voets T, Hummel B, van den Bergh A, Herijgers P, Nilius B, Flockerzi V, Schweda F & Freichel M (2010). Increased catecholamine secretion contributes to hypertension in TRPM4‐deficient mice. J Clin Invest 120, 3267–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederos y Schnitzler M, Storch U & Gudermann T (2011). AT1 receptors as mechanosensors. Curr Opin Pharmacol 11, 112–116. [DOI] [PubMed] [Google Scholar]
- Mederos y Schnitzler M, Storch U, Meibers S, Nurwakagari P, Breit A, Essin K, Gollasch M & Gudermann T (2008). Gq‐coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J 27, 3092–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G (2003). Constitutive activity and inverse agonists of G protein‐coupled receptors: a current perspective. Mol Pharmacol 64, 1271–1276. [DOI] [PubMed] [Google Scholar]
- Moreno‐Dominguez A, Colinas O, El‐Yazbi A, Walsh EJ, Hill MA, Walsh MP & Cole WC (2013). Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol 591, 1235–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Dominguez A, El‐Yazbi AF, Zhu HL, Colinas O, Zhong XZ, Walsh EJ, Cole DM, Kargacin GJ, Walsh MP & Cole WC (2014). Cytoskeletal reorganization evoked by Rho‐associated kinase‐ and protein kinase C‐catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. J Biol Chem 289, 20939–20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller KB, Bender SB, Hong K, Yang Y, Aronovitz M, Jaisser F, Hill MA & Jaffe IZ (2015). Endothelial mineralocorticoid receptors differentially contribute to coronary and mesenteric vascular function without modulating blood pressure. Hypertension 66, 988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranyi A, Derkach D, Erdodi F, Kiss A, Ito M & Hartshorne DJ (2005). Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett 579, 6611–6615. [DOI] [PubMed] [Google Scholar]
- Narayanan J, Imig M, Roman RJ & Harder DR (1994). Pressurization of isolated renal arteries increases inositol trisphosphate and diacylglycerol. Am J Physiol Heart Circ Physiol 266, H1840–H1845. [DOI] [PubMed] [Google Scholar]
- Nguyen Dinh CA & Touyz RM (2011). A new look at the renin–angiotensin system – focusing on the vascular system. Peptides 32, 2141–2150. [DOI] [PubMed] [Google Scholar]
- Nourian Z, Li M, Leo MD, Jaggar JH, Braun AP & Hill MA (2014). Large conductance Ca2+‐activated K+ channel (BKCa) α‐subunit splice variants in resistance arteries from rat cerebral and skeletal muscle vasculature. PLoS One 9, e98863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osol G, Laher I & Cipolla M (1991). Protein kinase C modulates basal myogenic tone in resistance arteries from the cerebral circulation. Circ Res 68, 359–367. [DOI] [PubMed] [Google Scholar]
- Osol G, Laher I & Kelley M (1993). Myogenic tone is coupled to phospholipase C and G protein activation in small cerebral arteries. Am J Physiol Heart Circ Physiol 265, H415–H420. [DOI] [PubMed] [Google Scholar]
- Parker TG, Packer SE & Schneider MD (1990). Peptide growth factors can provoke “fetal” contractile protein gene expression in rat cardiac myocytes. J Clin Invest 85, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Xu Y, Slayter HS & Izumo S (1993). Autocrine release of angiotensin II mediates stretch‐induced hypertrophy of cardiac myocytes in vitro . Cell 75, 977–984. [DOI] [PubMed] [Google Scholar]
- Schleifenbaum J, Kassmann M, Szijarto IA, Hercule HC, Tano JY, Weinert S, Heidenreich M, Pathan AR, Anistan YM, Alenina N, Rusch NJ, Bader M, Jentsch TJ & Gollasch M (2014). Stretch‐activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res 115, 263–272. [DOI] [PubMed] [Google Scholar]
- Schubert R, Lidington D & Bolz SS (2008). The emerging role of Ca2+ sensitivity regulation in promoting myogenic vasoconstriction. Cardiovasc Res 77, 8–18. [DOI] [PubMed] [Google Scholar]
- Schwartz MA (2010). Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol 2, a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slish DF, Welsh DG & Brayden JE (2002). Diacylglycerol and protein kinase C activate cation channels involved in myogenic tone. Am J Physiol Heart Circ Physiol 283, H2196–H2201. [DOI] [PubMed] [Google Scholar]
- Spassova MA, Hewavitharana T, Xu W, Soboloff J & Gill DL (2006). A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci USA 103, 16586–16591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch U, Blodow S, Gudermann T & Mederos y Schnitzler M (2015). Cysteinyl leukotriene 1 receptors as novel mechanosensors mediating myogenic tone together with angiotensin II type 1 receptors‐brief report. Arterioscler Thromb Vasc Biol 35, 121–126. [DOI] [PubMed] [Google Scholar]
- Storch U, Mederos y Schnitzler M & Gudermann T (2012). G protein‐mediated stretch reception. Am J Physiol Heart Circ Physiol 302, H1241–H1249. [DOI] [PubMed] [Google Scholar]
- Sun Z, Li Z & Meininger GA (2012). Mechanotransduction through fibronectin‐integrin focal adhesion in microvascular smooth muscle cells: is calcium essential? Am J Physiol Heart Circ Physiol 302, H1965–H1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Martinez‐Lemus LA, Hill MA & Meininger GA (2008). Extracellular matrix‐specific focal adhesions in vascular smooth muscle produce mechanically active adhesion sites. Am J Physiol Cell Physiol 295, C268–C278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CO, Hamner JW & Taylor JA (2013). The role of myogenic mechanisms in human cerebrovascular regulation. J Physiol 591, 5095–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Baukal AJ, Sandberg K, Bernstein KE, Balla T & Catt KJ (1996). Properties of AT1a and AT1b angiotensin receptors expressed in adrenocortical Y‐1 cells. Am J Physiol Endocrinol Metab 270, E831–E839. [DOI] [PubMed] [Google Scholar]
- Turner SR & Macdonald JA (2014). Novel contributions of the smoothelin‐like 1 protein in vascular smooth muscle contraction and its potential involvement in myogenic tone. Microcirculation 21, 249–258. [DOI] [PubMed] [Google Scholar]
- Walsh MP & Cole WC (2013). The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J Cereb Blood Flow Metab 33, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh MP, Thornbury K, Cole WC, Sergeant G, Hollywood M, McHale N (2011). Rho‐associated kinase plays a role in rabbit urethral smooth muscle contraction, but not via enhanced myosin light chain phosphorylation. Am J Physiol Renal Physiol 300, F73–F85. [DOI] [PubMed] [Google Scholar]
- Welsh DG, Morielli AD, Nelson MT & Brayden JE (2002). Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res 90, 248–250. [DOI] [PubMed] [Google Scholar]
- Welsh DG, Nelson MT, Eckman DM & Brayden JE (2000). Swelling‐activated cation channels mediate depolarization of rat cerebrovascular smooth muscle by hyposmolarity and intravascular pressure. J Physiol 527, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne BM, Chiao CW & Webb RC (2009). Vascular smooth muscle cell signaling mechanisms for contraction to angiotensin II and endothelin‐1. J Am Soc Hypertens 3, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Murphy TV, Ella SR, Grayson TH, Haddock R, Hwang YT, Braun AP, Peichun G, Korthuis RJ, Davis MJ & Hill MA (2009). Heterogeneity in function of small artery smooth muscle BKCa: involvement of the beta1‐subunit. J Physiol 587, 3025–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Flavahan S, Leung SW, Xu A, Vanhoutte PM & Flavahan NA (2015). Elevated pressure causes endothelial dysfunction in mouse carotid arteries by increasing local angiotensin signaling. Am J Physiol Heart Circ Physiol 308, H358–H363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Ernsberger P & Douglas JG (1993). A novel angiotensin receptor subtype in rat mesangium. Coupling to adenylyl cyclase. Hypertension 21, 1035–1038. [DOI] [PubMed] [Google Scholar]
- Zou H, Ratz PH & Hill MA (1995). Role of myosin phosphorylation and [Ca2+]i in myogenic reactivity and arteriolar tone. Am J Physiol Heart Circ Physiol 269, H1590–H1596. [DOI] [PubMed] [Google Scholar]
- Zou H, Ratz PH & Hill MA (2000). Temporal aspects of Ca2+ and myosin phosphorylation during myogenic and norepinephrine‐induced arteriolar constriction. J Vasc Res 37, 556–567. [DOI] [PubMed] [Google Scholar]
- Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T & Komuro I (2004). Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 6, 499–506. [DOI] [PubMed] [Google Scholar]