

Abstract
Key points
Optogenetics‐based defibrillation, a theoretical alternative to electrotherapy, involves expression of light‐sensitive ion channels in the heart (via gene or cell therapy) and illumination of the cardiac surfaces (via implanted LED arrays) to elicit light‐induced activations.
We used a biophysically detailed human ventricular model to determine whether such a therapy could terminate fibrillation (VF) and identify which combinations of light‐sensitive ion channel properties and illumination configurations would be effective.
Defibrillation was successful when a large proportion (> 16.6%) of ventricular tissue was directly stimulated by light that was bright enough to induce an action potential in an uncoupled cell.
While illumination with blue light never successfully terminated VF, illumination of red light‐sensitive ion channels with dense arrays of implanted red light sources resulted in successful defibrillation.
Our results suggest that cardiac expression of red light‐sensitive ion channels is necessary for the development of effective optogenetics‐based defibrillation therapy using LED arrays.
Abstract
Optogenetics‐based defibrillation has been proposed as a novel and potentially pain‐free approach to enable cardiomyocyte‐selective defibrillation in humans, but the feasibility of such a therapy remains unknown. This study aimed to (1) assess the feasibility of terminating sustained ventricular fibrillation (VF) via light‐induced excitation of opsins expressed throughout the myocardium and (2) identify the ideal (theoretically possible) opsin properties and light source configurations that would maximise therapeutic efficacy. We conducted electrophysiological simulations in an MRI‐based human ventricular model with VF induced by rapid pacing; light sensitisation via systemic, cardiac‐specific gene transfer of channelrhodopsin‐2 (ChR2) was simulated. In addition to the widely used blue light‐sensitive ChR2‐H134R, we also modelled theoretical ChR2 variants with augmented light sensitivity (ChR2+), red‐shifted spectral sensitivity (ChR2‐RED) or both (ChR2‐RED+). Light sources were modelled as synchronously activating LED arrays (LED radius: 1 mm; optical power: 10 mW mm–2; array density: 1.15–4.61 cm–2). For each unique optogenetic configuration, defibrillation was attempted with two different optical pulse durations (25 and 500 ms). VF termination was only successful for configurations involving ChR2‐RED and ChR2‐RED+ (for LED arrays with density ≥ 2.30 cm–2), suggesting that opsin spectral sensitivity was the most important determinant of optogenetic defibrillation efficacy. This was due to the deeper penetration of red light in cardiac tissue compared with blue light, which resulted in more widespread light‐induced propagating wavefronts. Longer pulse duration and higher LED array density were associated with increased optogenetic defibrillation efficacy. In all cases observed, the defibrillation mechanism was light‐induced depolarisation of the excitable gap, which led to block of reentrant wavefronts.
Key points
Optogenetics‐based defibrillation, a theoretical alternative to electrotherapy, involves expression of light‐sensitive ion channels in the heart (via gene or cell therapy) and illumination of the cardiac surfaces (via implanted LED arrays) to elicit light‐induced activations.
We used a biophysically detailed human ventricular model to determine whether such a therapy could terminate fibrillation (VF) and identify which combinations of light‐sensitive ion channel properties and illumination configurations would be effective.
Defibrillation was successful when a large proportion (> 16.6%) of ventricular tissue was directly stimulated by light that was bright enough to induce an action potential in an uncoupled cell.
While illumination with blue light never successfully terminated VF, illumination of red light‐sensitive ion channels with dense arrays of implanted red light sources resulted in successful defibrillation.
Our results suggest that cardiac expression of red light‐sensitive ion channels is necessary for the development of effective optogenetics‐based defibrillation therapy using LED arrays.
Abbreviations
- 3D
three dimensional
- AAV
adeno‐associated virus
- AP
action potential
- APD
action potential duration
- ChR2
channelrhodopsin‐2
- CL
cycle length
- HF
heart failure
- ICD
implantable cardioverter defibrillator
- LED
light emitting diode
- MRI
magnetic resonance imaging
- VF
ventricular fibrillation
Introduction
Ventricular fibrillation (VF) is the most common cause of sudden cardiac death (Rubart & Zipes, 2005). The only treatment for VF is defibrillation through delivery of powerful electrical shocks. Implantable cardioverter defibrillators (ICDs) are a widely used and effective therapy for patients at risk of ventricular arrhythmias (Kusumoto et al. 2014), but the sudden pain caused by collateral electrical activation of excitable tissue surrounding the heart (Jayam et al. 2005) following ICD shocks remains a major drawback (Pelletier et al. 2002). Therefore, there is still an urgent need for research into alternative VF treatment methods that do not require high‐energy shocks. Optogenetics is a method that could potentially enable selective stimulation of the heart.
Optogenetics requires expression of light‐sensitive proteins (opsins) in excitable tissue, via gene (Vogt et al. 2015) or cell (Jia et al. 2011) delivery. These proteins form ion channels or pumps that can be optically activated by light of a specific wavelength. Opsin expression in cardiac cells enables control of the cardiac action potential (AP) with light; illumination of cardiomyocytes expressing excitatory opsins elicits a depolarising current, which can evoke new APs. Potential cardiac applications of optogenetics stand to benefit from recent experimental advances. For example, targeted transfection of myocytes with blue‐light sensitive channelrhodopsin‐2 (ChR2) via a single injection of adeno‐associated virus (AAV) in live mice has been used to achieve robust, long‐term (≥ 10 weeks) and cardiomyocyte‐selective expression (Vogt et al. 2015). Moreover, progress in molecular optimisation of opsin properties has led to the emergence of new ChR2 variants with properties that would enhance light‐elicited excitation of the thick‐walled ventricles: opsins with enhanced light sensitivity, such as CatCh (Kleinlogel et al. 2011), increase the photocurrent elicited by a given light stimulus, while opsins with red‐shifted absorption spectrum, such as ReaChR (Lin et al. 2013) and ChRimson (Klapoetke et al. 2014), allow the use of longer wavelength light stimuli, which penetrate deeper in cardiac muscle than blue light.
Early proof‐of‐concept work suggests that reentrant activity in light‐sensitised cell monolayers can be terminated by optogenetic stimulation (Bingen et al. 2014). Development of an analogous optogenetics‐based therapy for the beating heart would require in vivo illumination, attainable via novel experimental devices, such as elastic membranes that allow the placement of light sources on cardiac surfaces (Xu et al. 2014). However, it remains unclear whether illumination of cardiac tissue photosensitised by opsin delivery can be used to terminate arrhythmias such as VF in humans. Specifically, it is unknown which opsin variant(s) should be used, how much illumination would be required, and whether therapeutic efficacy would be limited by the attenuation of light in the thick ventricular walls. In the absence of large animal in vivo models, computational simulations provide an alternative platform for answering such questions (Boyle et al. 2013).
Thus, in this study, we aim to (1) assess the feasibility of VF termination in the human heart via optogenetic stimulation and (2) quantify how the efficacy of this therapeutic approach would be affected by implementing theoretically possible opsin properties that result in deeper light penetration (i.e. increased photosensitivity and red‐shifted absorption spectrum). The latter goal is geared towards narrowing the scope of experimental investigation by steering researchers towards the most promising opsin characteristics for optogenetic defibrillation. To achieve these objectives, we employ electrophysiological simulations of VF in an MRI‐based model of the human ventricles.
Methods
Computational model of the failing human ventricles
We conducted computational simulations using a biophysically detailed patient‐based model of the human ventricles with realistic fibre orientation (Moreno et al. 2011), which was created from diffusion tensor MRI data using a previously described technique (Vadakkumpadan et al. 2010). Membrane kinetics of ventricular myocytes were represented by a human‐specific model (ten Tusscher & Panfilov, 2006). The model included transmural and apicobasal electrophysiological heterogeneity (Moreno et al. 2011; Bayer, 2014). Electrical propagation in the tissue was governed by the monodomain formulation (Vigmond et al. 2003; Plank et al. 2008). Cell‐ and tissue‐scale electrophysiological parameters (ion channel conductances and myocardial conductivities, respectively) were modified to represent remodelling associated with heart failure (HF), since such patients are at high risk of VF (Lane et al. 2005) and commonly receive ICDs (Kusumoto et al. 2014); these HF‐related changes were implemented as described previously (Moreno et al. 2011; Bayer, 2014) and include the following: (1) reduced transmural action potential duration (APD) gradient, due to epicardial AP prolongation (Glukhov et al. 2010); (2) reduced amplitude and slow‐recovering calcium transient due to deranged calcium handling (Lou et al. 2011); and (3) slowed myocardial conduction, due to down‐regulation of Cx43 expression (Glukhov et al. 2012).
ChR2 model variants
ChR2 photocurrent was represented by a detailed model of the widely used ChR2‐H134R variant (Williams et al. 2013); briefly, the ChR2 photo‐cycle was modelled as a four‐state Markov chain (Fig. 1 A) with light‐gated transitions between two closed states (non‐conducting) and two open states (permeable to cation flow). Additionally, we implemented theoretical ChR2 model variants with augmented light sensitivity (ChR2+) and red‐shifted absorption spectrum (ChR2‐RED); this was done to facilitate identification of opsin properties that could theoretically optimise defibrillation efficacy. In lieu of existing opsins with such properties (e.g. CatCh, ChRimson), for which photocycle dynamics have not yet been quantitatively characterised, we simulated hypothetical ChR2 variants by adjusting relevant parameters of the ChR2 model described above. For ChR2+ (Fig. 1 B), which we modelled after the ChR2 variant CatCh (Kleinlogel et al. 2011), we increased the closed‐to‐open rates (C1→O1 and C2→O2) by 120%; this increase ensured that the light power density required for ChR2+ half‐maximal activation matched the value measured experimentally for CatCh (0.4 mW mm–2) (Mattis et al. 2012). For ChR2‐RED (Fig. 1 C), we preserved ChR2‐H134R kinetics but adjusted the wavelength (λ) at which peak energy absorption occurred, shifting along the visible light spectrum from blue (λ = 480 nm) to red (λ = 669 nm). Finally, we simulated a theoretical fourth variant (ChR2‐RED+) that combined both of the above changes (Fig. 1 D).
Figure 1. Models of ChR2 variants .
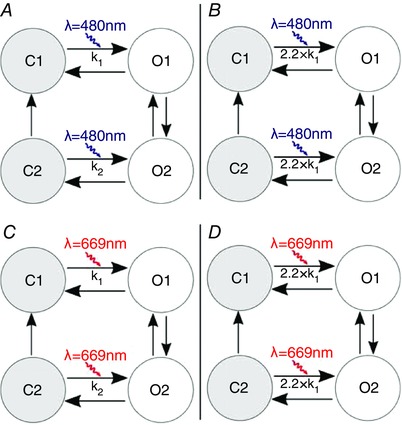
Photocycles of ChR2 variants: A, ChR2, modelled after ChR2‐H134R, as formulated by Williams et al.; B, ChR2+; C, ChR2‐RED; D, ChR2‐RED+.
Light sensitisation of failing ventricles
We used a previously described stochastic method (Boyle et al. 2013) to simulate delivery of opsin‐encoding genes to the failing ventricles via systemic gene delivery. As shown in Fig. 2 A, 58.2% of all simulated cells in the ventricles were modelled as opsin‐expressing with a diffuse spatial pattern (uniform random distribution); this was consistent with the average rate of opsin expression achieved in vivo via jugular vein injection in wild‐type mice (Vogt et al. 2015).
Figure 2. Models of opsin delivery and illumination .
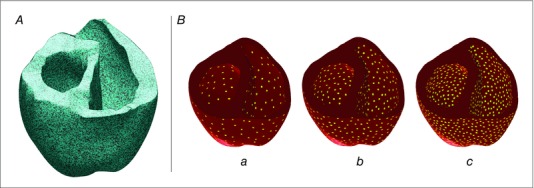
A, opsin distribution in the human ventricular model. Blue: opsin‐transfected tissue; black: tissue without opsins. B, Optrodes on the endocardial and epicardial surfaces in yellow. Three different optrode densities: a, 1.15 cm–2; b, 2.30 cm–2; and c, 4.61 cm–2.
Simulation of illumination
Light sources were modelled as grids of 1 mm‐radius optrodes on the endocardial and epicardial surfaces; this represented illumination by deformable arrays of light emitting diodes (LEDs) placed on elastic membranes, similar to devices recently developed by Xu et al. (2014). We used grids with three levels of optrode density (1.15, 2.30 and 4.61 cm–2, Fig. 2 B). A stochastic point repulsion algorithm (Turk, 1991) was used to ensure uniform inter‐optrode spacing.
To represent the attenuation of optical energy in cardiac tissue, which depends on both the wavelength of incident light and the optical properties of the tissue, we used a finite element approach to solve the steady state photon diffusion equation, assuming homogeneous absorption and isotropic scattering in the ventricles (Bishop et al. 2006; Boyle et al. 2015):
where E e is irradiance at each point r in the tissue, D is diffusivity and μa is the rate of light absorption; D and μa are wavelength‐dependent tissue properties. As described previously (Boyle et al. 2015), piecewise Dirichlet boundary conditions were imposed on the epicardial and endocardial surfaces, with E e = E e,max at sites directly in contact with optrodes and E e = 0 elsewhere. We used experimentally derived optical parameter values (Ding et al. 2001; Bishop et al. 2006) to calculate attenuation profiles for blue light (λ = 488 nm; D = 0.18 mm, μa = 0.52mm−1) and red light (λ = 669 nm; D = 0.34 mm, μa = 0.10 mm−1). This method allowed us to calculate the irradiance resulting from the additive effect of multiple overlapping light sources.
Initiation of ventricular fibrillation (VF)
To attain stable organ‐scale initial conditions, the ventricular apex was electrically paced at 60 beats min–1 (pulse duration, 5 ms; amplitude, 2× threshold) until steady state was achieved for all model variables (∼60 s). Then, to induce VF, the pacing cycle length (CL) was gradually shortened (50 ms decrements, 32 beats per pacing CL) from the sinus rate (CL = 1000 ms) until conduction block and reentry occurred (CL = 350 ms). The initial time (t = 0) in our optogenetic defibrillation simulations was 30 s after VF induction.
Simulation protocol for optogenetic defibrillation
We attempted to terminate VF using synchronised illumination from endocardial and epicardial optrode grids. In each simulation, we attempted defibrillation with a single light stimulus, either of short (25 ms) or of long (500 ms) duration; these durations were chosen to be shorter and longer, respectively, than the average time between successive AP upstrokes during VF (386.03 ± 37.16 ms). The optrodes were activated simultaneously and each individual optrode illuminated underlying tissue with E e,max = 10 mW mm–2; this value was consistent with illumination values used for cardiac optogenetics experiments in vitro (Abilez et al. 2011). To determine if the timing of the stimulus critically affected the outcome of defibrillation and thus to assess potential therapeutic efficacy, we varied the onset time of the light stimulus: we ran simulations with illumination at t = 0 (see above), 100, 200 or 300 ms.
A total of 96 3D ventricular simulations were conducted using the finite element method as implemented in the CARP Software package (Johns Hopkins University, Université de Bordeaux, Medizinische Universität Graz) (Vigmond et al. 2003, 2008; Plank et al. 2008) on a high‐performance computing cluster. Each simulation included one permutation of the following parameters:
One of four opsin variants: ChR2, ChR2+, ChR2‐RED or ChR2‐RED+
One of three optrode grid densities: 1.15, 2.30 or 4.61 cm–2
One of two light pulse durations: 25 or 500 ms
One of four light pulse timings: t stim = 0, 100, 200 or 300 ms
Each defibrillation simulation required ∼6.6 h of computing time on 30 Intel Xeon CPU cores (2.83 GHz) in parallel. Defibrillation was deemed successful if the reentrant activity was completely extinguished; simulation duration (2000 ms after the onset of each light stimulus) was long enough to ensure that post‐illumination activity following failed defibrillation attempts was indeed sustained arrhythmia.
Results
Calculation of irradiance in the tissue
For each light source configuration, we first calculated the irradiance (E e) delivered to every point in the ventricles when illumination (blue or red light) was applied at the endocardial and epicardial surfaces. Red light penetrated deeper in the myocardium than blue light, with exponential spatial decay rates of 1.84 and 0.58 mm, respectively. When the array with intermediate optrode density (2.30 cm–2, E e,max = 10 mW mm–2) was used to apply blue light (Fig. 3 A, middle), the mean irradiance that reached cells deep in the ventricles (≥ 5 mm from the surfaces) was very low (3.6 × 10−5 mW mm–2); in contrast, for red light illumination (Fig. 3 B, middle), average E e at the same depth was ∼90× stronger (E e = 0.032 mW mm–2).
Figure 3. Irradiance in the ventricles .
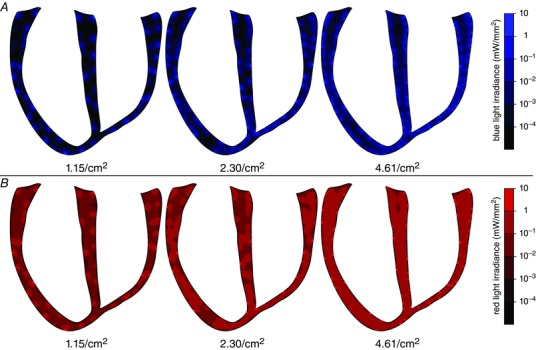
Profile of calculated irradiance in the ventricular tissue after stimulation by blue (A) or red (B) light source grids of density 1.15, 2.30 or 4.61 cm–2 on the endocardial and epicardial surfaces.
Whereas the irradiance delivered deep in the ventricular walls was predominantly determined by the wavelength of applied illumination, the amount of light delivered to myocytes nearer to the surface was more significantly affected by optrode array density. For the sparsest array (1.15 cm–2), a large proportion (87.7% and 75.0% for blue and red stimuli, respectively) of tissue in subsurface areas (≤1 mm depth) was weakly illuminated (E e< 1% of E e,max), because few myocytes in these regions were in the vicinity of light sources. In contrast, when illumination was applied using the densest light source array (4.61 cm–2), each subsurface area of the tissue was illuminated by multiple light sources. As a result, the proportions of weakly illuminated subsurface tissue were much lower (49.8% for blue light and 7.7% for red light).
Attempts to defibrillate with blue light
Stimulation with blue light did not terminate VF in the photosensitised ventricles for any optogenetic configuration (i.e. combination of optrode array density, illumination pulse duration, and opsin light sensitivity). A short (25 ms) blue light pulse delivered by the 1.15 cm–2 optrode array elicited a small number of localised activations in the ventricles photosensitised with ChR2 (compare Fig. 4 A and B, illustrating sustained VF in the absence of an optical stimulus). After the onset of illumination, ChR2‐mediated current was elicited in tissue underlying the optrodes, leading to local increases in transmembrane voltage (V m) (Fig. 4 C); however, most of these light‐induced depolarisations were below the necessary threshold to elicit propagating APs due to electrotonic coupling with non‐stimulated tissue deeper in the myocardial wall. Since few new propagating wavefronts were induced in the excitable tissue, reentry persisted. Similar results were observed for denser optrode grids (2.30 and 4.61 cm–2). More new activations were elicited following brief (25 ms) illumination of the epicardial and endocardial surfaces from these optrode arrays than from the sparse (1.15 cm–2) optrode array; nonetheless, these activations remained confined to the directly illuminated area and did not spread to surrounding excitable tissue, allowing the chaotic propagation of fibrillatory wavefronts to persist. Even when longer blue light stimuli (500 ms) were applied, optogenetic defibrillation was not successful, regardless of the illuminating array density; subthreshold depolarisations in tissue underlying the light sources lasted longer in this case, but were prevalent only in a thin layer beneath the illuminated surface (∼2 mm).
Figure 4. Defibrillation attempts with blue light .
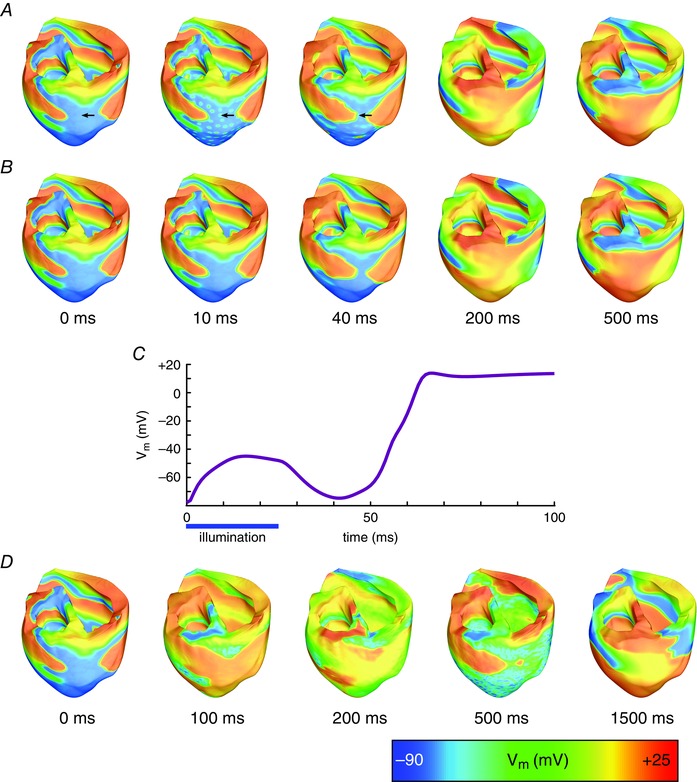
A, V m maps following illumination of ventricles with ChR2 via a sparse (1.15 LEDs cm–2) optrode array at time = 0 with stimulus duration 25 ms, compared with simulation of VF without a defibrillation attempt (B). C, V m at surface node denoted with arrow in A. D, defibrillation attempt with ChR2+ (enhanced light sensitivity) from a dense (4.61 LEDs cm–2) optrode array, at time = 0 with light pulse duration 500 ms. Videos S1–3 in ‘Supporting Information’ correspond to V m sequences in A, B and D, respectively.
When the ChR2+ model was incorporated in the ventricles, augmented light sensitivity resulted in a larger number of new activations near the epicardial and endocardial surfaces following 25 ms illumination. However, this did not improve defibrillation outcomes, since reentrant wavefronts were still able to propagate through the midmyocardium, in which optically induced activations did not occur. Even with long‐duration (500 ms) blue light illumination via the densest light source array (4.61 cm–2; Fig. 4 D), VF was not terminated. Large areas of the cardiac surfaces were depolarised, but light‐induced wavefronts did not propagate into the depths of the left ventricular (LV) wall. Consequently, reentry persisted in the midmyocardium of the LV free wall and, in some cases, propagated into the optogenetically depolarised surface regions, resulting in breakthrough patterns and continuation of VF.
Attempts to defibrillate with red light
In contrast to our findings for blue light, some defibrillation attempts for optogenetic configurations involving red light were successful; Table 1 contains a comprehensive list of outcomes for each configuration. Activations following a light stimulus delivered from a sparse (1.15 cm–2) array of LEDs were inadequate to extinguish VF with either ChR2‐RED or ChR2‐RED+. When denser optrode grids were used, there were some optogenetic configurations for which defibrillation outcome depended on the timing of illumination onset (i.e. success rates < 100% in Table 1); for one such configuration (25 ms pulse, ChR2‐RED, 4.61 cm–2 optrode array), two outcomes (corresponding to pulse onset times t stim = 0 or 300 ms) are highlighted in Fig. 5. For the first case (t stim = 0, Fig. 5 A), light‐elicited excitations occurred in the tissue up to a depth of 6 mm and led to widespread propagating ventricular activations; however, a small part of the tissue deep in the basal LV free wall was not excited (asterisk in Fig. 5 A, t = 20 ms). As a result, a wavefront propagated through the latter region, then along the mitral annulus (i.e. circular opening at top of LV) towards the ventricular septum and the posterior RV free wall (arrows in Fig. 5 A, t = 200–300 ms). The same wavefront then initiated persistent post‐illumination figure‐of‐eight reentry in the posterior wall of the heart, leading to sustained arrhythmia. In contrast to the chaotic, multi‐rotor activity in VF, the morphology of this arrhythmia was consistent with monomorphic ventricular tachycardia, because there was only a single re‐entrant wavefront and the same ventricular activation sequence occurred repetitively (compare t = 600 and t = 1000 in Fig. 5 A). In contrast, when simulations were repeated with a different stimulation onset time (t stim = 300 ms, Fig. 5 B), parts of the LV free wall were similarly unaffected by illumination (asterisk in Fig. 5 B, t = 20 ms); however, in this case, the wavefront that emerged post‐illumination was blocked by a pre‐existing activation (double lines in Fig. 5 B, t = 20 ms). Consequently, there was no post‐illumination reentry and the ventricles repolarised.
Table 1.
Defibrillation success rates for optogenetic configurations with red light
| Success percentages calculated for four defibrillation attempts | ||||
|---|---|---|---|---|
| with different light pulse timings (see text for details) at three | ||||
| different optrode densities (1.15, 2.30 or 4.61 cm–2). | ||||
| Optrode density | ||||
| Opsin | Illumination | |||
| model | duration | 1.15 cm–2 | 2.30 cm–2 | 4.61 cm–2 |
| ChR2‐RED | 25 ms | 0% | 25% | 50% |
| ChR2‐RED | 500 ms | 0% | 25% | 100% |
| ChR2‐RED+ | 25 ms | 0% | 50% | 100% |
| ChR2‐RED+ | 500 ms | 0% | 100% | 100% |
Figure 5. Timing‐sensitive defibrillation with ChR2‐RED .
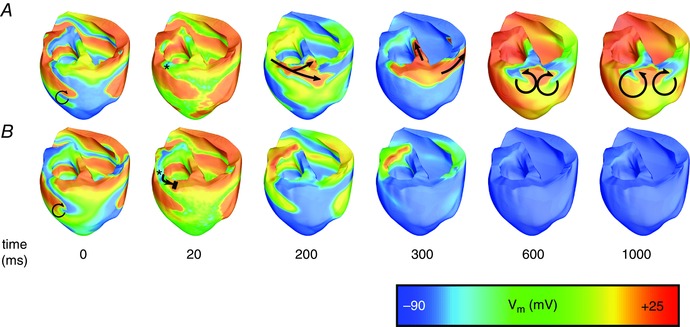
V m maps following illumination of ventricles transfected with ChR2‐RED (normal light sensitivity) via an optrode array with 4.61 LEDs cm–2, with stimulus duration 25 ms. A, stimulus onset at time = 0; B, stimulus onset at time = 300 ms. The time scale represents time since the onset of illumination in each case. Black arrows indicate propagation and re‐entry; double lines indicate conduction block. Asterisks indicate tissue deep in the basal LV free wall, as discussed in the text. Videos S4 and S5 in ‘Supporting Information’ correspond to V m sequences in A and B, respectively.
Illumination of the ventricles photosensitised with ChR2‐RED+ using the densest optrode array (4.61 cm–2) led to defibrillation regardless of the timing and duration of the red light pulse. When a short (25 ms) pulse was applied, it led to widespread depolarisation throughout the ventricles (Fig. 6 A) and terminated the propagation of fibrillatory wavefronts. For the example shown, the postero‐basal epicardium was already excited at the instant of illumination and did not undergo a light‐elicited AP (Fig. 6 B); consequently, this area repolarised faster than adjacent tissue following illumination and was subsequently re‐excited (Fig. 6 A, t = 200 ms) by an intramural wavefront that propagated through non‐excited tissue in the posterior septal wall (Fig. 6 C). This resulted in a single extra beat between the termination of VF and the return to electric quiescence. The same behaviour was observed following stimulation with different light pulse timings. When a longer (500 ms) pulse was applied, light‐induced depolarisation was similarly widespread (Fig. 6 D); sustained optogenetic current kept myocytes near the illuminated surfaces (depth < 9 mm) at V m ≈ –20 mV (see Fig. 6 D at t = 350 ms). Consequently, early tissue repolarisations, which led to post‐illumination ventricular activations in configurations with shorter light pulses, were inhibited. After the end of the 500 ms‐long light pulse, the ventricles uniformly repolarised, leading to instantaneous defibrillation after the end of optical stimulation.
Figure 6. Successful defibrillation with ChR2‐RED+ .
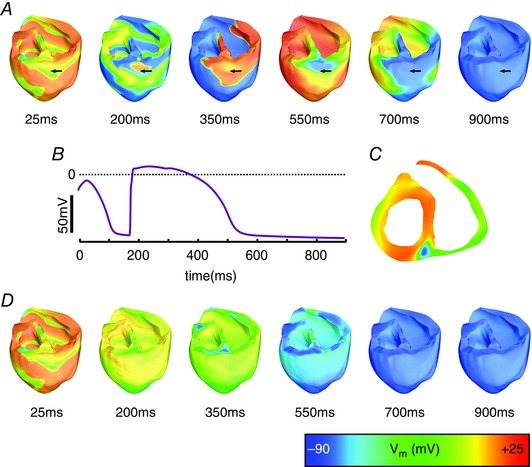
A, V m maps following illumination of ventricles with ChR2‐RED+ (enhanced light sensitivity) via a dense (4.61 cm–2) optrode array, with light pulse duration 25 ms and onset at 0 ms. B, V m at surface node denoted with arrows in A. C, cutaway view (same level as the arrows in A) at time = 100 ms in the same simulation. D, V m maps following stimulation with the same configuration as in A above, but with 500 ms light pulse duration. Videos S6 and S7 in ‘Supporting Information’ correspond to V m sequences in A and D, respectively.
Estimation of illumination requirement for defibrillation
Our results suggested that defibrillation was achieved when light‐induced depolarisation elicited new wavefronts that propagated into non‐activated ventricular tissue, resulting in elimination of the excitable gap. We observed that the likelihood of defibrillation success was higher when the number of propagating wavefronts generated by opsin‐mediated current was larger. To quantify this observation, we calculated, for each optogenetic configuration, the proportion of tissue that was subjected to an intrinsically suprathreshold optical stimulus (i.e. a pulse strong enough to elicit an AP in an electrically uncoupled light‐sensitive cell model). First, threshold irradiance values were determined in single myocyte models for each combination of pulse duration and light sensitivity; then we identified the proportion of tissue that was illuminated with at least that value in the ventricles model. Comparison of the resulting data (Table 2) with defibrillation outcomes (Table 1) provided a first order approximation of the minimum amount of light‐excited tissue necessary to ensure robust (i.e. insensitive to pulse timing) optogenetic defibrillation (25 ms pulse: ≥ 34.5% of the tissue; 500 ms pulse: ≥ 16.6%). It should be noted that this is an approximation of the amount of excited tissue in the ventricles, as described in Methods; due to strong electrotonic effects in the 3D tissue, activation thresholds are not expected to be homogeneous.
Table 2.
Proportion of light‐activated ventricular tissue
| Percentages of light‐activated ventricular tissue at three different optrode densities (1.15, 2.30 or 4.61 cm–2). | |||||
|---|---|---|---|---|---|
| Optrode Density | |||||
| Opsin model | Illumination duration | 1.15 cm−2 | 2.30 cm−2 | 4.61 cm−2 | Calculated illumination threshold |
| ChR2 | 25 ms | 1.37% | 2.76% | 5.54% | 0.08 mW mm–2 |
| ChR2 | 500 ms | 1.48% | 2.99% | 5.99% | 0.07 mW mm–2 |
| ChR2+ | 25 ms | 2.08% | 4.18% | 8.47% | 0.04 mW mm–2 |
| ChR2+ | 500 ms | 2.45% | 4.94% | 10.0% | 0.03 mW mm–2 |
| ChR2‐RED | 25 ms | 2.68% | 5.64% | 13.87% | 0.08 mW mm–2 |
| ChR2‐RED | 500 ms | 3.05% | 6.47% | 16.6% | 0.07 mW mm–2 |
| ChR2‐RED+ | 25 ms | 5.35% | 12.1% | 34.5% | 0.04 mW mm–2 |
| ChR2‐RED+ | 500 ms | 7.17% | 17.0% | 46.8% | 0.03 mW mm–2 |
Percentages of light‐activated ventricular tissue at three different optrode densities (1.15, 2.30 or 4.61 cm–2).
Discussion
This study utilised biophysically detailed computational modelling to assess whether ventricular fibrillation can be terminated with a therapy based on optogenetics and to identify how opsin and illumination properties relate to the efficacy of optogenetics‐based defibrillation. The main findings are as follows: first, optogenetics‐based therapy comprising opsin delivery to the heart and illumination from light sources on the cardiac surfaces terminated fibrillation in a model of the failing human ventricles for some of the simulated configurations. Second, this type of optogenetics‐based defibrillation depended on eliciting widespread propagating wavefronts and its efficacy was correlated with the volume of ventricular tissue directly excited by light; red‐shifted opsin absorption spectrum, augmented opsin light sensitivity, denser light source distribution, and prolonged illumination were factors that increased this volume and subsequently led to successful defibrillation. In particular, opsin sensitivity to red light was the primary determinant of defibrillation success, as blue light illumination did not lead to termination of arrhythmia for any of the simulated optogenetic configurations.
Mechanisms of optogenetic defibrillation
The mechanism of optogenetics‐based defibrillation in this study is the initiation of propagating wavefronts of excitation via light‐induced depolarisation of photosensitised cardiac tissue, which renders the ventricular myocardium refractory and eliminates the excitable gap.
Since ChR2 is a strongly rectifying channel, induced photocurrents are very weak at membrane potentials equal to or greater than the ChR2 reversal potential of 0 mV (Williams et al. 2013); as such, illumination of tissue that is already depolarised has minimal effects and the effect of optogenetic stimulation is most prominent in regions that are near resting V m (i.e. repolarised between APs) at the instant of light pulse onset. A prolonged stimulus can therefore be more effective than a short one because it allows for excitation of tissue that was refractory during the early stages of illumination; this ensures that all of the illuminated tissue will be subjected to optogenetically induced current.
Several similarities between optogenetics‐based defibrillation and electrical defibrillation stand out. We observed that, in a manner similar to electrical defibrillation, optogenetic configurations associated with higher energy requirements (i.e. more light sources and/or longer pulse durations) have increased probability of success. As in electrical defibrillation with near‐threshold shocks (Chattipakorn et al. 2004; Constantino et al. 2010), new wavefront propagations occur after optical stimuli that only excite a limited portion of the tissue; depending on the pre‐stimulus state, such propagations can lead to either one or more extra beats followed by quiescence or to sustained post‐shock reentry resembling tachycardia. Conversion of VF to tachycardia following low energy electrical stimulation has been reported before (Rantner et al. 2013), as part of a two‐phase defibrillation protocol; a similar dual light pulse optogenetic defibrillation protocol could potentially lead to termination of reentry.
There are similarities between the mechanisms leading to optogenetics‐based defibrillation failure and those underlying electrical defibrillation failure. Our observations indicate that residual propagating wavefronts after light stimulation are often located in the LV wall, due to the insufficient illumination of the thick LV wall myocardium. Likewise, post‐shock activations in the LV are a well‐documented cause of electrical defibrillation failure (Chattipakorn et al. 2001; Chattipakorn et al. 2003) and can also be attributed to the thickness of the LV wall (Constantino et al. 2010). Furthermore, following long optical stimuli, we noticed that, while the ventricular walls are depolarised, arrhythmic activity persists in the LV mid‐myocardium (Fig. 5), behaviour akin to post‐shock tunnel propagation in electrical defibrillation (Constantino et al. 2010).
A different optogenetic defibrillation mechanism has been reported in experiments featuring illumination of rat myocyte monolayers transfected with CatCh (Bingen et al. 2014). Stimulation with a 500 ms‐long low‐energy blue light pulse led to reduced excitability near the spiral wave functional core; this caused the organising centre of reentry to drift and eventually collide with a monolayer boundary, thereby terminating arrhythmia. We did not observe a similar behaviour in the 3D model of the tissue; following blue light illumination, organising centres of spiral waves appeared largely unchanged; in contrast, after high‐energy red light illumination, spiral waves were extinguished before the end of the 500 ms‐long stimulus. These differences can be attributed to the higher illumination energy used in our simulations, as well as the different wave dynamics in 3D tissue stemming from stronger electrotonic coupling.
Requirements of optogenetics‐based defibrillation
Our results demonstrate that the efficacy of light‐based VF termination depends on the amount of tissue excited. As has been shown in the past (Entcheva, 2013; Karathanos et al. 2014) and as the results presented in this study confirm, the low penetration of blue light in cardiac tissue limits the extent of tissue activation in the mid‐myocardium, thereby diminishing the efficacy of potential therapeutic applications in the heart. However, as we show in this study, this shortcoming of optogenetics‐based defibrillation can be mitigated through the use of opsins with red‐shifted absorption wavelength and increased light sensitivity. Indeed, red light sensitivity was identified as the most crucial optogenetic parameter affecting the success of light‐based defibrillation.
Enhanced light sensitivity alone does not sufficiently increase the number of light‐elicited activations to achieve defibrillation, as illustrated by the poor efficacy of defibrillation attempts involving ChR2+. However, combined with mutations conferring red‐light sensitivity (ChR2‐RED+), it greatly increases the number of activations elicited deep in the ventricular walls, leading to successful defibrillation.
In addition to opsin modifications, sufficient illumination is necessary for optogenetics‐based defibrillation. Successful termination of VF was achieved with simultaneous illumination of the epicardial and endocardial surfaces with optrode arrays of density ≥ 2.30 cm–2. The optical energy these LED arrays deliver to the tissue can easily be calculated by multiplying the total LED surface with the illuminated irradiance (10 mW mm–2) and the duration of the light pulses; for stimuli of duration 25 and 500 ms, this energy is 0.52 and 10.43 J, respectively. However, since LEDs have energy losses in the form of heat, higher electrical energy input is required to achieve this amount of optical energy. According to published results for a blue LED illumination device designed for optogenetic neural stimulation (Kim et al. 2013), this efficiency may be as low as 2.5%. However, research on LED illumination is progressing rapidly (Tsao et al. 2015) and modern experimental designs boast efficiencies ≥ 50% (Jahangir et al. 2014).
Towards clinical application: limitations and challenges
Here, we have shown that opsin variants with enhanced light sensitivity and red‐shifted absorption spectrum are necessary to achieve ventricular defibrillation. Two ChR2 variants with red‐shifted absorption spectrum have already been engineered: ChRimson (Klapoetke et al. 2014) and ReaChR (Lin et al. 2013). Notably, both of these opsins have some characteristics that differ from the theoretical ChR2‐RED variant used in the present study. ChRimson generates leak current (Klapoetke et al. 2014) in the absence of illumination, making it a suboptimal choice for optogenetic applications in the heart because it could lead to diastolic depolarisation and potentially pro‐arrhythmic conduction slowing due to baseline Na+ channel inactivation. ReaChR has significantly slower kinetics than ChR2, and thus it may not be suitable for excitation via short light pulses. Opsins with high light sensitivity also tend to exhibit a slower response than the theoretical ChR2+ model used in this study (Mattis et al. 2012). Further research may lead to elimination of the drawbacks in existing opsin variants or to the design of new opsins with characteristics similar to ChR2‐RED and ChR2‐RED+.
Expression of these opsins in the heart can be achieved via gene delivery with viral vectors. Preliminary studies have established that gene delivery using AAV vectors can be safe, persistent and efficacious (Wasala et al. 2011). However, the prevalence of antibodies that target AAV vectors in a significant proportion of the population (Halbert et al. 2006) may be a hindrance to their widespread utilisation for gene delivery (Gwathmey et al. 2011).
Finally, devices capable of reliably delivering light to the heart in the manner simulated in this study are not currently clinically available. Experimental devices with this capability have been used in Langendorff‐perfused rabbit hearts (Xu et al. 2014), but more research is needed to assess whether they could be safely and effectively deployed in living, beating human hearts. Furthermore, we determined that simultaneous illumination from epicardial and endocardial light sources is required for successful termination of VF. Design and implantation of a device that can deliver light both underneath the pericardium and inside the ventricular chambers would pose an additional biomedical engineering challenge that will need to be overcome if optogenetic defibrillation is to become a clinical reality.
Limitations
Heart failure is a disease that affects the myocardium in many different ways; in our study we used a previously developed comprehensive model of heart failure‐induced changes in cardiomyocyte membrane kinetics. However, some aspects of heart failure are not explicitly represented in our model, such as T‐tubule dysregulation (Ibrahim et al. 2011). Since ChR2 has been shown to localise to T‐tubules when expressed in cardiomyocytes (Bruegmann et al. 2010; Vogt et al. 2015), this dysregulation may limit the effect of opsin gene delivery in failing hearts.
In addition, the effects of gene delivery to the human heart are not fully known; as a result, cardiac optogenetics modelling requires the use of parameters derived from in vitro experiments or in vivo animal models. In particular, the level of opsin expression that can be achieved following viral gene delivery in humans and, consequently, the total conductance of the opsin channels is unknown. In the absence of definitive values, we adopted the maximum conductance (0.4 mS cm–2) that was implemented in the ChR2 model we used, which had been calculated via experiments in HEK‐293 cells (Williams et al. 2013). However, preliminary results from viral gene delivery to the murine heart suggest that the real photocurrent level might be lower (Vogt et al. 2015). By fitting our model to ChR2 current traces in that experiment, we calculated a maximum conductance value of 0.17 mS cm–2. This discrepancy between data acquired from different sources constitutes a limitation in our modelling endeavour. Nonetheless, the computational framework we utilise is flexible and allows easy implementation of new experimental data as it becomes available.
Conclusions
Our study represents the first attempt to assess whether optogenetic termination of arrhythmias in humans is possible. Utilising a previously developed computational framework, we demonstrated that a light‐based therapy is capable of terminating VF and we identified some of the requirements for the further development of this technology. We believe that computational modelling can guide the evolution of the emerging field of optogenetics‐based defibrillation.
Additional information
Competing interests
None declared.
Author contributions
T.K.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing. J.B.: provision of study materials or patients, collection and assembly of data; D.W.: provision of study materials or patients, collection and assembly of data. P.B.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing. N.T.: conception and design, manuscript writing. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the Whitaker International Program administered by the Institute for International Education (to J.D.B.); the Fondation Lefoulon Delalande Institut de France (to J.D.B.); and the National Heart, Lung, and Blood Institute at the National Institutes of Health (DP1 HL123271 to N.A.T.).
Translational perspective
We used computational modelling to assess whether ventricular fibrillation can be terminated using light and concluded that optogenetics‐based defibrillation is theoretically feasible. In our study, we considered three categories of issues related to the implementation of optogenetics‐based defibrillation therapies in humans. (1) Molecular optimisation of opsins: we showed that modifications that will render opsins sensitive to longer light wavelengths (e.g. red light) are necessary in order for such a therapy to work. (2) Gene therapy to selectively deliver these opsins to the heart: experimental results in mice are promising, but it is unknown whether photosensitization of the human heart can be achieved in a safe and complication‐free manner. (3) Devices to illuminate the heart: we identified that optogenetics‐based defibrillation required illumination from numerous light sources, evenly distributed on the endocardial and epicardial walls of the heart. Such clinical devices are not currently available. Even though newly designed devices using elastic membranes are promising, they are still at an experimental stage and it is unknown if and when their use in human hearts will be feasible.
Supporting information
Video S1 Failed defibrillation with ChR2 and a short light stimulus
Video S2 Ventricular fibrillation control
Video S3 Failed defibrillation with ChR2+ and a long light stimulus
Video S4 Failed defibrillation with ChR2‐RED and a short light stimulus
Video S5 Successful defibrillation with ChR2‐RED and a short light stimulus
Video S6 Successful defibrillation with ChR2‐RED+ and a short light stimulus
Video S7 Successful defibrillation with ChR2‐RED+ and a long light stimulus
P. M. Boyle and N. A. Trayanova contributed equally to this work.
References
- Abilez OJ, Wong J, Prakash R, Deisseroth K, Zarins CK & Kuhl E (2011). Multiscale computational models for optogenetic control of cardiac function. Biophys J 101, 1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer JD (2014). Mechanisms of microvolt T‐wave alternans in human heart failure. PhD thesis, Johns Hopkins University.
- Bingen BO, Engels MC, Schalij MJ, Jangsangthong W, Neshati Z, Feola I, Ypey DL, Askar SF, Panfilov AV, Pijnappels DA & de Vries AA (2014). Light‐induced termination of spiral wave arrhythmias by optogenetic engineering of atrial cardiomyocytes. Cardiovasc Res 104, 194–205. [DOI] [PubMed] [Google Scholar]
- Bishop MJ, Rodriguez B, Eason J, Whiteley JP, Trayanova N & Gavaghan DJ (2006). Synthesis of voltage‐sensitive optical signals: application to panoramic optical mapping. Biophys J 90, 2938–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PM, Karathanos TV, Entcheva E & Trayanova NA (2015). Computational modeling of cardiac optogenetics: Methodology overview & review of findings from simulations. Comput Biol Med 65, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PM, Williams JC, Ambrosi CM, Entcheva E & Trayanova NA (2013). A comprehensive multiscale framework for simulating optogenetics in the heart. Nat Commun 4, 2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruegmann T, Malan D, Hesse M, Beiert T, Fuegemann CJ, Fleischmann BK & Sasse P (2010). Optogenetic control of heart muscle in vitro and in vivo. Nat Methods 7, 897–900. [DOI] [PubMed] [Google Scholar]
- Chattipakorn N, Banville I, Gray RA & Ideker RE (2001). Mechanism of ventricular defibrillation for near‐defibrillation threshold shocks: a whole‐heart optical mapping study in swine. Circulation 104, 1313–1319. [DOI] [PubMed] [Google Scholar]
- Chattipakorn N, Banville I, Gray RA & Ideker RE (2004). Effects of shock strengths on ventricular defibrillation failure. Cardiovasc Res 61, 39–44. [DOI] [PubMed] [Google Scholar]
- Chattipakorn N, Fotuhi PC, Chattipakorn SC & Ideker RE (2003). Three‐dimensional mapping of earliest activation after near‐threshold ventricular defibrillation shocks. J Cardiovasc Electrophysiol 14, 65–69. [DOI] [PubMed] [Google Scholar]
- Constantino J, Long Y, Ashihara T & Trayanova NA (2010). Tunnel propagation following defibrillation with ICD shocks: hidden postshock activations in the left ventricular wall underlie isoelectric window. Heart Rhythm 7, 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Splinter R & Knisley SB (2001). Quantifying spatial localization of optical mapping using Monte Carlo simulations. IEEE Trans Biomed Eng 48, 1098–1107. [DOI] [PubMed] [Google Scholar]
- Entcheva E (2013). Cardiac optogenetics. Am J Physiol Heart Circ Physiol 304, H1179–H1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhov AV, Fedorov VV, Kalish PW, Ravikumar VK, Lou Q, Janks D, Schuessler RB, Moazami N & Efimov IR (2012). Conduction remodeling in human end‐stage nonischemic left ventricular cardiomyopathy. Circulation 125, 1835–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N & Efimov IR (2010). Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res 106, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwathmey JK, Yerevanian AI & Hajjar RJ (2011). Cardiac gene therapy with SERCA2a: from bench to bedside. J Mol Cell Cardiol 50, 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Miller AD, McNamara S, Emerson J, Gibson RL, Ramsey B & Aitken ML (2006). Prevalence of neutralizing antibodies against adeno‐associated virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: Implications for gene therapy using AAV vectors. Hum Gene Ther 17, 440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M, Gorelik J, Yacoub MH & Terracciano CM (2011). The structure and function of cardiac t‐tubules in health and disease. Proc Biol Sci 278, 2714–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahangir S, Schimpke T, Strassburg M, Grossklaus KA, Millunchick JM & Bhattacharya P (2014). Red‐emitting (λ = 610 nm) In0.51Ga0.49 N/GaN disk‐in‐nanowire light emitting diodes on silicon. IEEE J Quantum Electron 50, 530–537. [Google Scholar]
- Jayam V, Zviman M, Jayanti V, Roguin A, Halperin H & Berger RD (2005). Internal defibrillation with minimal skeletal muscle activation: a new paradigm toward painless defibrillation. Heart Rhythm 2, 1108–1113. [DOI] [PubMed] [Google Scholar]
- Jia Z, Valiunas V, Lu Z, Bien H, Liu H, Wang HZ, Rosati B, Brink PR, Cohen IS & Entcheva E (2011). Stimulating cardiac muscle by light: cardiac optogenetics by cell delivery. Circ Arrhythm Electrophysiol 4, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karathanos TV, Boyle PM & Trayanova NA (2014). Optogenetics‐enabled dynamic modulation of action potential duration in atrial tissue: feasibility of a novel therapeutic approach. Europace 16 Suppl 4, iv69–iv76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TI, McCall JG, Jung YH, Huang X, Siuda ER, Li Y, Song J, Song YM, Pao HA, Kim RH, Lu C, Lee SD, Song IS, Shin G, Al‐Hasani R, Kim S, Tan MP, Huang Y, Omenetto FG, Rogers JA & Bruchas MR (2013). Injectable, cellular‐scale optoelectronics with applications for wireless optogenetics. Science 340, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey‐Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, Wang J, Xie Y, Yan Z, Zhang Y, Chow BY, Surek B, Melkonian M, Jayaraman V, Constantine‐Paton M, Wong GK & Boyden ES (2014). Independent optical excitation of distinct neural populations. Nat Methods 11, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinlogel S, Feldbauer K, Dempski RE, Fotis H, Wood PG, Bamann C & Bamberg E (2011). Ultra light‐sensitive and fast neuronal activation with the Ca2+‐permeable channelrhodopsin CatCh. Nat Neurosci 14, 513–518. [DOI] [PubMed] [Google Scholar]
- Kusumoto FM, Calkins H, Boehmer J, Buxton AE, Chung MK, Gold MR, Hohnloser SH, Indik J, Lee R, Mehra MR, Menon V, Page RL, Shen WK, Slotwiner DJ, Stevenson LW, Varosy PD, Welikovitch L; Heart Rhythm Society; American College of Cardiology; American Heart Association (2014). HRS/ACC/AHA expert consensus statement on the use of implantable cardioverter‐defibrillator therapy in patients who are not included or not well represented in clinical trials. J Am Coll Cardiol 64, 1143–1177. [DOI] [PubMed] [Google Scholar]
- Lane RE, Cowie MR & Chow AW (2005). Prediction and prevention of sudden cardiac death in heart failure. Heart 91, 674–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Knutsen PM, Muller A, Kleinfeld D & Tsien RY (2013). ReaChR: a red‐shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat Neurosci 16, 1499–1508.23995068 [Google Scholar]
- Lou Q, Fedorov VV, Glukhov AV, Moazami N, Fast VG & Efimov IR (2011). Transmural heterogeneity and remodeling of ventricular excitation‐contraction coupling in human heart failure. Circulation 123, 1881–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis J, Tye KM, Ferenczi EA, Ramakrishnan C, O'Shea DJ, Prakash R, Gunaydin LA, Hyun M, Fenno LE, Gradinaru V, Yizhar O & Deisseroth K (2012). Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods 9, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JD, Zhu ZI, Yang PC, Bankston JR, Jeng MT, Kang C, Wang L, Bayer JD, Christini DJ, Trayanova NA, Ripplinger CM, Kass RS & Clancy CE (2011). A computational model to predict the effects of class I anti‐arrhythmic drugs on ventricular rhythms. Sci Transl Med 3, 98ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier D, Gallagher R, Mitten‐Lewis S, McKinley S & Squire J (2002). Australian implantable cardiac defibrillator recipients: quality‐of‐life issues. Int J Nurs Pract 8, 68–74. [DOI] [PubMed] [Google Scholar]
- Plank G, Zhou L, Greenstein JL, Cortassa S, Winslow RL, O'Rourke B & Trayanova NA (2008). From mitochondrial ion channels to arrhythmias in the heart: computational techniques to bridge the spatio‐temporal scales. Philos Trans A Math Phys Eng Sci 366, 3381–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantner LJ, Tice BM & Trayanova NA (2013). Terminating ventricular tachyarrhythmias using far‐field low‐voltage stimuli: mechanisms and delivery protocols. Heart Rhythm 10, 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubart M & Zipes DP (2005). Mechanisms of sudden cardiac death. J Clin Invest 115, 2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Tusscher KHWJ & Panfilov AV (2006). Alternans and spiral breakup in a human ventricular tissue model. Am J Physiol Heart Circ Physiol 291, H1088–H1100. [DOI] [PubMed] [Google Scholar]
- Tsao JY, Han J, Haitz RH & Pattison PM (2015). The Blue LED Nobel Prize: Historical context, current scientific understanding, human benefit. Ann Phys 527, A53–A61. [Google Scholar]
- Turk G (1991). Generating textures on arbitrary surfaces using reaction‐diffusion. In SIGGRAPH ‘91. Proceedings of the 18th Annual Conference on Computer Graphics and Interactive Techniques, pp. 289–298. ACM, New York.
- Vadakkumpadan F, Arevalo H, Prassl AJ, Chen J, Kickinger F, Kohl P, Plank G & Trayanova N (2010). Image‐based models of cardiac structure in health and disease. Wiley Interdiscip Rev Syst Biol Med 2, 489–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigmond EJ, Hughes M, Plank G & Leon LJ (2003). Computational tools for modeling electrical activity in cardiac tissue. J Electrocardiol 36 Suppl, 69–74. [DOI] [PubMed] [Google Scholar]
- Vigmond EJ, Weber dos Santos R, Prassl AJ, Deo M & Plank G (2008). Solvers for the cardiac bidomain equations. Prog Biophys Mol Biol 96, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt CC, Bruegmann T, Malan D, Ottersbach A, Roell W, Fleischmann BK & Sasse P (2015). Systemic gene transfer enables optogenetic pacing of mouse hearts. Cardiovasc Res 106, 338–343. [DOI] [PubMed] [Google Scholar]
- Wasala NB, Shin JH & Duan D (2011). The evolution of heart gene delivery vectors. J Gene Med 13, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JC, Xu J, Lu Z, Klimas A, Chen X, Ambrosi CM, Cohen IS & Entcheva E (2013). Computational optogenetics: empirically‐derived voltage‐ and light‐sensitive channelrhodopsin‐2 model. PLoS Comput Biol 9, e1003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Gutbrod SR, Bonifas AP, Su Y, Sulkin MS, Lu N, Chung HJ, Jang KI, Liu Z, Ying M, Lu C, Webb RC, Kim JS, Laughner JI, Cheng H, Liu Y, Ameen A, Jeong JW, Kim GT, Huang Y, Efimov IR & Rogers JA (2014). 3D multifunctional integumentary membranes for spatiotemporal cardiac measurements and stimulation across the entire epicardium. Nat Commun 5, 3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1 Failed defibrillation with ChR2 and a short light stimulus
Video S2 Ventricular fibrillation control
Video S3 Failed defibrillation with ChR2+ and a long light stimulus
Video S4 Failed defibrillation with ChR2‐RED and a short light stimulus
Video S5 Successful defibrillation with ChR2‐RED and a short light stimulus
Video S6 Successful defibrillation with ChR2‐RED+ and a short light stimulus
Video S7 Successful defibrillation with ChR2‐RED+ and a long light stimulus