

Abstract
Apoptosis plays a critical role in the development of myocardial infarction. Cardiomyocytes are enriched with mitochondria and excessive mitochondrial fission can trigger cellular apoptosis. Recently, the mitochondrial ubiquitin ligase (MITOL), localized in the mitochondrial outer membrane, was reported to play an important role in the regulation of mitochondrial dynamics and apoptosis. However, the underlying mechanism of its action remains uncertain. The present study was aimed at uncovering the role of MITOL in the regulation of cardiomyocyte apoptosis. Our results showed that MITOL expression was up‐regulated in cardiomyocytes in response to apoptotic stimulation. Mitochondrial ubiquitin ligase overexpression blocked dynamin‐related protein 1 accumulation in the mitochondria, and attenuated the mitochondrial fission induced by hydrogen peroxide. Conversely, MITOL knockdown sensitized cardiomyocytes to undergo mitochondrial fission, resulting in subsequent apoptosis. These findings suggest that MITOL plays a protective role against apoptosis in cardiomyocytes, and may serve as a potential therapeutic target for apoptosis‐related cardiac diseases.
Keywords: MITOL, apoptosis, mitochondrial fission, cardiomyocytes, doxorubicin, hydrogen peroxide
Introduction
Apoptosis is essential for normal development and maintenance of tissue homeostasis 1, 2. In the cardiovascular system, for example, apoptosis participates in shaping the cardiac and vascular structures during early morphogenesis and in regulating the growth of established and differentiated cardiovascular tissues at later developmental stages 2. However, there is growing body of evidence, which shows that excessive apoptosis is related to many cardiovascular diseases such as myocardial infarction, cardiomyopathy, cardiac hypertrophy and anthracycline‐induced cardiotoxicity, etc. 3, 4, 5, 6. Reactive oxygen species play an important role in triggering apoptosis 7, 8, but the molecular mechanism by which they exert their effects remains to be fully understood.
Cardiomyocytes are enriched with mitochondria, which play an essential role in various cellular phenomena including ATP synthesis, lipid and iron metabolism, calcium buffering and cell death 9, 10. It has been recently demonstrated that the mitochondrial morphology is an important determinant of mitochondrial function 2, 11, 12. Mitochondria constantly undergo fusion and fission, which are necessary for the maintenance of organelle fidelity 13, 14, 15. At the same time, growing evidence has shown that abnormal mitochondrial fusion and fission also participate in the regulation of apoptosis 10, 11. Mitochondrial fusion is able to inhibit apoptosis, while mitochondrial fission is involved in the initiation of apoptosis 11, 13, 16. Although mitochondrial malfunction has been shown to be involved in brain and skeletal muscle disorders 17, 18, 19, 20, it remains largely unknown as to whether the abnormal mitochondrial fission and/or fusion play a role in regulating cardiomyocyte survival and death.
The mitochondrial ubiquitin ligase (MITOL), also known as MARCH5/RNF153 was recently reported to play a functional role in mitochondria 21. It can potentially modulate mitochondrial fission as well as mitochondrial morphology 21, 22. Mitochondrial ubiquitin ligase is characterized by its four transmembrane domains for the binding of mitochondrial fission proteins namely, human mitochondrial Fission 1 (hFis1), dynamin‐related protein 1 (Drp1) and mitofusin 2 23, 24, 25. However, whether MITOL plays a functional role in the mitochondrial dynamics and apoptosis of cardiomyocytes remains unknown. Accordingly, the current study was aimed at uncovering the role MITOL in the regulation of cardiomyocyte apoptosis induced by hydrogen peroxide (H2O2).
Materials and methods
Cell cultures and treatment
Mouse HL‐1 cardiomyocytes, kindly provided by Dr. William C. Claycomb, were cultured in Claycomb media supplemented with 10% foetal bovine serum (Sigma‐Aldrich, St. Louis, MO, USA), 0.1 mol/l norepinephrine (Sigma‐Aldrich), 2 mmol/l L‐glutamine (Invitrogen, Carlsbad, CA, USA) and penicillin/streptomycin (Invitrogen) in a humidified 5% CO2 incubator at 37°C 26. Primary rat cardiac myocytes (Lonza, Walkersville, MD, USA) were cultured in Rat Cardiac Myocyte Growth Media (RCGM; Lonza) containing horse serum, foetal bovine serum and gentamicin/amphotericin‐B, further supplemented with 200 μM 5‐bromo‐2′‐deoxyuridine in a humidified 5% CO2 incubator at 37°C. Cardiomyocytes were treated with identical concentration of H2O2 plus ferrous sulphate for 1 hr and further cultured in normal culture medium without H2O2 and ferrous sulphate as we have previously described 27.
Construction of MITOL expression vector
Myc‐DDK‐tagged ORF clone of Mus musculus membrane‐associated ring finger (C3H4)5 (MITOL) cDNA was cloned into pCMV6‐Entry (Origene, Rockville, MD, USA) according to the manufacturer's instruction. β‐galactosidase (β‐gal) was used as a control.
Construction of MITOL RNA interference (RNAi) vectors
For HL‐1 cells, the MITOL RNAi sense sequence was 5′‐AGGAGCATTTAAGGTTTACTTCAA ACAGC‐3′, and the antisense sequence was 5′‐GCTGTTTGAAGTAAACCTTAAATG CTCCT‐3′. The scramble MITOL RNAi sense sequence was 5′‐GCACTACCAGAGCTAACTCAGATAGTACT‐3′, and the antisense sequence was 5′‐AGTACTATCTGAGTTAGCTCTGGTAGTGC‐3′. They were cloned into the pGFP‐V‐RS shRNA retroviral vector (Origene) according to the manufacturer's instructions.
For primary neonatal rat cardiomyocyte, the MITOL‐shRNA sense sequence was 5′‐CTAAGTGGGTTCACCAGGCTTGTCTACAA‐3′, and the antisense sequence was 5′‐TTGTAGACAAGCCTGGTGAACCCACTTAG‐3′. The scramble MITOL‐shRNA sense sequence was 5′‐GCACTACCAGAGCTAACTCAGATAGTACT‐3′, and the antisense sequence was 5′‐AGTACTATCTGAGTTAGCTCTGGTAGTGC‐3′. They were cloned into the pGFP‐C‐shRNA lentiviral vector (Origene) according to the manufacturer's instructions.
Plasmid transfection
Using Lipofectamine 2000 (Invitrogen), we transfected the cells with plasmids expressing MITOL, MITOL‐siRNA, MITOL‐shRNA vectors or empty/scrambled siRNA/shRNA vectors according to the manufacturer's instructions.
Preparation of mitochondrial fractions
Mitochondrial fractions were prepared as we described 28. Briefly, cells were washed twice with PBS and the pellet was suspended in 0.2 ml of buffer A [20 mM HEPES pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM Ethylene Glycol Tetraacetic Acid (EGTA), 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM dithiothreitol (DTT), 0.1 mM PhenylMethane Sulfonyl Fluoride (PMSF), 250 mM sucrose] containing a protease inhibitor cocktail. The cells were homogenized by 12 strokes in a Dounce homogenizer. The homogenates were centrifuged twice at 750 × g for 5 min. at 4°C. The supernatants were centrifuged at 10,000 × g for 15 min. at 4°C to collect mitochondria‐enriched heavy membranes.
Analysis of mitochondrial fission
Mitochondrial fission was analysed by staining mitochondria as we and others have described earlier with some modification 29, 30. Briefly, cells were plated onto the coverslips coated with 0.01% poly‐l‐lysine. After treatment, they were stained for 20 min. with 0.02 μM MitoTracker Red CMXRos (Molecular Probes, Eugene, OR, USA). Mitochondria were imaged using a laser scanning confocal microscope (Zeiss LSM710 META, Dublin, CA, USA). To quantitatively analyse cells with mitochondria fission, those cells with disintegrated mitochondria were taken as mitochondrial fission. The percentage of cells with fragmented mitochondria relative to the total number of cells was presented as the mean ± S.E.M. of at least three independent experiments, counted by an observer blinded to the experimental conditions. A range of 100–150 cells in 20–30 random fields were counted.
Immunoblot analysis
Immunoblotting was carried out as previously described 31. Cells were lysed for 1 hr at 4°C in a lysis buffer (20 mM Tris pH 7.5, 2 mM EDTA, 3 mM EGTA, 2 mM DTT, 250 mM sucrose, 0.1 mM phenylmethylsulfonyl fluoride, 1% Triton X‐100) containing a protease inhibitor cocktail. Samples were subjected to 12% SDS‐PAGE and transfected to PVDF membrane (Millipore, Billerica, MA, USA). Equal protein loading was controlled by Ponceau Red staining of membranes. Blots were probed using primary antibodies, followed by horseradish peroxidase‐conjugated secondary antibodies. Anti‐MITOL polyclonal antibody was from Lifespan Biosciences. Anti‐cleaved‐PARP, anti‐Actin, anti‐β‐tubulin and anti‐Drp1 antibody were from Cell Signalling Technology Inc (Danvers, MA, USA). Anti‐Bid, ‐bad ‐caspase‐3 antibodies were from Santa Cruz Biotechnology Inc (Dallas, TX, USA). Antigen‐antibody complexes were visualized by enhanced chemiluminescence. The protein band intensity was quantified by ImageJ (National Institutes of Health, Bethesda, MD, USA) using protocol written by Luke Miller, November 2010 (http://www.lukemiller.org/ImageJ_gel_analysis.pdf). Briefly, the density of each sample was first quantified with image J, then the percent value of each sample and that of standard was calculated. Finally, the relative density was calculated by dividing the percent value of each sample by the percent value of each standard.
DNA fragmentation and apoptosis assays
DNA fragmentation was monitored using the cell death detection ELISA kit (Roche, Branford, CT, USA) as we have described elsewhere 28. Briefly, the anti‐histone monoclonal antibody was added to the 96 well ELISA plates and incubated overnight at 4°C. After recoating and three rinses, the cytoplasmic fractions were added and incubated for 90 min. at room temperature. After three washes, bound nucleosomes were detected by the addition of anti‐DNA peroxidase monoclonal antibody and reacted for 90 min. at room temperature. After the addition of the substrate, the optical density was determined at 405 nm using an ELISA reader. For apoptosis analysis, a terminal deoxynucleotidyl transferase‐mediated Dutp nick‐end‐labelling (TUNEL) kit (Clontech, Mountain View, CA, USA) was used according to the kit's instructions. One hundred and fifty to two hundred cells were counted in 20–30 random fields in each group.
Detection of caspase‐3 and ‐9 activities
Caspase activity was detected using caspase‐3 and ‐9 colorimetric assay kits (R&D Systems, Minneapolis, MN, USA). The assay procedures were followed according to the kit instructions. Protein concentration was determined using a Bio‐Rad (Hercules, CA, USA) protein assay kit.
Statistical analysis
Paired data were evaluated by Student's t‐test. A one‐way anova was used for multiple comparisons. A value of P < 0.05 was considered significant.
Results
MITOL is up‐regulated in cardiomocytes upon apoptosis induction by H2O2 and doxorubicin
To test whether MITOL participates in the regulation of mitochondrial fission in cardiomyocytes, we treated cardiomyocytes with H2O2 or Dox to induce apoptosis and mitochondrial fission. We found that the expression levels of MITOL in mitochondria were increased upon H2O2 exposure (Fig. 1A and B). Meanwhile, PAPR and caspase‐3 cleavage also increased in a same manner to H2O2 exposure (Fig. 1A and B), suggesting that MITOL could be a component in the apoptosis cascades induced by H2O2. Similarly, the expression levels of MITOL were increased upon Dox treatment in a dose‐ and time‐dependent manner (Fig. 2A, and B). Concomitantly, we observed an increase in the cleaved‐PARP (Fig. 2C, and D), suggesting that apoptosis occurred upon treatment with Dox. To further confirm the occurrence of apoptosis, we analysed for the levels of expression of other apoptotic factors. Our results demonstrated that both caspase‐3 and Bid were cleaved. Strikingly, we observed a significant increase in the cleavage of Drp1 after Dox treatment (Fig. 2E). Thus, it appears that alteration in the levels of expression of MITOL is also associated with Dox‐induced apoptosis in cardiomyocytes.
Figure 1.
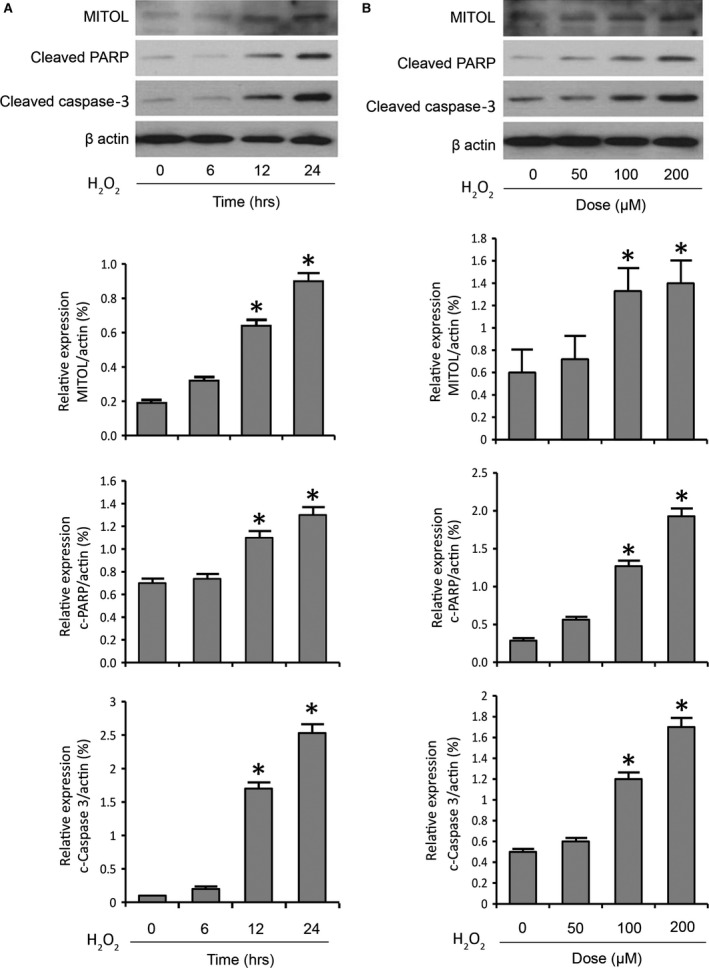
Hydrogen peroxide exposure leads to a time‐ and dose‐dependent up‐regulation of MITOL. (A) HL‐1 cells were stimulated with 200 μM hydrogen peroxide (H2O2) and then harvested at the indicated time for immunoblotting. (B) HL‐1 cells were stimulated with the indicated doses of H2O2 and then harvested for immunoblotting. (A and B) Immunoblots showing MITOL expression, PARP cleavage and caspase‐3 cleavage upon treatment of cells with H2O2. β‐actin served as a loading control. The densitometry data were expressed as the mean ± S.E.M. of three independent experiments. The relative expression level of protein was determined by dividing the percent value of specific protein to that of standard. *P < 0.05 versus non‐treated control.
Figure 2.
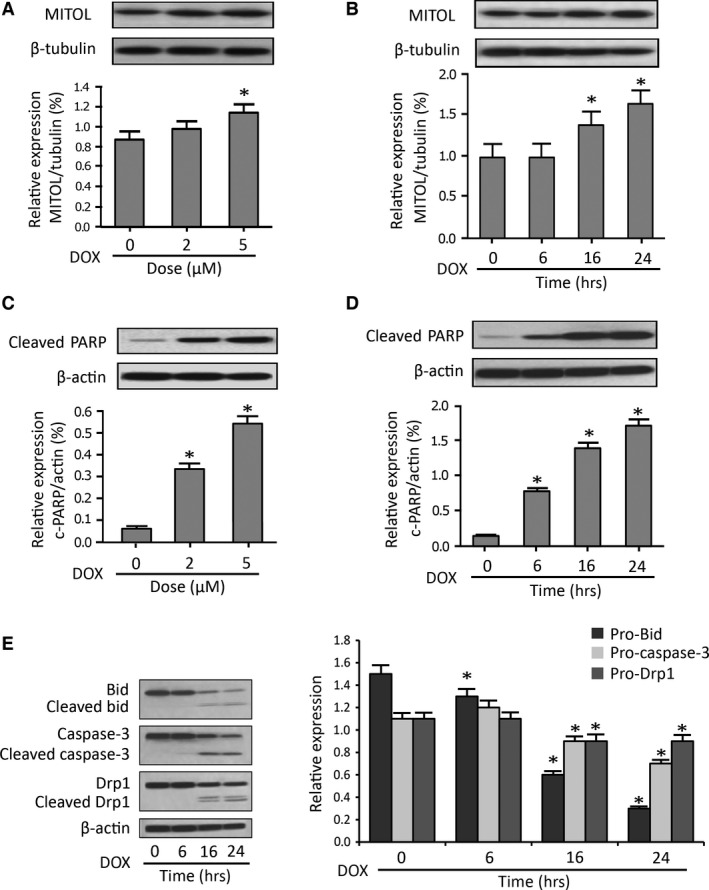
Doxorubicin induces a dose‐ and time‐ dependent up‐regulation of MITOL. (A and C) HL‐1 cells were stimulated with the indicated doses of Doxorubicin (DOX), and harvested for immunoblotting. (B, D and E) HL‐1 cells were stimulated with 5 μM Doxorubicin (DOX) and then harvested at the indicated time for immunoblotting. (A and B) It shows MITOL expression upon treatment with Dox. (C and D) It shows PARP cleavage upon treatment with DOX. (E) Immunoblots showing Bid, caspase‐3 and Drp1 expression and cleavage upon treatment of cells with Dox. β‐tubulin and β‐actin served as a loading control. The densitometry data were expressed as the mean ± S.E.M. of three independent experiments. The relative expression level of protein was determined by dividing the percent value of specific protein to that of standard. *P < 0.05 versus non‐treated control.
Overexpression of MITOL prevents mitochondrial fission and apoptosis
To determine if MITOL plays a critical role in mitochondrial fission, we expressed exogenous MITOL in cardiomyocytes. Transfection of cardiomyocytes with a MITOL expression vector resulted in elevated levels of MITOL expression (Fig. 3A and Fig. S1A, upper panel). Treatment with 200 μM of H2O2 caused mitochondrial fission; however, overexpression of MITOL inhibited mitochondrial fission as revealed by the mitochondrial morphology (Fig. 3B). Concomitantly, overexpression of MITOL could reduce the percentage of cells with mitochondrial fission (Fig. 3C). These data suggested that MITOL could prevent mitochondrial fission.
Figure 3.
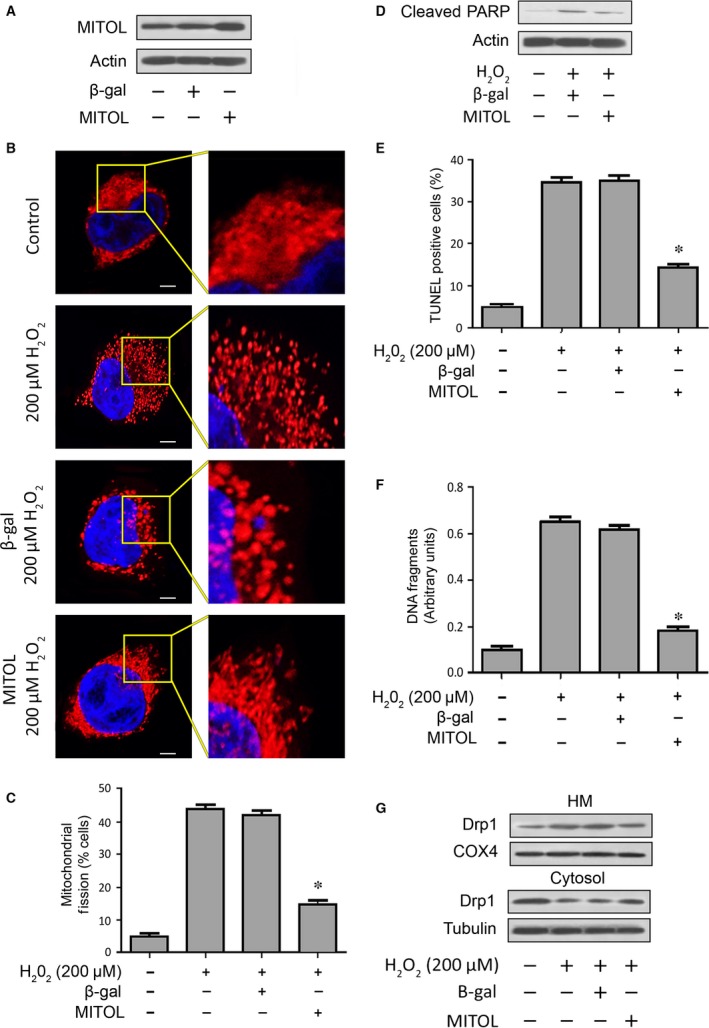
Enforced expression of exogenous MITOL prevents mitochondrial fission and apoptosis. (A) Analysis of MITOL expression. Immunoblots show overexpression of MITOL in HL‐1 cells. (B and C) Enforced expression of exogenous MITOL inhibits mitochondrial fission induced by hydrogen peroxide (H2O2). HL‐1 cells were treated with H2O2. (B) It shows mitochondrial morphology, bar = 2 μm. Cells with mitochondrial fission (C). *P < 0.05 versus H2O2 alone. (D) Enforced expression of MITOL inhibits PARP cleavage. Cleaved‐PARP was analysed by immunoblotting. (E) Enforced expression of MITOL attenuates apoptosis. HL‐1 cells were treated with H2O2. Apoptosis was analysed by TUNEL assay. *P < 0.05 versus H2O2 alone. Data are expressed as the mean ± S.E.M. of three independent experiments. (F) Enforced expression of exogenous MITOL attenuates DNA fragmentation. HL‐1 cells with and without exogenous MITOL expression were treated with H2O2. DNA fragments were analysed using the cell death detection ELISA. *P < 0.05 versus H2O2 alone. (G) Enforced expression of MITOL inhibits Drp1 accumulation in mitochondria. HM = mitochondria‐enriched heavy membranes.
To determine if exogenous MITOL expression affected apoptosis, we analysed several apoptotic events. Our results showed that PARP cleavage was reduced (Fig. 3D and Fig. S1A, lower panel). We quantitatively analysed apoptosis by employing TUNEL staining and cell death ELISA, both of which specifically detect apoptosis. Mitochondrial ubiquitin ligase could attenuate apoptosis as indicated by reduced TUNEL staining (Fig. 3E) and DNA fragmentation (Fig. 3F). These data suggested that exogenous MITOL is able to inhibit apoptosis in cardiomyocytes.
Finally, we detected the distribution of Drp1 and found that Drp1 accumulation in mitochondria was attenuated by MITOL (Fig. 3G and Fig. S1B). Thus, it suggests that exogenous MITOL could prevent Drp1 translocation.
Knockdown of MITOL sensitizes cardiomyocytes to mitochondrial fission and apoptosis
To further investigate the role of MITOL in the regulation of mitochondrial fission, we detected the cell fate upon knockdown of MITOL. To this end, we produced and used an RNAi construct to knock down MITOL. As shown in Figure 4A and Figure S2, the siRNA could reduce MITOL expression. In cells expressing MITOL, exposure to H2O2 (at a low dose) led to no significant alterations in mitochondrial morphology; however, the same low dose of H2O2 caused mitochondrial fission upon knockdown of MITOL (Fig. 4B). Consistently, upon knockdown of MITOL, low dose of H2O2 caused a significant increase in the number of cells undergoing mitochondrial fission (Fig. 4C). Moreover, cleaved‐PARP (Fig. 4D and Fig. S2), the percentages of TUNEL positive cells (Fig. 4E) and DNA fragments were significantly elevated (Fig. 4F), indicating an increase in apoptosis. These data suggested that endogenous MITOL participates in the inhibition of mitochondrial fission and apoptosis.
Figure 4.
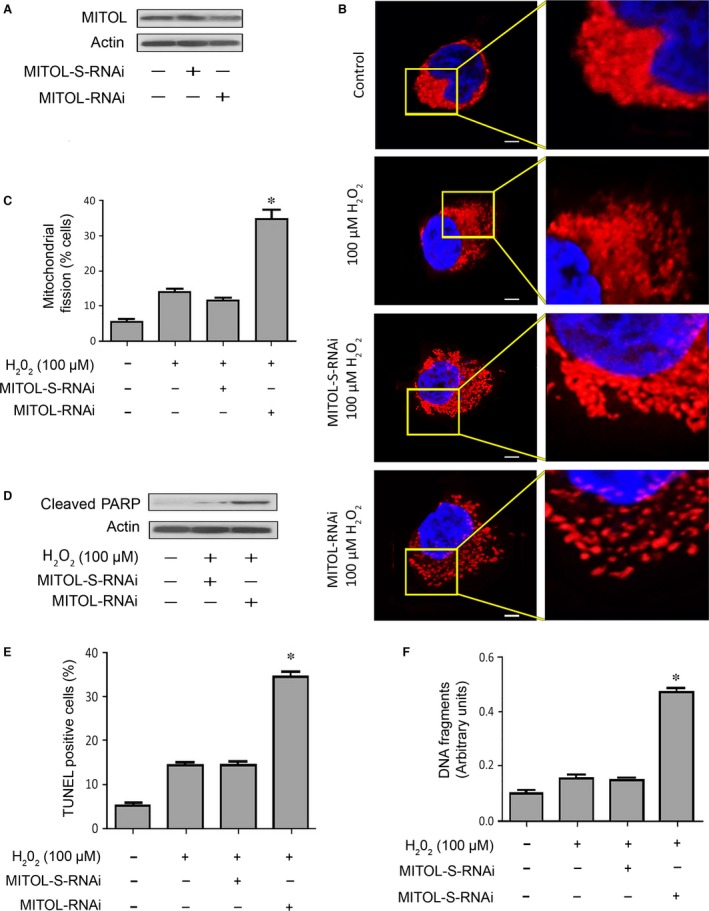
Knockdown of MITOL sensitizes cardiomyocytes to mitochondrial fission and apoptosis. (A) Immunoblot shows MITOL knockdown in HL‐1 cells. (B and C) Knockdown of MITOL sensitizes cells to undergo mitochondrial fission. HL‐1 cells were exposed to H2O2. (B) It shows mitochondrial morphology, bar = 2 μm. (C) It shows cells with mitochondrial fission; *P < 0.05 versus H2O2 alone. (D) Immunoblot shows cleaved‐PARP. (E) Knockdown of MITOL sensitizes cells to undergo apoptosis. Apoptosis was analysed by TUNEL assay. *P < 0.05 versus H2O2 alone. (F) Knockdown of MITOL sensitizes cells to undergo DNA fragmentation. DNA fragments were analysed using the cell death detection ELISA. *P < 0.05 versus H2O2 alone. Data were expressed as the mean ± S.E.M. of three independent experiments.
We further confirmed the effect of MITOL knockdown in primary neonatal rat cardiac myocytes. When MITOL‐shRNA was used to knockdown endogenous MITOL, a higher percentage of cells underwent mitochondrial fission upon H2O2 exposure, compared to negative control and scramble shRNA‐treated groups (Fig. 5A and B). Concomitantly, cell death ELISA showed that knockdown of MITOL increased H2O2‐induced cardiomyocyte apoptosis (Fig. 5C). Therefore, these findings further support the anti‐apoptotic effect of MITOL in primary cardiomyocytes.
Figure 5.
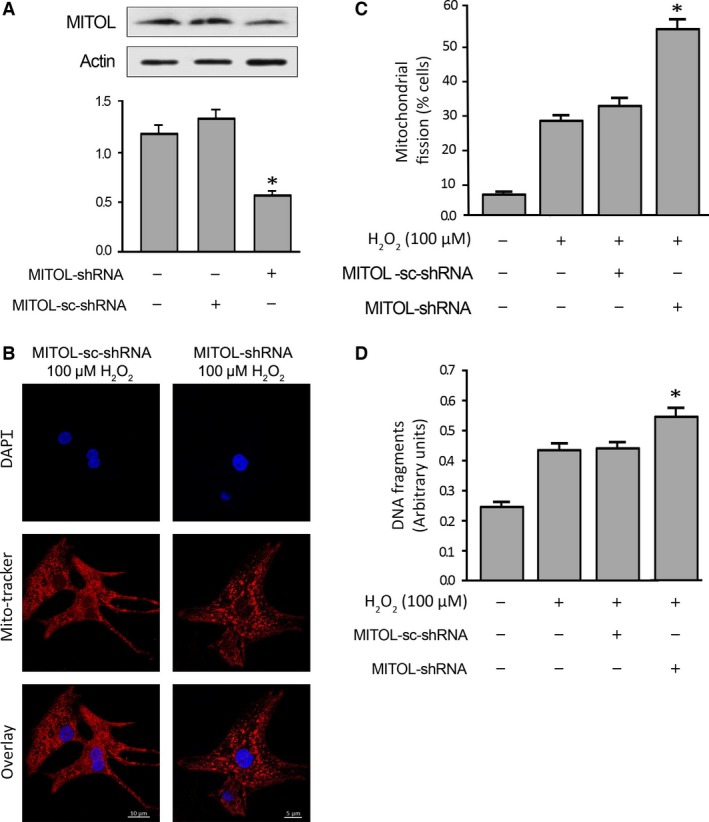
Knockdown of MITOL increases hydrogen peroxide‐induced mitochondrial fission and apoptosis in primary cardiomyocytes. (A, upper panel) Immunoblot shows MITOL knockdown in primary cardiomyocytes; (lower panel) the densitometry data were expressed as the mean ± S.E.M. of three independent experiments. The relative expression level of protein was determined by dividing the percent value of specific protein to that of standard. *P < 0.05 versus negative and scramble controls. (B and C) Knockdown of MITOL promotes mitochondrial fission. (B) It shows mitochondrial morphology. (C) It shows cells with mitochondrial fission; *P < 0.05 versus H2O2 alone. (D) Knockdown of MITOL promotes apoptosis. Apoptosis‐related DNA fragmentation was analysed using the cell death detection ELISA. *P < 0.05 versus H2O2 alone. Data were expressed as the mean ± S.E.M. of three independent experiments.
MITOL prevents caspase‐3 and caspase‐9 activation
Apoptosis is executed by activated caspases, we therefore analysed for the activities of caspase‐3 and caspase‐9, two caspases that can be activated by mitochondrial apoptotic pathway. As shown in Figure 6A and B, H2O2 could activate caspase‐3 and caspase‐9, respectively, and enforced expression of exogenous MITOL led to a reduction in the activities of these caspases. Furthermore, inhibition of caspase‐3 and caspase‐9 by their inhibitors could attenuate apoptosis (Fig. 6C), suggesting that they played a critical role in cardiomyocyte apoptosis. These findings further support that MITOL regulates the Drp1‐related mitochondrial fission and its downstream apoptotic pathway.
Figure 6.
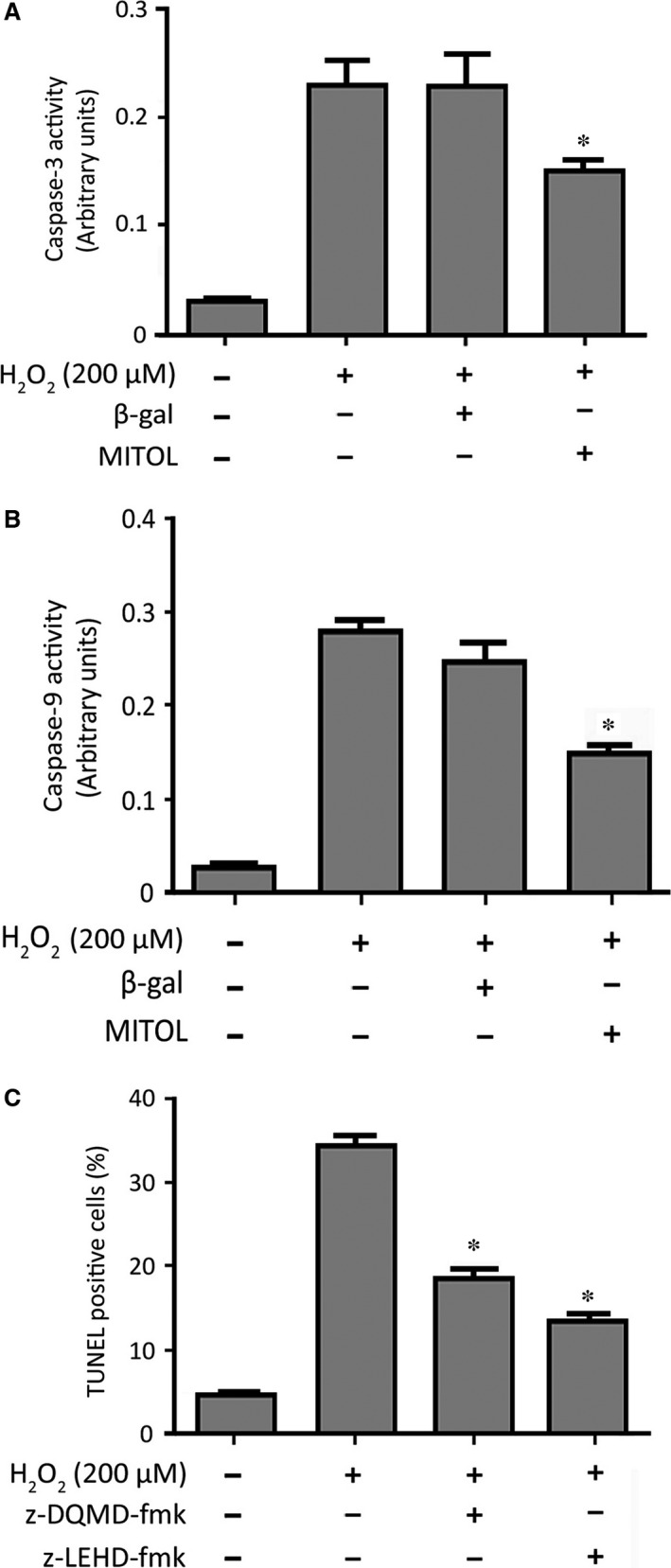
MITOL inhibits caspase‐3 and caspase‐9 activation. (A and B) It shows Caspase‐3 and Caspase‐9 activity, respectively; *P < 0.05 versus H2O2 alone. (C) Inhibition of caspase‐3 or caspase‐9 attenuates apoptosis. HL‐1 cells were treated with the inhibitor of caspase‐3 (Z‐DQMD‐FMK) or caspase‐9 (Z‐LEHD‐FMK) at 100 μM and then exposed to 200 μM of H2O2. Apoptosis was analysed by TUNEL assay. *P < 0.05 versus H2O2 alone. Data were expressed as the mean ± S.E.M. of three independent experiments.
Discussion
Complex molecular mechanisms regulate apoptosis in cardiomyocytes 32; and participate in the regulation of a variety of cardiac diseases 3, 4, 5, 33, 34. Amongst those mechanisms is mitochondrial fusion and fission events, which controls apoptosis 10. Mitochondrial ubiquitin ligase has been shown to be involved in mitochondrial dynamics and mitochondrial quality control 21, 23, 35, 36. However, the exact role of MITOL in cardiomyocyte mitochondrial function is still obscure. In this study, we found that MITOL attenuates mitochondrial fission and apoptosis induced by H2O2 or Dox, most likely by inhibiting Drp1 accumulation in mitochondria. Mitochondrial ubiquitin ligase, an ubiquitin ligase, is characterized by its four transmembrane domains that help anchor the protein in the mitochondrial outer membrane 23, 24, 25. Mitochondrial ubiquitin ligase interacts with both fission proteins (hFis1 and Drp1) as well as fusion proteins 23, 24, 37. Mitochondrial ubiquitin ligase plays a critical role in regulating mitochondrial dynamic by regulating the function of mitochondrial fission and fusion proteins 21, 22, 23. Dynamin‐related protein 1 is one of the key proteins in the control of mitochondrial fission 13, 29, 38 and is involved in cytochrome c release from mitochondria into the cytosol 29. Cytochrome c binds to apoptosis protease‐activating factor 1 and procaspase‐9 to form apoptosomes resulting in the activation of caspase‐9 39, 40.
In the current study, we found that MITOL overexpression inhibited Drp1 accumulation, and attenuated mitochondrial fission and apoptosis. Conversely, MITOL knockdown induced the cells to undergo mitochondrial fission and apoptosis. It has been noted that the MITOL‐dependent Drp1 regulation tends to vary by cell conditions including cell cycle phase or nutritional status 23. For instance, MITOL knockdown in mouse embryonic fibroblasts showed similar regulatory effect as has been shown in our current study 23. But in human HeLa cells, a completely opposite effect was noted in that MITOL was required for Drp1‐dependent mitochondrial division, and inhibition of MITOL leads to mitochondrial fusion in HeLa cells 24, 37. Furthermore MITOL is required for degradation of mitochondrial fusion protein Mfn1 in LNCaP prostate cancer cells treated with CGP37157 (CGP), an inhibitor of mitochondrial calcium efflux, and resulted in mitochondrial fission. Comparably, knockdown of MITOL reduced Mfn1 degradation, which in turn elevated Mfn1 levels and thus, promoted mitochondrial fusion 41. Another interesting study, using an in‐vitro neuronal cell (RGC5) exposed to glaucoma‐relevant stress conditions, found that mitochondrial fission was significantly blocked in cell expressing inactive MITOL and did significantly delay the cell death 42. These evidence further substantiate the contrasting function of MITOL in different cell types under various cellular stress conditions.
The specific roles of MITOL in mitochondria have not yet been fully elucidated. It is reported that MITOL is able to eliminate misfolded proteins localized in mitochondria, such as mutant superoxide dismutase 1 43, and mutant short chain acyl CoA dehydrogenase 44, which exacerbate neuronal disorders. Thus, MITOL plays an important role in protecting neuronal cell death by degrading the accumulated denatured proteins in mitochondria 23. It is of note that mitochondrial fission and fusion are controlled by a complex molecular mechanism in which a variety of proteins such as optic atrophy 1, hFis1, and mitochondrial division 1 are involved 14, 16, 45. It is critical for future studies to investigate whether these molecules participate in the regulation of mitochondrial fission process in the heart. Therefore, it is also necessary to elucidate whether ubiquitination by MITOL is required for the regulation of mitochondrial dynamic. Interestingly, we noticed that mitochondrial morphology in HL‐1 cell lines in fusion state were packed into a network rather than long thin filamentous configuration, which is commonly seen in cardiac fibroblasts (Fig. S3). This morphological discrepancy highlights that mitochondrial configuration in different cell types can be varied depending on their functions and histological backgrounds 10, 30, 46, 47.
In conclusion, this study reveals that MITOL is involved in the mitochondrial fission machinery of cardiomyocyte apoptosis. Mitochondrial ubiquitin ligase attenuates the mitochondrial fission induced by H2O2 and blocks Drp1 accumulation in the mitochondria of cardiomyocytes. Thus, MITOL may serve as a novel therapeutic target for apoptosis‐related cardiac diseases.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Author contribution
Designed the experiments: PL, JW and LHHA. Performed the experiments: LHHA and JW. Analysed the data: LHHA, JW and PL. Wrote the paper: LHHA, JW, PL and BP.
Supporting information
Figure S1 (A, upper panel) Quantitative densitometry of immunoblot for expression levels of MITOL (corresponded to Fig. 3A, *P < 0.05 versus negative control or β‐gal) and (lower panel) PARP cleavage (corresponded to Fig. 3D, *P < 0.05 versus negative control or β‐gal treated with 200 μM H2O2).
Figure S2 (upper panel) Quantitative densitometry of immunoblot for expression levels of MITOL (corresponded to Fig. 4A, *P < 0.05 versus MITOL‐S‐RNAi) and (lower panel) PARP cleavage (corresponded to Fig. 4D, *P < 0.05 versus non‐treated control or MITOL‐S‐RNAi treated with 100 μM H2O2).
Figure S3 Mitochondrial morphology during fusion state in non‐treated rat primary cardiac fibroblast (A), rat primary cardiomyocyte (B) and HL‐1 cells (C).
Acknowledgements
We would like to thank Dr. William C. Claycomb (Departments of Biochemistry and Molecular Biology, Louisiana State University Medical Center, New Orleans, LA 70112) for providing HL‐1 murine cardiomyocyte cell line. This work was supported by a grant 5R01HL102202 from the National Institutes of Health, Bethesda, MD, and a grant from American Cancer Society (120861).
References
- 1. Cotter TG, Lennon SV, Glynn JG, et al Cell death via apoptosis and its relationship to growth, development and differentiation of both tumour and normal cells. Anticancer Res. 1989; 10: 1153–9. [PubMed] [Google Scholar]
- 2. James TN. Normal and abnormal consequences of apoptosis in the human heart. From postnatal morphogenesis to paroxysmal arrhythmias. Circulation. 1994; 90: 556–73. [PubMed] [Google Scholar]
- 3. Anversa P, Cheng W, Liu Y, et al Apoptosis and myocardial infarction. Basic Res Cardiol. 1998; 93 (Suppl. 3): 8–12. [DOI] [PubMed] [Google Scholar]
- 4. Narula J, Haider N, Virmani R, et al Apoptosis in myocytes in end‐stage heart failure. N Engl J Med. 1996; 335: 1182–9. [DOI] [PubMed] [Google Scholar]
- 5. Frohman MA. Role of mitochondrial lipids in guiding fission and fusion. J Mol Med (Berl). 2015; 93: 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu W, Wang X, Mei Z, et al Chronic stress promotes the progression of pressure overload‐induced cardiac dysfunction through inducing more apoptosis and fibrosis. Physiol Res. 2014; 22: 22. [DOI] [PubMed] [Google Scholar]
- 7. Chen Z, Jiang H, Wan Y, et al H2O2‐induced secretion of tumor necrosis factor‐alpha evokes apoptosis of cardiac myocytes through reactive oxygen species‐dependent activation of p38 MAPK. Cytotechnology. 2012; 64: 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hosseinzadeh L, Behravan J, Mosaffa F, et al Curcumin potentiates doxorubicin‐induced apoptosis in H9c2 cardiac muscle cells through generation of reactive oxygen species. Food Chem Toxicol. 2011; 49: 1102–9. [DOI] [PubMed] [Google Scholar]
- 9. McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006; 16: R551–60. [DOI] [PubMed] [Google Scholar]
- 10. Cosentino K, Garcia‐Saez AJ. Mitochondrial alterations in apoptosis. Chem Phys Lipids. 2014; 181: 62–75. [DOI] [PubMed] [Google Scholar]
- 11. Suen D‐F, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008; 22: 1577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nasrallah CM, Horvath TL. Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol. 2014; 10: 650–8. [DOI] [PubMed] [Google Scholar]
- 13. Tanaka A, Youle RJ. A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol Cell. 2008; 29: 409–10. [DOI] [PubMed] [Google Scholar]
- 14. Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007; 8: 870–9. [DOI] [PubMed] [Google Scholar]
- 15. Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012; 46: 265–87. [DOI] [PubMed] [Google Scholar]
- 16. Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006; 22: 79–99. [DOI] [PubMed] [Google Scholar]
- 17. Keeney PM, Xie J, Capaldi RA, et al Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006; 26: 5256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corral‐Debrinski M, Horton T, Lott MT, et al Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992; 2: 324–9. [DOI] [PubMed] [Google Scholar]
- 19. Schapira AHV, Cooper JM, Dexter D, et al Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990; 54: 823–7. [DOI] [PubMed] [Google Scholar]
- 20. Mann VM, Cooper JM, Krige D, et al Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson's disease. Brain. 1992; 115: 333–42. [DOI] [PubMed] [Google Scholar]
- 21. Yonashiro R, Ishido S, Kyo S, et al A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006; 25: 3618–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagashima S, Yanagi S. [Role of MITOL in mitochondrial dynamics and diseases]. Seikagaku. 2014; 86: 63–7. [PubMed] [Google Scholar]
- 23. Nagashima S, Tokuyama T, Yonashiro R, et al Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J Biochem. 2014; 155: 273–9. [DOI] [PubMed] [Google Scholar]
- 24. Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007; 178: 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakamura N, Kimura Y, Tokuda M, et al MARCH‐V is a novel mitofusin 2‐ and Drp1‐binding protein able to change mitochondrial morphology. EMBO Rep. 2006; 7: 1019–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Claycomb WC, Lanson NA Jr, Stallworth BS, et al HL‐1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998; 95: 2979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li YZ, Lu DY, Tan WQ, et al p53 initiates apoptosis by transcriptionally targeting the antiapoptotic protein ARC. Mol Cell Biol. 2008; 28: 564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li PF, Dietz R, von Harsdorf R. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c‐independent apoptosis blocked by Bcl‐2. EMBO J. 1999; 18: 6027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frank S, Gaume B, Bergmann‐Leitner ES, et al The role of dynamin‐related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001; 1: 515–25. [DOI] [PubMed] [Google Scholar]
- 30. Wang JX, Li Q, Li PF. Apoptosis repressor with caspase recruitment domain contributes to chemotherapy resistance by abolishing mitochondrial fission mediated by dynamin‐related protein‐1. Cancer Res. 2009; 69: 492–500. [DOI] [PubMed] [Google Scholar]
- 31. Murtaza I, Wang HX, Feng X, et al Down‐regulation of catalase and oxidative modification of protein kinase CK2 lead to the failure of apoptosis repressor with caspase recruitment domain to inhibit cardiomyocyte hypertrophy. J Biol Chem. 2008; 283: 5996–6004. [DOI] [PubMed] [Google Scholar]
- 32. Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. 2013; 19: 546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwarz K, Simonis G, Yu X, et al Apoptosis at a distance: remote activation of caspase‐3 occurs early after myocardial infarction. Mol Cell Biochem. 2006; 281: 45–54. [DOI] [PubMed] [Google Scholar]
- 34. Caroppi P, Sinibaldi F, Fiorucci L, et al Apoptosis and human diseases: mitochondrion damage and lethal role of released cytochrome C as proapoptotic protein. Curr Med Chem. 2009; 16: 4058–65. [DOI] [PubMed] [Google Scholar]
- 35. Sugiura A, Nagashima S, Tokuyama T, et al MITOL regulates endoplasmic reticulum‐mitochondria contacts via Mitofusin2. Mol Cell. 2013; 51: 20–34. [DOI] [PubMed] [Google Scholar]
- 36. Yonashiro R, Kimijima Y, Shimura T, et al Mitochondrial ubiquitin ligase MITOL blocks S‐nitrosylated MAP1B‐light chain 1‐mediated mitochondrial dysfunction and neuronal cell death. Proc Natl Acad Sci USA. 2012; 109: 2382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Park YY, Lee S, Karbowski M, et al Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin‐related protein 1 and mitofusin 1. J Cell Sci. 2010; 123: 619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007; 177: 439–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reubold TF, Eschenburg S. A molecular view on signal transduction by the apoptosome. Cell Signal. 2012; 24: 1420–5. [DOI] [PubMed] [Google Scholar]
- 40. Park HH. Structural features of caspase‐activating complexes. Int J Mol Sci. 2012; 13: 4807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Choudhary V, Kaddour‐Djebbar I, Alaisami R, et al Mitofusin 1 degradation is induced by a disruptor of mitochondrial calcium homeostasis, CGP37157: a role in apoptosis in prostate cancer cells. Int J Oncol. 2014; 44: 1767–73. [DOI] [PubMed] [Google Scholar]
- 42. Fang L, Hemion C, Goldblum D, et al Inactivation of MARCH5 prevents mitochondrial fragmentation and interferes with cell death in a neuronal cell model. PLoS ONE. 2012; 7: e52637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yonashiro R, Sugiura A, Miyachi M, et al Mitochondrial ubiquitin ligase MITOL ubiquitinates mutant SOD1 and attenuates mutant SOD1‐induced reactive oxygen species generation. Mol Biol Cell. 2009; 20: 4524–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Livnat‐Levanon N, Glickman MH. Ubiquitin‐proteasome system and mitochondria ‐ reciprocity. Biochim Biophys Acta. 2011; 2: 80–7. [DOI] [PubMed] [Google Scholar]
- 45. Berman SB, Pineda FJ, Hardwick JM. Mitochondrial fission and fusion dynamics: the long and short of it. Cell Death Differ. 2008; 15: 1147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang K, Zhou LY, Wang JX, et al E2F1‐dependent miR‐421 regulates mitochondrial fragmentation and myocardial infarction by targeting Pink1. Nat Commun. 2015; 6: 7619. [DOI] [PubMed] [Google Scholar]
- 47. Wang K, Long B, Zhou LY, et al CARL lncRNA inhibits anoxia‐induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR‐539‐dependent PHB2 downregulation. Nat Commun. 2014; 5: 3596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A, upper panel) Quantitative densitometry of immunoblot for expression levels of MITOL (corresponded to Fig. 3A, *P < 0.05 versus negative control or β‐gal) and (lower panel) PARP cleavage (corresponded to Fig. 3D, *P < 0.05 versus negative control or β‐gal treated with 200 μM H2O2).
Figure S2 (upper panel) Quantitative densitometry of immunoblot for expression levels of MITOL (corresponded to Fig. 4A, *P < 0.05 versus MITOL‐S‐RNAi) and (lower panel) PARP cleavage (corresponded to Fig. 4D, *P < 0.05 versus non‐treated control or MITOL‐S‐RNAi treated with 100 μM H2O2).
Figure S3 Mitochondrial morphology during fusion state in non‐treated rat primary cardiac fibroblast (A), rat primary cardiomyocyte (B) and HL‐1 cells (C).