

Abstract
Key points
Nitric oxide is essential in the vascular adaptation to pregnancy, as knockout mice lacking nitric oxide synthase (NOS3) have abnormal utero‐placental perfusion, hypertension and growth restriction.
We previously showed with ex vivo studies on transgenic animals lacking NOS3 that adverse intrauterine environment alters fetal programming of vascular reactivity in adult offspring.
The current research shows that altered vascular reactivity correlates with higher blood pressure in vivo.
Our data suggest that higher blood pressure depends on both genetic background (NOS3 deficiency) and uterine environment, becomes more evident with age (> 7 postnatal weeks), activity and stress, is gender specific (preponderant among males), and can be affected by the sleep–awake cycle.
In utero or early postnatal life (< 7 weeks), before onset of hypertension, may represent a potential window for intervention to prevent future cardiovascular disorders.
Abstract
Nitric oxide is involved in the vascular adaptation to pregnancy. Using transgenic animals, we previously showed that adverse intrauterine environment alters vascular reactivity in adult offspring. The aim of our study was to determine if altered vascular programming is associated with abnormal blood pressure (BP) profiles in vivo. Mice lacking a functional endothelial nitric oxide synthase (KO, NOS3−/−) and wild‐type mice (WT, NOS3+/+) were crossbred to generate homozygous NOS3−/− (KO), maternally derived heterozygous NOS3+/− (KOM: mother with adverse intrauterine environment from NOS3 deficiency), paternally derived heterozygous NOS3+/− (KOP: mother with normal in utero milieu) and NOS3+/+ (WT) litters. BP was measured in vivo at 7, 14 and 21 weeks of age. After univariate analysis, multivariate population‐averaged linear regression models were used to identify factors affecting BP. When compared to WT offspring, systolic (SBP), diastolic (DBP) and mean (MAP) BP progressively increased from KOP, to KOM, and peaked among KO (P < 0.001), although significance was not reached for KOP. Higher BP was also associated with male gender, older age (> 7 postnatal weeks), higher locomotor activity, daytime recordings, and recent blood pressure transducer insertion (P < 0.001). Post hoc analysis showed that KOM had higher SBP than KOP (P < 0.05). Our study indicates that adverse intrauterine environment contributes, along with multiple other factors, to account for hypertension; moreover, in utero or early postnatal life may represent a possible therapeutic window for prevention of cardiovascular disease later in life.
Keywords: aging, gender differences, fetal programming, hypertension, murine animal model
Key points
Nitric oxide is essential in the vascular adaptation to pregnancy, as knockout mice lacking nitric oxide synthase (NOS3) have abnormal utero‐placental perfusion, hypertension and growth restriction.
We previously showed with ex vivo studies on transgenic animals lacking NOS3 that adverse intrauterine environment alters fetal programming of vascular reactivity in adult offspring.
The current research shows that altered vascular reactivity correlates with higher blood pressure in vivo.
Our data suggest that higher blood pressure depends on both genetic background (NOS3 deficiency) and uterine environment, becomes more evident with age (> 7 postnatal weeks), activity and stress, is gender specific (preponderant among males), and can be affected by the sleep–awake cycle.
In utero or early postnatal life (< 7 weeks), before onset of hypertension, may represent a potential window for intervention to prevent future cardiovascular disorders.
Abbreviations
- BP
blood pressure
- DBP
diastolic blood pressure
- HR
heart rate
- KO
knockout
- KOM
maternally derived heterozygous offspring
- KOP
paternally derived heterozygous offspring
- MAP
mean arterial pressure
- NO
nitric oxide
- NOS3
nitric oxide synthase
- SBP
systolic blood pressure
Introduction
In 1986 Barker and Osmond proposed that the intrauterine environment impacts a fetus well beyond the perinatal period, demonstrating an association between low birth weight and the occurrence of hypertension, cardiovascular disease and diabetes later in life. Barker's hypothesis, which was subsequently named the ‘Developmental origin of health and disease’, suggests that insults to the fetus during critical periods of development lead to fetal programming and produce adaptive changes in the fetal anatomy, physiology and metabolism that have long‐term consequences (Barker et al. 1989, 2000; Barker, 1997).
The long‐term health of the offspring depends on a gene–environment interaction, as it is determined by both the intrauterine environment, which is influenced by the maternal genetic makeup, and the genetic makeup of the fetus/offspring, which in turn includes some of the same genes inherited from the mother. Therefore, a genetic trait that regulates vascular function in the mother may indirectly affect the fetus by influencing uteroplacental perfusion, as well as directly contribute to the long‐term health of the offspring if inherited by the fetus (Barker et al. 1989, 2000; Barker, 1997).
Epidemiological studies have suggested that both the ‘in utero’ environment and genetic factors play a fundamental role in the fetal origins of adult cardiovascular diseases (Huxley et al. 2002). To study these complex interactions, we used a transgenic mouse model lacking the endothelial nitric oxide synthase gene (NOS3), the rate‐limiting enzyme responsible for nitric oxide (NO) production from l‐arginine in endothelial cells (Furchgott, 1984; Ignarro, 1990). NO is a potent smooth muscle relaxant and an essential modulator of vascular tone (Palmer et al. 1987; Moncada et al. 1991); it also contributes to the maintenance of adequate uteroplacental perfusion through its vasodilatatory properties. Inhibition of NOS3 during pregnancy results in maternal hypertension and fetal growth restriction (Pallares & Gonzalez‐Bulnes, 2008; Tsukimori et al. 2008; Wagenseil et al. 2010). This animal model is well suited to study the contribution of each variable on fetal vascular programming and its long‐term effects, as it combines an adverse uterine environment with a maternal genetic predisposition to abnormal vascular function.
In prior work, transgenic male and female mice lacking a functional NOS3 gene [NOS3−/− or knockout (KO)] were crossbred with wild‐type mice [NOS3+/+ or wild type (WT)] to generate heterozygous offspring (NOS3+/−) that developed either in a mother lacking a functional NOS3 (maternally derived heterozygous offspring; NOS3+/− KOM) or in normal wild‐type mothers (paternally derived heterozygous offspring; NOS3+/− KOP). We have previously demonstrated that the abnormal uterine environment in which KOM offspring developed (KO mother) altered fetal and postnatal growth resulting in abnormal adult vascular function as compared to KOP offspring, which are genomically similar to KOM but developed in a normal uterine environment (WT mother). When compared to WT and KOP offspring, KO and KOM showed increased contractile response to phenylephrine and deficient endothelium‐dependent relaxation due to an impaired nitric oxide pathway, as such differences were abolished by incubation with l‐NAME, a non‐selective NOS inhibitor. Different genetic background could not entirely account for the different vascular reactivity profiles as KOM showed vascular responses similar to KO offspring while KOP had vascular responses similar to WT offspring. We also showed the progression of vascular dysfunction over time in the offspring, and characterized the gender differences (Longo et al. 2005; Chiossi et al. 2011). We hypothesized that these changes in vascular reactivity will ultimately lead to hypertension at some time in the life‐course of the offspring. Our objective in the current study was to determine the effect of an adverse intrauterine environment on the blood pressure (BP) of genomically similar offspring. We also intended to define the contributions of time, sex and diurnal variation to BP. Determining the temporal change and the most important factors contributing to hypertension would identify a potential therapeutic window and targets for future studies aiming to prevent cardiovascular disease later in life.
Methods
Ethical approval
Approval for the study was obtained from the Institutional Animal Care and Use Committee (IACUC).
Animals
Mature cycling female and male mice that are homozygous for disruption of the NOS3 gene (NOS3 knockout, strain B6.129P2‐Nos3tm1Unc, stock no. 002684, KO) and their age‐matched wild‐type controls (NOS3 wild type, strain C57BL/6J, stock no. 000664, WT) were purchased from Jackson Laboratory (Bar Harbor, ME, USA). The mice were housed separately in temperature‐ and humidity‐controlled quarters with constant 12:12 h light–dark cycles in the animal care facility at the University of Texas Medical Branch. Female and male KO mice and their WT controls were crossbred to obtain KO, WT, KOM and KOP litters. At least one animal per litter underwent genotype confirmation via PCR, as indicated by the standard protocol from Jackson Laboratory: identification of a 337 bp band would be indicative of WT, detection of a 258 bp band would be specific for KO, while heterozygous offspring would have both bands. There were three to four litters in each group; one or two offspring were randomly taken from each litter for BP recordings. Only one male and one female were sampled from the same litter. Different litters were homogeneous in size, as they were selected only if they consisted of six to eight animals. First‐generation offspring were killed using CO2 inhalation after BP recording, according to the IACUC and the American Veterinary Medical Association guidelines: animals were placed in a chamber containing room air, followed by a gradual fill of CO2. All surgical procedures were carried out by trained personnel according to the IACUC guidelines.
BP transducer insertion and haemodynamic measurements
In vivo BP was measured by telemetry in conscious unrestrained WT, KO, KOM and KOP offspring at 7, 14 and 21 weeks of age for up to 1 week [median 4 days, interquartile range (IQR): 2 – 7 days]. Animals were anaesthetized with a mixture of ketamine 100 mg kg−1 (Ketalar; Parke‐Davis, Morris Plains, NJ, USA) and xylazine 10 mg kg−1 (Gemini; Rugby, Rockville Center, NY, USA) injected intraperitoneally. After anaesthetic induction, animals were moved to a 37°C warming plate during surgery. Adequate anaesthesia was confirmed by observing the loss of the righting and palpebral reflexes, by assessing muscular tone, and by the response to painful stimulation. The left common carotid artery was located and carefully isolated through a vertical midline skin incision along the neck. A 0.4 mm catheter was then introduced into the carotid artery through a small incision in the vessel wall. The catheter was then tunnelled to a telemetric encased in a miniature capsule. The body of the transducer (PA‐C10 model, Data Sciences, St. Paul, MN, USA) was secured in a subcutaneous pouch. The neck incision was closed with 6‐0 silk. After surgery, animals were administered buprenorphine (Reckitt Benckiser Pharmaceuticals Inc., Richmond, VA, USA) subcutaneously at 0.1 mg kg−1 for postoperative pain, they were kept warm and monitored closely until fully recovered from anaesthesia. Animals were single housed after transducer insertion; data recording started 36 h after recovery from surgery.
The total number of activity counts reported by the telemetry system over each 30 s sample period was used as an index of locomotor activity, while BP was measured in mmHg. Systolic BP (SBP), diastolic BP (DBP), heart rate (HR) and locomotor activity were collected and averaged over 6 h‐periods. Mean arterial pressure (MAP) was calculated as (SBP + 2 × DBP)/3 from the recorded data. As haemodynamic recording was completed, offspring were killed as previously described.
Statistical analysis
Univariate analysis used descriptive statistics to characterize animals’ SBP, DBP, MAP and HR according to their genotype, age, sex, locomotor activity, time of haemodynamic recordings (daytime vs nighttime) and time from transducer insertion. The univariate approach provided data on how the aforementioned variables could affect haemodynamic parameters when considered independently from each other; moreover, it did not account for repeated measurements within each study animal. Normally distributed continuous variables were compared with one‐way ANOVA, or Student's t test as appropriate, and expressed as mean ± standard error of the mean (SEM), while associations between continuous variables were measured with Spearman's correlation. P ≤ 0.05 was considered statistically significant.
Variables significantly different on the univariate approach were included in multivariate analyses to identify determinants of SBP, DBP, MAP and HR. Multivariate analyses investigated if each variable significantly affected BP and HR when concomitantly studied with the others, instead of being evaluated independently. As cardiovascular parameters were repeatedly measured on each study animal at different time points, longitudinal models were utilized to overcome violation of the independence assumption. Population‐averaged linear regression models with an exchangeable correlation matrix and a robust variance estimator were used to study covariates affecting BP and HR. Each offspring was considered as a study unit in our population: inference on the entire population was based on a multivariate approach that analysed the haemodynamic recordings of all the study units, while controlling for confounding. Interactions between animal type and sex, and between animal type and age were investigated and noted when significant.
Post hoc analysis after multivariate estimation was performed to compare mean BP and HR within different study groups, instead of using a specific subpopulation as a fixed reference for comparisons. Therefore, the Wald test was used to assess if mean haemodynamic parameters differed according to genotype, age and sex.
Quasilikelihood under the independence model criterion (QIC) and its simplified version (QICU) were used to determine the best‐working correlation structure (Van Vliet & Chafe, 2007; Cui, 2007): the final models presented the smallest QIC and QICU values. Statistical analyses were performed using Stata 13.1 (StataCorp, College Station, TX, USA).
Results
Table 1 describes the genotype, age and sex of the animals undergoing telemetry, and it also indicates the median number of haemodynamic recordings for each study subgroup.
Table 1.
Genotype, age and sex of the animals undergoing haemodynamic recordings
| WT | KOP | KOM | KO | Total | |
|---|---|---|---|---|---|
| Male 7 weeks | |||||
| No. of animals | 8 | 4 | 6 | 4 | 22 |
| Observations (median IQR) | 29 (27–37) | 27 (17–28) | 27 (27–31) | 31 (24–37) | |
| Male 14 weeks | |||||
| No. of animals | 8 | 5 | 7 | 5 | 25 |
| Observations (median IQR) | 38 (29–42) | 27 (27–27) | 29 (29–33) | 31 (31–31) | |
| Male 21 weeks | |||||
| No. of animals | 7 | 4 | 4 | 5 | 20 |
| Observations (median IQR) | 32 (31–35) | 29 (28–30) | 25 (13–35) | 28 (28–47) | |
| Female 7 weeks | |||||
| No. of animals | 7 | 6 | 7 | 5 | 25 |
| Observations (median IQR) | 27 (17–28) | 28 (5–41) | 39 (27–43) | 23 (23–23) | |
| Female 14 weeks | |||||
| No. of animals | 8 | 7 | 4 | 4 | 23 |
| Observations (median IQR) | 38 (31–44) | 32 (32–39) | 27 (20–29) | 24 (18–31) | |
| Female 21 weeks | |||||
| No. of animals | 6 | 5 | 5 | 4 | 20 |
| Observations (median IQR) | 31 (30–35) | 32 (32–39) | 31 (31–33) | 35 (33–37) | |
| Total | 44 | 31 | 33 | 27 | 135 |
The number of vital sign assessments for the animals in each study subgroup is presented as median and interquartile range (IQR).
Univariate analysis showed that the lowest SBP, DBP and MAP were measured among WT offspring; a progressive increase in BP recordings was detected among KOP, followed by KOM, reaching the highest values among KO offspring (P < 0.001). BP also increased with age, male sex and in daytime (P < 0.001). Seven‐week‐old animals had lower SBP, DBP and MAP than older mice (P < 0.001), while only SBP and DBP were lower among 14‐ when compared to 21‐week‐old animals (P < 0.001). The lowest HR was observed at night, and among KO, older (> 7 postnatal weeks) as well as male offspring (P < 0.001). Haemodynamic parameters increased with animals’ locomotor activity and dropped with time from transducer insertion (P < 0.05) (Table 2).
Table 2.
Characteristics of the study population: univariate analysis
| SBP | P value | DBP | P value | MAP | P value | HR | P value | |
|---|---|---|---|---|---|---|---|---|
| Animal type (n = 135) | ||||||||
| WT (n = 44) | 120.8 ± 0.4 | 91.9 ± 0.4 | 101.5 ± 0.4 | 564.7 ± 1.5 | ||||
| KOP (n = 31) | 124.5 ± 0.5 | 95.9 ± 0.5 | 105.1 ± 0.5 | 566.8 ± 1.8 | ||||
| KOM (n = 33) | 130.2 ± 0.6 | 100.7 ± 0.6 | 110.5 ± 0.6 | 566.4 ± 1.9 | ||||
| KO (n = 27) | 138.7 ± 0.8 | 110.3 ± 0.8 | 119.8 ± 0.8 | 535.9 ± 2.8 | ||||
| < 0.001*, 1a | < 0.001*, 1a | < 0.001*, 1a | < 0.001*, 1b | |||||
| Age (n = 135) | ||||||||
| 7 weeks (n = 47) | 121.3 ± 0.5 | 93.7 ± 0.4 | 102.9 ± 0.4 | 570.4 ± 1.6 | ||||
| 14 weeks (n = 48) | 129.2 ± 0.5 | 101.3 ± 0.5 | 110.6 ± 0.5 | 556.7 ± 1.7 | ||||
| 21 weeks (n = 40) | 130.9 ± 0.5 | 99.5 ± 0.5 | 109.9 ± 0.5 | 554 ± 1.8 | ||||
| < 0.001*, 2a | < 0.001*, 2a | < 0.001*, 2b | < 0.001*, 2b | |||||
| Gender (n = 135) | ||||||||
| Female (n = 68) | 124.7 ± 0.4 | 95.5 ± 0.3 | 105.2 ± 0.3 | 566.2 ± 1.5 | ||||
| Male (n = 67) | 129.8 ± 0.5 | 101.2 ± 0.5 | 110.6 ± 0.4 | 554 ± 1.3 | ||||
| < 0.001† | < 0.001† | < 0.001† | < 0.001† | |||||
| Time (n = 135) | ||||||||
| Day | 128.8 ± 0.4 | 99.8 ± 0.4 | 109.5 ± 0.4 | 563.3 ± 1.4 | ||||
| Night | 125.6 ± 0.4 | 96.8 ± 0.4 | 106.3 ± 0.4 | 557.1 ± 1.4 | ||||
| < 0.001† | < 0.001† | < 0.001† | 0.001† | |||||
| Time from transducer insertion (n = 135) | −0.2 | < 0.001# | −0.11 | < 0.001# | –0.15 | < 0.001# | –0.1 | <0.001# |
| Activity (n = 135) | 0.03 | 0.03# | 0.06 | < 0.001# | 0.05 | < 0.001# | 0.3 | < 0.001# |
SBP, systolic blood pressure; DBP: diastolic blood pressure; MAP, mean arterial pressure; HR, heart rate. n = number of animals. Time from transducer insertion is expressed as a progressive number defining each 6 h interval after transducer placement. Activity: number of activity counts reported by the telemetry system over a 30 sample period. Data presented indicate how SBP, DBP, MAP and HR vary according to each categorical study variable (animal type, age, sex and time of recording; summarized by mean ± SEM) or how haemodynamic parameters correlate with the continues variables (time from transducer insertion, and activity; summarized by correlation coefficients).
*One‐way ANOVA followed by Bonferroni post hoc test: only significantly different comparisons are listed.
1aWT vs. KOP P < 0.001, WT vs. KOM P < 0.001, WT vs. KO P < 0.001, KOP vs. KOM P < 0.001, KOP vs. KO P < 0.001, KOM vs. KO P < 0.001.
1bWT vs. KO P < 0.001, KOP vs. KO P < 0.001, KOM vs. KO P < 0.001.
2a7 weeks vs. 14 weeks P < 0.001, 7 weeks vs. 21 weeks P < 0.001, 14 weeks vs. 21 weeks P = 0.03.
2b7 weeks vs. 14 weeks P < 0.001, 7 weeks vs. 21 weeks P < 0.001.
†Student's t test.
#Spearman's correlation.
Multivariate analysis confirmed the findings on the univariate approach. KOM and KO offspring had higher mean SBP, DBP and MAP than WT (P < 0.001). Higher SBP, DBP and MAP were detected during daytime recordings, as well as among older (> 7 postnatal weeks) and male animals (P < 0.001). KO had lower mean HR than WT (P = 0.02). BP and HR also rose with physical activity, and decreased with time from transducer insertion (P < 0.001) (Table 3). No significant interactions were found between animal type and sex, or between animal type and age.
Table 3.
Predictors of haemodynamic profiles: multivariate analysis
| SBP | DBP | MAP | HR | |||||
|---|---|---|---|---|---|---|---|---|
| β coefficient (95% CI) | P value | β coefficient (95% CI) | P value | β coefficient (95% CI) | P value | β coefficient (95% CI) | P value | |
| Animal type | ||||||||
| WT | 1* | – | 1* | – | 1* | – | 1* | – |
| KOP | 3.6 (–0.2 to 7.4) | 0.06 | 3.9 (–0.6 to 8.4) | 0.09 | 3.5 (–0.5 to 7.5) | 0.09 | 0.4 (–17.4 to 18.3) | 0.9 |
| KOM | 9.9 (4.5 to 15.2) | <0.001 | 7.8 (2.5 to 13) | 0.004 | 8.5 (3.6 to 13.4) | <0.001 | –5.1 (–22 to 11.7) | 0.5 |
| KO | 15.8 (9.9 to 21.8) | <0.001 | 16.5 (9.9 to 23.1) | <0.001 | 16.3 (10.2 to 22.4) | <0.001 | –31.9 (–57.7 to –6) | 0.02 |
| Age | ||||||||
| 7 weeks | 1* | – | 1* | – | 1* | – | – | – |
| 14 weeks | 10.1 (5.6 to 14.6) | <0.001 | 9.2 (4.6 to 13.8) | <0.001 | 9.5 (5.2 to 13.9) | <0.001 | – | – |
| 21 weeks | 11.8 (7.2 to 16.3) | <0.001 | 5.9 (1 to 10.8) | 0.02 | 7.7 (3.3 to 12.1) | 0.001 | – | – |
| Sex | ||||||||
| Female | 1* | – | 1* | – | 1* | – | – | – |
| Male | 7.8 (3.1 to 12.6) | 0.001 | 6.2 (2.9 to 9.5) | <0.001 | 6.7 (3 to 10.4) | <0.001 | – | – |
| Time | ||||||||
| Day | 1* | – | 1* | – | 1* | – | – | – |
| Night | –1.9 (–2.7 to –1.1) | <0.001 | –1.7 (–2.4 to –1) | <0.001 | –1.8 (–2.5 to –1.1) | <0.001 | – | – |
| Time from transducer insertion | –0.4 (–0.6 to –0.3) | <0.001 | –3 (–0.4 to –0.2) | <0.001 | –0.3 (–0.4 to –0.2) | <0.001 | –0.7 (–1.1 to –0.4) | <0.001 |
| Activity | 0.9 (0.8 to 1) | <0.001 | 0.8 (0.7 to 0.9) | <0.001 | 0.9 (0.8 to 1) | <0.001 | 3.8 (2.7 to 4.8) | <0.001 |
SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure; HR, heart rate. n = number of animals. Time from transducer insertion is expressed as a progressive number defining each 6 h interval after transducer placement. Activity: number of activity counts reported by the telemetry system over a 30 s sample period. The table presents the population‐averaged linear regression models investigating the determinants of SBP, DBP, MAP and HR. The β coefficient indicates the increase (positive value) or decrease (negative value) in mean blood pressure (expressed in mmHg) or heart rate (expressed in beats per minute) for a unitary change in the independent variables if covariates are continuous, or when compared to the reference group (*) in the case of categorical covariates. Each multivariate model is presented in a separate column, and was built on a population of 135 offspring. Quasilikelihood under the independence model for multivariate analysis: QIC = 1,140,805, QICU = 1,140,672 for SBP model, QIC = 1,173,284, QICU = 1,173,158 for DBP model, QIC = 1,086,101, QICU = 1,085,978 for MAP model, for HR model QIC = 14,362,332, QICU = 14,362,235.
Post hoc analysis showed that KOM offspring had higher BP than KOP, which were genomically similar but developed in a normal uterine environment, although significance was reached for SBP, but not for DBP or MAP (P < 0.05) (Fig. 1). The analysis of age and sex confirmed previous findings: SBP, DBP and MAP were significantly higher among older (7 weeks vs 14 and 21 weeks, P < 0.05) (Fig. 2), and male offspring (P < 0.05) (Fig. 3). Finally, KO showed the lowest mean HR when compared to other genotypes (P < 0.05) (Fig. 4)
Figure 1. Blood pressure and offspring genotype .
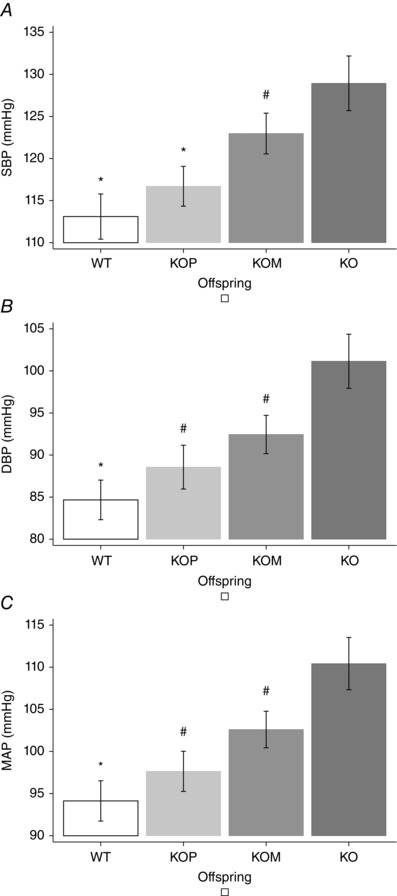
A, systolic blood pressure. B, diastolic blood pressure. C, mean arterial pressure. * P < 0.05 vs KOM and KO; # P < 0.05 vs KO, post hoc analysis after multivariate estimation using Wald test: differences in mean BP values are displayed according to animals’ genotype, after controlling for age, sex, locomotor activity, time of haemodynamic recordings and time from transducer insertion. SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure. Data presented as mean ± SEM.
Figure 2. Blood pressure and offspring age .
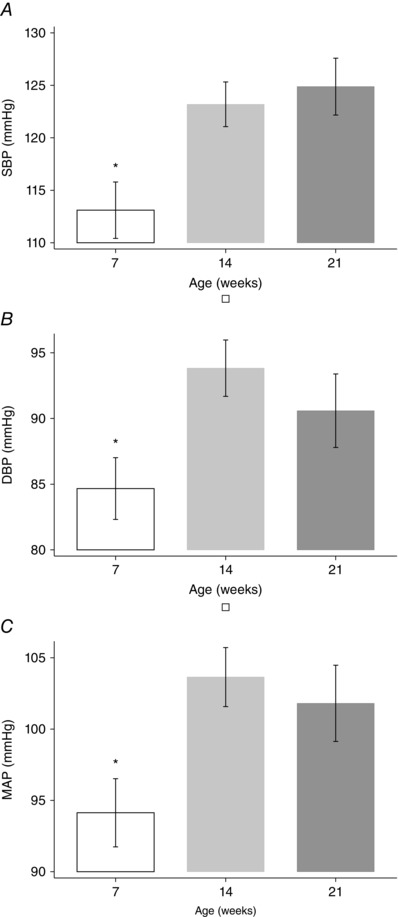
A, systolic blood pressure. B, diastolic blood pressure. C, mean arterial pressure. * P < 0.05 vs 14 and 21 weeks, post hoc analysis after multivariate estimation using Wald test: differences in mean BP values are displayed according to animals’ age, after controlling for genotype, sex, locomotor activity, time of haemodynamic recordings and time from transducer insertion. SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure. Data presented as mean ± SEM.
Figure 3. Blood pressure and offspring gender .
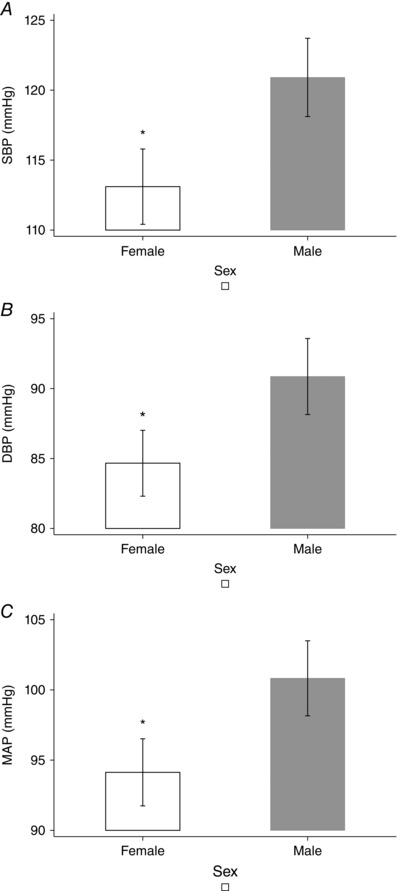
A, systolic blood pressure. B, diastolic blood pressure. C, mean arterial pressure. * P < 0.05, post hoc analysis after multivariate estimation using Wald test: differences in mean BP values are displayed according to animals’ sex, after controlling for age, genotype, locomotor activity, time of haemodynamic recordings and time from transducer insertion. SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure. Data presented as mean ± SEM.
Figure 4. Heart rate and offspring genotype .
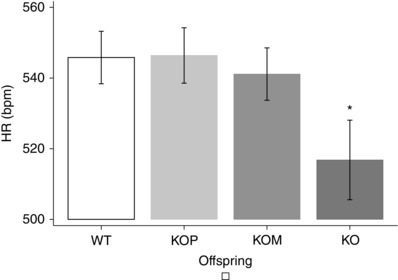
* P < 0.05 vs WT, KOP, KOM, post hoc analysis after multivariate estimation using Wald test: differences in mean HR values are displayed according to animals’ genotype, after controlling for age, sex, locomotor activity, time of haemodynamic recordings and time from transducer insertion. SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure. Data presented as mean ± SEM.
Discussion
In a transgenic mouse model deficient in endothelial NOS, the propensity for hypertension was affected by both genetic background and uterine environment, after controlling for age, sex, sleep–awake cycle, locomotor activity and time from BP transducer insertion.
In the multivariate analysis, SBP, DBP and MAP progressively increased from KOM (heterozygous for functional NOS3 gene, developing in an adverse uterine environment) to KO (lacking functional NOS3 genes, developing in an adverse uterine environment) when compared to WT mice (homozygous for functional NOS3 gene, developing in a normal uterine environment). Also, KOP offspring (heterozygous for functional NOS3 gene, developing in a normal uterine environment) had higher haemodynamic profiles than WT animals, but significance was not reached (Table 3). Subgroup analysis demonstrated that KOM had higher SBP, DBP and MAP than KOP, although only SBP reached statistical significance (P < 0.05) (Fig. 1). These results confirm previous work indicating that maternal NOS3 genotype affects an offspring's BP (Pan, 2001).
Our in vivo findings support previous ex vivo studies demonstrating the pivotal role of altered uterine environment in developmental programming of BP later in life (Longo et al. 2005; Chiossi et al. 2011). KO and KOM offspring had altered vascular profiles as opposed to WT and KOP, showing increased vascular reactivity and impaired endothelium‐dependent relaxation, all of which are considered risk factors for hypertension. Our prior results also highlighted the dominant effect of the intrauterine environment over genetic background as the heterozygous KOM offspring that developed in an abnormal uterine environment had vascular responses similar to the complete knockout KO, while their genomically similar KOP heterozygous offspring, which developed in a normal uterine environment, had vascular responses similar to the control WT. In an attempt to evaluate the mechanisms responsible for this developmental programming of vascular function, we previously evaluated NOS3 gene expression in the vasculature of these animals and found no differences between KOP and KOM (Costantine et al. 2008). To exclude genetic imprinting as a potential mechanism, we have also shown that the abnormal vascular phenotype of the adult KOM offspring (born to a KO mother) was abolished when KOM embryos were transferred to develop in WT surrogate mothers (Longo et al. 2003). These results suggest that the differences in vascular profile between heterozygous offspring are related to the uterine environment in which the pups develop, rather than genetics. Structural and cellular changes characteristic of uterine artery remodelling during pregnancy and gestational adaptation are markedly reduced in NOS3‐deficient mice, contributing to the poor utero‐placental perfusion and pregnancy outcome (Van der Heijden et al. 2005). In fact, NOS3 KO dams have significantly reduced cardiac output and altered uterine vascular reactivity profiles as compared to wild‐type mice, highlighting the differences in uterine environment between the KOM and KOP pups (Huang et al. 1995; Shesely et al. 1996; Kulandavelu et al. 2006; Kusinski et al. 2012).
Several mechanisms have been previously proposed and are thought to lead to abnormal fetal vascular programming. One putative mechanism involves structural changes in resistant vessels. Arterial wall stiffness is associated with adult hypertension. As vessel wall development is affected by blood flow and elastin synthesis (Li et al. 1998), hypertension in the adult may therefore be the result of decreased arterial compliance originating in fetal life secondary to reduced elastin deposition (Bendeck et al. 1994). In fact, elastin knockout mice manifest high BP (Wagenseil et al. 2010). Adverse structural vascular changes are known to occur with ageing, and this may explain the lower BP at 7 postnatal weeks but higher BP at 14 and 21 weeks in our animal model.
An adverse fetal environment may also lead to altered renal structure and function, leading to hypertension in adulthood (Barker et al. 1989; Geelhoed et al. 2009). Animal models support this hypothesis as well. Specifically, in the NOS3 knockout mouse model, we have shown that the glomeruli of 14‐week‐old KOM and KO offspring are decreased and have altered structure compared with KOP and WT offspring (Longo et al. 2004). Others have shown that KO mice have altered renal function associated with accelerated glomerular and tubulointerstitial injury with a loss of glomerular and peritubular capillaries (Nakayama et al. 2009). These changes, associated with the endothelial dysfunction seen in these KO and KOM mice, may lead to hypertension. Because nephrogenesis in mice is not completed until 2 weeks after birth, further haemodynamic changes related to the postnatal environment may contribute to the abnormal renal and vascular development occurring later in life.
Previous studies addressed the influence of the maternal NOS expression on an offspring's BP, without appropriately considering the effect of ageing (Van Vliet & Chafe, 2007). Our multivariate analysis evaluated the change over time between the various offspring, and underlined the potentiating of the effects of adverse uterine environment with ageing. As the offspring aged, the effect of fetal programming on cardiovascular function became more pronounced. Our blood pressure data are consistent with our prior studies showing that age amplified the effect of the uterine environment on vascular reactivity later in life. In fact, at 7 weeks of age, only KO offspring showed higher contractile response to phenylephrine (PE), whereas at 14 and 21 weeks both KO and KOM had significantly higher PE‐induced vascular contractility than WT and KOP (Longo et al. 2005; Chiossi et al. 2011). Together, our data underline that the full extent of developmental programming may not be apparent until later in life (Rammos et al. 2014), and provide insight about a potential window in utero or in the first postnatal weeks when interventions may be directed to prevent cardiovascular disease.
Important sex differences in BP profiles have been observed, as male offspring had higher BP than female offspring (Table 3, Fig. 3). Our findings are in line with previous studies documenting that KO male offspring had higher responses to contractile agents and lower relaxant responses at any age, whereas the differences in female offspring became evident only later in life (Chiossi et al. 2011). The increased arterial pressure seen in male offspring has been observed in different animal models of developmental programming (Musha et al. 2006). Sex‐specific variations may be related to differences in oestrogen concentrations or to the protective effect of the endothelium derived hyperpolarizing factor (EDHF). Indeed, female sex has been known to be protective against atherosclerosis and hypertension. Oestrogens improve vascular tone by decreasing myointimal smooth muscle cell proliferation (Sader & Celermajer, 2002), inhibiting tumour necrosis factor‐α‐induced apoptosis in endothelial cells (Mabley et al. 2005), and inducing NOS3 in different tissues (Zhu et al. 2002; Villar et al. 2008). Targeted disruption of both NOS and COX‐1 genes results in elevated BP in male but not in female mice, suggesting that EDHF may contribute to BP regulation in the latter group (Scotland et al. 2005). This mechanism seems to be maintained in this transgenic NOS3 model during offspring vascular programming, although this is speculative and requires further investigation (Scotland et al. 2005).
We showed a circadian rhythmicity of SBP, DBP and MAP (Table 3). Our findings confirm previous work in mice documenting BP changes over a 24 h period, with a peak during the day and nadir during the night. Although circadian rhythmicity was related to the expression of genes relevant to catecholamine synthesis, demonstrating an association between BP variations and sympathetic tone (Agarwai, 2010), higher daytime BP could also be related to more pronounced physical activity (Table 3).
Acute stress was found to produce MAP and HR changes related to an increased sympathetic activity and a reduction in BP buffering in a murine animal model (Farah et al. 2004); moreover, the adaptive sympathetic activation in response to stress led to the association between chronic stress and hypertension in mice (Costantine et al. 2009; Ruohonen et al. 2009). We demonstrated that BP declined with time from transducer insertion: along with a decrease in postoperative pain and stress, sympathetic activation probably dropped with normalization of haemodynamic parameters (Table 3). Studies addressing developmental programming may need to evaluate BP at different times of the day and under various activity and stress states.
As BP equals cardiac output × peripheral vascular resistance, and cardiac output is the product of stroke volume and heart rate, BP is also ultimately affected by heart rate. Although complex compensatory mechanisms interact to maintain cardiovascular haemostasis, KO offspring had significantly higher BP at lower HR, compared with KOM, KOP and WT (Table 3, Fig. 4), suggesting that their adverse cardiovascular profile is more likely to be related to altered peripheral vascular resistances, rather than altered cardiac function, a speculation supported by our previous in vitro vascular reactivity studies (Longo et al. 2005; Chiossi et al. 2011). Instead, we confirmed that HR depends on sympathetic tone, as it directly correlated with locomotor activity, and it was inversely associated with time from transducer insertion (Table 3).
Among the strengths of our study, the multivariate statistical approach allowed us to assess the concomitant contribution of different BP determinants. Our study also had some weaknesses: the study groups had different numbers, haemodynamic recordings had different durations and our study may not have the power to detect small differences among the variables of interest.
In conclusion, we demonstrated that while the impact of an abnormal uterine environment on fetal programming of cardiovascular function occurs early in life, its phenotype becomes more evident with age and activity, and depends on sex. Future studies should include long‐term follow up under various conditions. In utero or early postnatal life, before the phenotype becomes evident, may represent a potential window for nutritional, behavioural or pharmacological interventions to prevent long‐term adverse cardiovascular outcomes. Our findings emphasize the complexity of mechanisms involved in fetal cardiovascular development, suggesting that further research is needed to identify the pathways involved in the development of hypertension, and to develop effective prevention strategies.
Additional information
Competing interests
The authors report no conflict of interests.
Funding
The study was supported by NHLBI R01 HL080558‐02 grant, from the National Heart, Lung and Blood Institute.
Author contributions
Mice were housed in the animal care facility at the University of Texas Medical Branch, while BP transducer insertion, and haemodynamic recording were performed at the Maternal and Fetal Medicine Laboratory, Department of Obstetrics and Gynecology, University of Texas Medical Branch, Galveston, TX. The contribution of each author was as follows. GC: design of the work; data acquisition, analysis and interpretation; drafting the manuscript. MC: data acquisition, analysis and interpretation; revising the manuscript for important intellectual content. ET: data acquisition, analysis and interpretation; revising the manuscript for important intellectual content. GH: conception and design of the work; revising the manuscript for important intellectual content. GS: conception and design of the work; revising the manuscript for important intellectual content. ML: conception and design of the work; data acquisition, analysis and interpretation; revising the manuscript for important intellectual content. All authors approved the final version of the manuscript, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Moreover, all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
References
- Agarwai R (2010). Regulation of circadian blood pressure; from mice to astronauts. Curr Opin Neprol Hypertens 19, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ (1997). Maternal nutrition, fetal nutrition, and disease in later life. Nutrition 13, 807–813. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Golding J, Kuh D & Wadsworth ME (1989). Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 298, 564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Shiell AW, Barker EW & Law M (2000). Growth in utero and blood pressure levels in the next generation. J Hypertens 18, 843–846. [DOI] [PubMed] [Google Scholar]
- Bendeck MP, Keeley FW & Langille BL (1994). Perinatal accumulation of arterial wall constituents: relation to hemodynamic changes at birth. Am J Physiol Heart Circ Physiol 267, H2268–H2279. [DOI] [PubMed] [Google Scholar]
- Chiossi G, Costantine MM, Tamayo E, Hankins GD, Saade GR & Longo M (2011). Effect of age and gender on the progression of adult vascular dysfunction in a mouse model of fetal programming lacking endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol 301, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantine M, Yin H, Tamayo E & Longo M (2008). Fetal origin of adult diseases: genetic imprinting versus developmental programming. Am J Obstet Gynecol 199, S21. [Google Scholar]
- Costantine MM, Ferrari F, Chiossi G, Tamayo E, Hankins GD, Saade GR & Longo M (2009). Effect of intrauterine fetal programming on response to postnatal shaker stress in endothelial nitric oxide knockout mouse model. Am J Obstet Gynecol 201, 301e1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J (2007). QUIC program and model selection GEE analyses. Stata J 7, 209–220. [Google Scholar]
- Farah VM, Joaquim LF, Bernatova I & Morris M (2004). Acute and chronic stress influence blood pressure variability in mice. Physiol Behav 83, 135–142. [DOI] [PubMed] [Google Scholar]
- Furchgott RF (1984). The role of endothelium in the responses of vascular smooth muscle to drugs. Annu Rev Pharmacol Toxicol 24, 175–197. [DOI] [PubMed] [Google Scholar]
- Geelhoed JJ, Verburg BO, Nauta J, Lequin M, Hofman A, Moll HA, Witteman JC, van der Heijden AJ, Steegers EA & Jaddoe VW (2009). Tracking and determinants of kidney size from fetal life until the age of 2 years: the Generation R Study. Am J Kidney Dis 53, 248–258. [DOI] [PubMed] [Google Scholar]
- Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, & Fishman MC (1995). Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377, 239–242. [DOI] [PubMed] [Google Scholar]
- Huxley R, Neil A & Collins R (2002). Unravelling the fetal origins hypothesis: is there really an inverse association between birth weight and subsequent blood pressure? Lancet 360, 659–665. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ (1990). Biosynthesis and metabolism of endothelium‐derived nitric oxide. Annu Rev Pharmacol Toxicol 30, 535–560. [DOI] [PubMed] [Google Scholar]
- Kulandavelu S, Qu D & Adamson SL (2006). Cardiovascular function in mice during normal pregnancy and in the absence of endothelial NO synthase. Hypertension 47, 1175–1182. [DOI] [PubMed] [Google Scholar]
- Kusinski LC, Stanley JL, Dilworth MR, Hirt CJ, Andersson IJ, Renshall LJ, Baker BC, Baker PN, Sibley CP, Wareing M & Glazier JD (2012). eNOS knockout mouse as a model of fetal growth restriction with an impaired uterine artery function and placental transport phenotype. Am J Physiol Regul Integr Comp Physiol 303, R86–93. [DOI] [PubMed] [Google Scholar]
- Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E & Keating MT (1998). Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276–280. [DOI] [PubMed] [Google Scholar]
- Longo M, Jain V, Vedernikov YP, Bukowski R, Garfield RE & Hankins GD, Anderson GD & Saade GR (2005). Fetal origins of adult vascular dysfunction in mice lacking endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol 288, 1114–1121. [DOI] [PubMed] [Google Scholar]
- Longo M, Langenveld J, Vedernikov Y & Saade GR (2003). Abnormal vascular function in offspring of endothelial nitric oxide synthase knockout mice: genetic factors or fetal programming by uterine environment? Am J Obstet Gynecol 189, S91. [DOI] [PubMed] [Google Scholar]
- Longo M, Lu F, Snyder R, Anderson GD, Hankins GDV & Saade GR (2004). Abnormal renal development in a mouse model of fetal vascular programming. Am J Obstet Gynecol 191, S31. [Google Scholar]
- Mabley JG, Horváth EM, Murthy KG, Zsengellér Z, Vaslin A, Benko R, Kollai M & Szabó C (2005). Gender differences in the endotoxin‐induced inflammatory and vascular responses: potential role of poly (ADP‐ribose) polymerase activation. J Pharmacol Exp Ther 315, 812–820. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RMJ & Higgs EA (1991). Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 43, 109–142. [PubMed] [Google Scholar]
- Musha Y, Itoh S, Hanson MA & Kinoshita K (2006). Does estrogen affect the development of abnormal vascular function in offspring of rats fed a low‐protein diet in pregnancy? Pediatr Res 59, 784–789. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Sato W, Kosugi T, Zhang L, Campbell‐Thompson M, Yoshimura A, Croker BP, Johnson RJ & Nakagawa T (2009). Endothelial injury due to eNOS deficiency accelerates the progression of chronic renal disease in the mouse. Am J Physiol Renal Physiol 296, F317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallares P & Gonzalez‐Bulnes A (2008). Intrauterine growth retardation in endothelial nitric oxide synthase‐deficient mice is established from early stages of pregnancy. Biol Reprod 78, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Palmer RMJ, Ferrige AG & Moncada S (1987). Nitric oxide release accounts for the biological activity of endothelium‐derived relaxing factor. Nature 327, 524–526. [DOI] [PubMed] [Google Scholar]
- Pan W (2001). Akaike's information criterion in generalized estimating equations. Biometrics 57, 120–125. [DOI] [PubMed] [Google Scholar]
- Rammos C, Hendgen‐Cotta UB, Deenen R, Deenen R, Pohl J, Hinzmann C, Kelm M & Rassaf T (2014). Age‐related vascular gene expression profiling in mice. Mech Ageing Dev 135, 15–23. [DOI] [PubMed] [Google Scholar]
- Ruohonen ST, Savontaus E, Rinne P, Rosmaninho‐Salgado J, Cavadas C, Ruskoaho H, Koulu M & Pesonen U (2009). Stress‐induced hypertension and increased sympathetic activity in mice overexpressing neuropeptide Y in noradrenergic neurons. Neuroendocrinology 89, 351–360. [DOI] [PubMed] [Google Scholar]
- Sader MA & Celermajer DS (2002). Endothelial function, vascular reactivity and gender differences in the cardiovascular system. Cardiovasc Res 53, 597–604. [DOI] [PubMed] [Google Scholar]
- Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, Hobbs AJ & Ahluwalia A (2005). Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase‐1 double‐knockout mice: key role for endothelium‐derived hyperpolarizing factor in the regulation of blood pressure in vivo . Circulation 111, 796–803. [DOI] [PubMed] [Google Scholar]
- Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC & Smithies O (1996). Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA 93, 13176–13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukimori K, Komatsu H, Fukushima K, Kaku T, Nakano H & Wake N (2008). Inhibition of nitric oxide synthetase at mid‐gestation in rats is associated with increases in arterial pressure, serum tumor necrosis factor‐alpha, and placental apoptosis. Am J Hypertens 21, 477–481. [DOI] [PubMed] [Google Scholar]
- Van der Heijden OWH, Essers YPG, Fazzi G, Peeters LLH, De Mey GR, & Van Eys JM (2005). Uterine artery remodeling and reproductive performance are impaired in endothelial nitric oxide synthase‐deficient mice. Biol Reprod 72, 1161–1168. [DOI] [PubMed] [Google Scholar]
- Van Vliet BN & Chafe LL (2007). Maternal endothelial nitric oxide synthase genotype influences offspring blood pressure and activity in mice. Hypertension 49, 556–562. [DOI] [PubMed] [Google Scholar]
- Villar IC, Hobbs AJ & Ahluwalia A (2008). Sex differences in vascular function: implication of endothelium‐derived hyperpolarizing factor. J Endocrinol 197, 447–462. [DOI] [PubMed] [Google Scholar]
- Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A & Mecham RP (2010). The importance of elastin to aortic development in mice. Am J Physiol Heart Circ Physiol 299, H257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, Gustafsson JA & Mendelsohn ME (2002) Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science 18, 505–508. [DOI] [PubMed] [Google Scholar]