

Abstract
Risk stratification in the context of sudden cardiac death has been acknowledged as one of the major challenges facing cardiology for the past four decades. In recent years, the advent of high performance computing has facilitated organ‐level simulation of the heart, meaning we can now examine the causes, mechanisms and impact of cardiac dysfunction in silico. As a result, computational cardiology, largely driven by the Physiome project, now stands at the threshold of clinical utility in regards to risk stratification and treatment of patients at risk of sudden cardiac death. In this white paper, we outline a roadmap of what needs to be done to make this translational step, using the relatively well‐developed case of acquired or drug‐induced long QT syndrome as an exemplar case.
Abbreviations
- aLQTS
acquired long QT syndrome
- CICR
calcium‐induced calcium release
- CiPA
comprehensive in vitro proarrhythmic assay
- EC
excitation–contraction
- ECG
electrocardiogram
- EP
electrophysiology
- HPC
high performance computing
- HR
heart rate
- ICD
implantable cardioverter defibrillator
- IPSC
induced pluripotent stem cell
- SCD
sudden cardiac death
- SR
sarcoplasmic reticulum
- TdP
torsades de pointes
The challenge
Sudden cardiac death (SCD), which is most commonly caused by cardiac arrhythmias, accounts for ∼10% of all deaths in developed countries (de Vreede‐Swagemakers et al. 1997; Deo & Albert, 2012). Patients who suffer a SCD have some underlying cardiac pathology, even if they were unaware of it at the time. While implantable cardioverter defibrillators (ICDs) appear very effective at reducing the incidence of SCD, they remain expensive, and are not devoid of significant adverse effects such as inappropriate shocks, broken leads requiring invasive replacement, infections, and surgical complications, particularly in paediatric patients (Rosenqvist et al. 1998; Schimpf et al. 2003; Olde Nordkamp et al. 2015). Therefore, in order to optimise the use of ICDs, there is a critical need to correctly identify the patients who are at most risk and so most likely to derive benefit. At present our ability to identify these patients is poor and largely based on non‐specific associations such as the extent of contractile dysfunction in patients with heart failure (Stevenson & Epstein, 2003), or altered cardiac autonomic function (Nagahara et al. 2008). In the modern era of genomics‐inspired precision medicine, surely we can do better? In the last 20 years we have made great strides in understanding the molecular and cellular events underlying cardiac arrhythmias (Keating & Sanguinetti, 2001; George, 2013). The challenge now is to translate this knowledge into improved prediction and prevention of cardiac arrhythmias and SCD in patients.
Integrating the activity of the dozens of different types of ion channels and electrogenic transporters in multiple different cell types that underlie the electrical signals recorded on the electrocardiogram (ECG) is an extraordinarily complex task. Indeed, over 50 years ago Denis Noble, co‐founder of the Human Physiome project (Hunter et al. 2002; Hunter & Borg, 2003), recognised that it could only be achieved using high performance computing (HPC). Today's computers are many orders of magnitude more sophisticated and modelling of cardiac electrical activity is probably the most advanced of all the different components of the Human Physiome project. Even so, computational cardiology is not yet sufficiently advanced that it can be directly applied in the clinic. However, the establishment of a multitude of start‐up companies in this space suggests that we could be on the cusp of a revolution. Perhaps the strongest evidence to suggest that this may be the case is the Food and Drug Administration in the USA investing significant resources in the comprehensive in vitro proarrhythmia assay (CiPA) initiative. This new paradigm has in silico modelling as one of its core components for the pre‐clinical assessment of the proarrhythmic risk of all new drugs prior to clinical development (Sager et al. 2014; Fermini et al. 2016).
In this white paper, we will discuss the requirements for computational cardiology to become a reality in the context of predicting risk of cardiac arrhythmias. As a first step, we propose a roadmap for the development of such application and then discuss recent progress with the CiPA initiative and risk stratification for drug‐induced or acquired long QT syndrome (aLQTS) as an exemplar case.
A proposed roadmap for computational cardiology
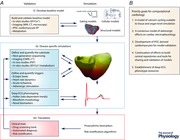
Arrhythmias are often discussed as falling into three groups: (1) triggered activities caused by early or delayed after depolarisations that may manifest as tachyarrhythmias; (2) altered automaticity resulting in either brady‐ or tachycardia; and (3) re‐entrant, where circus movement around areas of anatomical or functional block manifests as monomorphic/polymorphic ventricular tachycardia or fibrillation, depending on the stability and anchoring of the rotor (Antzelevitch & Burashnikov, 2011; Qu & Weiss, 2015). More recently, discussion has focused on the concepts of underlying substrates (that may be either structural or electrical in nature) that permit re‐entry and how they may interact with triggers in the genesis and maintenance of sustained arrhythmias (Kalin et al. 2010). The challenge in developing computational approaches to risk stratification for arrhythmias is to accurately define what we mean by substrates and triggers, and then to quantify these factors such that we can use them to generate mathematical models that can be validated against quantifiable outcomes. Lastly, and more importantly, the models then need to be able to make quantitative predictions that can be tested against the endpoint of clinical concern: proarrhythmia (see Abstract figure).
Defining and quantifying substrates for arrhythmogenesis
Arrhythmias are emergent properties of intact tissue. Nevertheless, tissues are made up of cells and molecules and so quantifying tissue level properties will also require quantifying the genetic/molecular as well as cellular/intercellular components that make up tissues. For example, consider the scar tissue that forms after a myocardial infarct. At a tissue level, the extent of the scar tissue should be imaged as accurately as possible, as well as the relative distribution of fibroblasts versus extracellular matrix within the scar and border zone. Within these regions it is also important to understand the degree of remodelling of electrical and calcium handling properties, as well as the extent and spatial heterogeneity of sympathetic denervation (Li et al. 2004; Gardner et al. 2015). Ideally, one should also be aware of the genetic makeup of patients in order to comprehend potential impact on their baseline electrophysiological profiles. Adding further complexity, dynamic changes to the substrate such as variation in extracellular electrolytes, alterations in blood flow and supply of oxygen and nutrients secondary to acute episodes of ischaemia are important as they may explain, at least partly, why the same triggers can initiate arrhythmias on some occasions but not others. Finally, when thinking about dynamic changes it is also important to consider timescales. The above modifications are likely to occur over minutes to days. However, more rapid changes such as changes in heart rate (HR) can also alter how triggers will interact with the substrate.
Defining and quantifying triggers for cardiac arrhythmias
It has long been recognised that an appropriately timed premature beat, i.e. a trigger delivered during the so‐called ‘vulnerable window’, has the propensity to initiate an arrhythmia (Mines, 1914). Indeed, the programmed application of premature, often multiple beats, forms the basis of most invasive electrophysiology (EP) studies aimed at determining the inducibility of arrhythmias (Doherty et al. 1983; Hummel et al. 1994). One challenge though is defining the conditions that determine a trigger in the setting of a dynamic substrate. For example, an ectopic beat delivered at a given coupling interval may not initiate an arrhythmia when the underlying HR is fast, but may do so when the underlying HR is slower. Furthermore, an ectopic beat of a given coupling interval and underlying HR may be more or less proarrhythmic depending on the prevailing autonomic tone. Fortuitously this is precisely where computational approaches can be most useful. In such complex scenarios, the very large number of combinations of variables may be prohibitive to test in wet lab experiments. However, with sufficient computational power hundreds and thousands of combinations of variables can be readily tested in silico and then only the critical variable combinations tested in situ or in vivo.
Developing mathematical models
When it comes to quantifying each variable and incorporating them into mathematical models, another factor that we need to consider in the face of such a challenging and complex phenomenon, is how comprehensive do these models need to be – and to what extent can they be simplified? Bearing in mind the dictum that ‘all models are wrong but some are useful’ (Box, 1976) – we need to include enough information for models to be useful, but no more than is appropriate for the task, given the constraints of computational load as well as limitations in properly constraining the model. Fortunately, this is a challenge that has been embraced over many decades, starting with the pioneering work of Denis Noble (Noble, 1962). As a result, we now have a wealth of model frameworks from the single‐channel level, through to the whole‐heart to work with. Nevertheless, if we are going to reach the summit of developing models that have genuine clinical utility then understanding the limitations of each component of each model, and determining where simplification can and should (and indeed should not) be made, is always going to be critical. Inevitably, this will be an iterative process. It will be critical to use data that provide quantitative limits on substrates and triggers, to ensure that the models ultimately will produce predictions that are relevant to arrhythmias.
A particularly good example of an area in which model development and the assessment of the most appropriate level of model complexity merits close attention is calcium handling and excitation–contraction (EC) coupling. Calcium signalling is tightly coupled to the electrical properties of the heart, such that any disturbance to the electrical system will affect calcium signalling, and vice versa. Therefore, in understanding both the genesis and functional consequences of rhythm disturbances that can lead to SCD, this is a critical area of focus. There has been a huge amount of work exploring the molecular and cellular basis of calcium handling in isolated cardiac myocytes and its contribution to EC coupling (Bers, 2001, 2002). However, modelling calcium handling even at a single cell level is much more complex than describing the electrical properties of the cell. In the case of modelling intracellular calcium concentrations, both spatial gradients and microarchitecture of the subcellular compartment are potentially just as important as temporal changes, making this a much more computationally challenging problem. Over the years, modelling approaches to this phenomenon have evolved as our knowledge of intracellular calcium handling has grown. Starting with models where intracellular calcium exists purely as a function of calcium influx through the plasma membrane (Beeler & Reuter, 1977), we have seen the addition of intracellular compartments (the sarcoplasmic reticulum) that sequester and release calcium (DiFrancesco & Noble, 1985), linking of these compartments to membrane calcium currents to describe calcium‐induced calcium release (CICR) (Luo & Rudy, 1994) and addition of diffusion restricted sub‐compartments of the cytoplasm approximating the environment of the junctional SR–T‐tubule interface (Jafri et al. 1998). The modelling of calcium homeostasis has been subsequently refined to include more accurate representations of calcium removal mechanisms and the dependence of CICR on SR load (Shannon et al. 2004; Grandi et al. 2010), the kinetics of phosphorylation of calcium handling proteins by CaMKII (Iribe et al. 2006), and updates of L‐type calcium current and Na+–Ca2+ exchanger based on data from human ventricular tissue (O'hara et al. 2011). In all of these models, calcium concentration is spatially homogenised in each compartment, i.e. takes the same value everywhere in the compartment at a given time. More recently, microdomain models have been developed that include structural as well as molecular descriptions of the dyadic cleft to varying degrees of accuracy (Schendel & Falcke, 2010; Schendel et al. 2011; Cannell et al. 2013; Stern et al. 2013), that can be scaled up to describe whole cytoplasmic calcium concentrations (Higgins et al. 2007; Restrepo et al. 2008; Rovetti et al. 2010; Gaur & Rudy, 2011; Nivala et al. 2015; Vierheller et al. 2015). This reductionist approach necessitates the simulation of tens of thousands of dyads per cell, requiring significant computational times between ten minutes to many hours for a single beat of an individual cell, depending on the level of detail included (Nivala et al. 2012; Vierheller et al. 2015). As a result, while this certainly represents a gold standard for simulation of calcium dynamics in single cells, it presents a problem for consideration of larger scale tissue or organ level simulations that will likely require implementation of multiscale computational strategies. This example therefore embodies perfectly the conundrum described above, of selecting models of appropriate level of complexity for the task at hand. It also highlights an area where particular focus should be applied in advancing numerical techniques or taking advantage of new, more powerful parallel computing hardware (Tuan et al. 2011; Chai et al. 2013), such that more complete descriptions of calcium handling can be incorporated into tissue level simulations.
Model validation
Once we have established the required models, thorough validation is critical, and this must use datasets independent of those used to generate the models in the first place. At the cellular level, one area of growth that will no doubt play a central role in validating cellular models is the use of cardiomyocytes derived from human induced pluripotent stem cells (IPSC) (Takahashi & Yamanaka, 2006; Narsinh et al. 2011). IPSC derived cardiomyocytes from patients with normal as well as diseased hearts present a platform for studying cardiac diseases (Carvajal‐Vergara et al. 2010; Moretti et al. 2010) or drug effects (Sinnecker et al. 2014) in a more faithful biological context. Critically, these ‘hearts in a dish’ maintain many intrinsic disease characteristics and genetic background of the patient, and so represent a powerful tool to study the mechanisms of disease, or the actions of drugs, without the ethical and practical implications of collecting human cardiac tissue. However, this technology is still in its relative infancy. In particular, issues exist around the relative immaturity of these cells and how well they reflect the function and morphology of the adult ventricular myocyte in relation to electrophysiology, calcium handling and responses to adrenergic stimulation for example (Laflamme & Murry, 2011). It is certainly true that efforts are already well underway to tackle this issue. Aside from long term culture on the order of months to years (Otsuji et al. 2010), more recent efforts have focused on efforts to mimic more closely the environment of the developing heart and drive these cells to a more adult phenotype such as 3D culture and mechanical stimulation (Nunes et al. 2013; Mihic et al. 2014), electrical stimulation (Eng et al. 2016) and co‐culture with non‐cardiomyocyte cells (Kim et al. 2010; Saini et al. 2015). Culturing of IPSC derived cardiomyocytes on microgrooved substrates has proved particularly effective in promoting more adult‐like cellular morphology as well sarcomeric organisation, that in turn is important for defining the subcellular spatial restrictions necessary for normal calcium homeostasis. Under these conditions, cells showed calcium transients that more faithfully reflected those observed in adult myocytes (Rao et al. 2013). These approaches have therefore shown a great deal of promise in promoting both electrical and morphological maturity, yet there is no doubt that these cells are still more fetal than adult in nature. Despite this, the technology is rapidly being embraced – most notably as part of the CiPA initiative (Sager et al. 2014). As such, it is critical that issues surrounding maturation of IPSC derived cardiomyocytes are tackled such that this rapidly emerging field can play a central role in the validation and construction of cellular electrophysiological models.
Ultimately though, sudden cardiac death represents the final common endpoint of abnormal electrical signalling in the organ, i.e. is not solely a cellular phenomenon. First and foremost therefore, any computational model must be able to reproduce, at a quantitative level, the electrical signals recorded from patients. In this regard, we foresee a very important role for deep ECG phenotyping and specifically for high‐resolution analysis of Holter ECGs that provide information on how the electrical signalling in the heart responds to a wide range of physiological stimuli. Until recently, the analysis of Holter ECGs has been limited (due to the relatively large volumes of data acquired compared to the computational power available to analyse them). Recent increases in computer processing power and storage capacity have largely obviated this problem, but there is still a lot that can and should be done to develop better software packages for analysing multi‐lead Holter ECG datasets, which will be very useful for validating computational models. This again should be seen as an iterative process and indeed one can envisage that this process will lead to identification of better biomarkers of cardiac electrical activity that in themselves may provide better means for stratifying risk of SCD.
Drug‐induced arrhythmias: a case study for in silico risk prediction
In the past 15 years, a range of structurally unrelated non‐cardiovascular drugs have been withdrawn from the market due to adverse effects on cardiac repolarisation and risk of heart rhythm disturbances – so called acquired or drug‐induced long QT syndrome (aLQTS) (Wood & Roden, 2004). These drugs include antihistamines, antibiotics, antipsychotics and most recently the analgesic propoxyphene, which was prescribed to an estimated 10 million patients in the US at the time of its withdrawal in 2010. The aLQTS is characterised by delayed repolarisation, prolongation of the QT interval on the surface electrocardiogram (ECG) and a markedly increased risk of a potentially lethal ventricular arrhythmia named torsades de pointes (TdP) (Wood & Roden, 2004; Kannankeril et al. 2010). aLQTS can be caused by drugs that block any of the ionic currents that contribute to repolarisation of the heart. In practice, however, the vast majority of drugs that can cause aLQTS do so by inhibiting hERG channels (Perrin et al. 2008 b). This observation led to the current suite of guidelines relating to cardiac safety that were issued in 2005: recommending that, at a minimum, all compounds are subject to an in vitro evaluation of hERG block together with assessment of in vivo QT interval prolongation in an appropriate animal model (ICH S7B) and an assessment of QT prolongation in humans (ICH E14) (Food and Drug Administration, HHS, 2005 a,b).
These guidelines have several important shortcomings. First amongst them is that while the tests are very sensitive, and have been successful in eliminating dangerous drugs, they are believed to be insufficiently specific. Neither hERG block nor QT prolongation are good predictors of risk of TdP (Sager et al. 2014). There is also growing concern that this approach has led to the premature termination of the development of drugs based on their hERG affinity, rather than propensity to actually cause arrhythmia. In the context of the roadmap outlined in the Abstract figure, potency of hERG block and QT prolongation do not provide sufficiently accurate markers of the proarrhythmogenic substrate to be useful for quantifying risk. These shortcomings are acknowledged by the regulatory agencies, which has led to the establishment of CiPA. This initiative is set to change the regulatory guidelines around cardiac safety testing to a multi‐ion channel, rather than hERG‐centric approach, that is driven by in silico risk prediction (Sager et al. 2014; Fermini et al. 2016). This new approach will take into account detailed characterisation of molecular pharmacology including ion channel–drug interaction kinetics as well as multichannel block profiles with the idea that simulations of cardiac electrophysiology based on this mechanistic insight will better identify proarrhythmic drugs.
However, while CiPA focused on the use of single cell modelling for prediction of proarrhythmic propensity, in reality the genesis of arrhythmia in the intact heart involves so much more than solely the molecular interaction of ligand and target. Drugs that block the hERG channel both delay repolarisation and increase the dispersion of repolarisation across the heart (Kannankeril et al. 2010). The delayed repolarisation is considered to increase the probability of reactivation of L‐type calcium channels leading to early after‐depolarisations (EADs) that could act as the ‘trigger’ for arrhythmia (January & Riddle, 1989). Additionally, the increased transmural repolarisation heterogeneity produces regions of functional block and slow conduction that provide an intramural substrate for re‐entry (Akar & Rosenbaum, 2003). Although we understand in principle the tissue level mechanisms of arrhythmogenesis in aLQTS, the link between hERG block and the emergent arrhythmia is complex. However, the processes that need to be in place to interrogate this in silico are relatively well established, meaning the aLQTS example is an ideal illustration of the computational risk prediction pipeline outlined in the Abstract figure. The specifics of model development, defining and quantifying substrates and identification of novel risk biomarkers from multiscale models in relation to aLQTS are discussed below.
Development and optimisation of models for in silico risk prediction in aLQTS
An important step in the pursuit of effective in silico risk prediction is the selection and optimisation of the molecular and cellular models used for studying the action of pharmaceutical compounds. At the cellular scale, we need to reach a consensus on an appropriate action potential model. This is a critical step given the dramatic range in action potential morphology that exists between published models (Cooper et al. 2016) as well as the intrinsic properties of the models, such as forward/reverse rate dependence, that must be considered in understanding how individual models respond to external perturbations (Cummins et al. 2014). In either case, variation in maximal current densities (maximal ‘conductances’ in the case of ion channels) is a major source of discrepancy between predictions from different models. Unfortunately, most of these conductances are not easy to measure directly, particularly from healthy human cardiomyocytes, and have to be inferred from action potential recordings and cell‐level behaviour. While some recent models of the ventricular action potential have been based on data from healthy human hearts (O'hara et al. 2011), there is still work to be done in tuning these models for the range of physiological stressors that are critical in arrhythmogenesis such as their responses to adrenergic stimulation. In this regard, the use of IPSC derived cardiomyocytes (see Model validation section, above) will no doubt contribute to providing the datasets necessary for this ongoing model development and validation.
Equally important in determining model predictions is the choice of model for each ion current, specifically concerning channel gating kinetics in control conditions. Here, most models use Hodgkin–Huxley or Markov model structures for the ion channel states and transitions. This is not a simple task. For example, there are at least seven different published model structures for the I Kr current alone (Noble et al. 1998; Lu et al. 2001; Piper et al. 2003; Adeniran et al. 2011; Bett et al. 2011; Di Veroli et al. 2013 b). When we consider different parameterisations of these model structures (different transition rates and their voltage dependence), there are over thirty published model variants for I Kr. There is therefore uncertainty in both the model structure/equations and also the parameters which should be used (see Mirams et al. (2016): another white paper in this issue). Quantifying this uncertainty will be an important goal in the coming years, and will be necessary to place an appropriate level of trust in model predictions (Johnstone et al. 2015). As part of this effort, we should quantify which experiments provide information on parameters and structures: a whole sub‐field of statistics/control theory called ‘optimal experimental design’ is dedicated to this task, but only a few steps have been taken so far in applying this to cardiac modelling (Dokos & Lovell, 2004; Fink & Noble, 2009; Groenendaal et al. 2015). It is also worth noting that a perfect fit to training data is not a sign that the model will be predictive in future, particularly if multiple model structures and/or parameter sets provide an equally good fit (Fink et al. 2011). Models should always be evaluated in the context of predicting an outcome that they were not trained to replicate.
Each of these factors, the selection of cellular and molecular models, have important bearing in terms of evaluating predictions of drug‐induced proarrhythmic risk, such as those envisaged by CiPA, where in silico predictions are currently based mostly on fits to standardised in vitro datasets. In this regard, a gold‐standard model for use in computational evaluation of proarrhythmic risk may be the largest gap in our knowledge. Careful choice and further calibration and validation of ion current and action potential models remains one of the fundamental challenges for computational physiology in the coming years that is necessary to predict proarrhythmia associated with acquired LQTS more accurately. To ensure transparency and engender confidence in such computational approaches, we need to publish: (i) training data; (ii) calibration/fitting and selection algorithms that give rise to the final model; and (iii) validation data and performance metrics. This approach is in line with the general trend within science of moving towards ‘open data’ with a view to ensuring reproducibility, especially in computational science. In this regard, platforms such as Zenodo (hosted at CERN), datahub.org and researchcompendia.org provide the infrastructure for publishing and sharing of scientific data and models while many discipline specific repositories have also been built in recent years (see e.g. NIH Data repositories: https://www.nlm.nih.gov/NIHbmic/nih_data_sharing_repositories.html). Of particular relevance to the Physiome community, the CellML effort allows us to share model equations and parameters easily and provides a forum for model curation to ensure consistency of implementation between groups (Lloyd et al. 2008), while the recently established Cardiac Electrophysiology Web Lab (Cooper et al. 2016) allows functional curation of CellML encoded models, letting users compare and contrast how different models behave in response to a suite of virtual protocols. Perhaps the research field in which the discussion around data/model sharing is most advanced is genomics. The Broad Institute recently published white paper discussing the international standards and computational needs to share and integrate data in a secure, controlled and interpretable manner (Alliance, 2014). This can serve as a worthy template for the action that should be taken by the Physiome community which should be encouraged and indeed mandated by funding agencies and consortium heads to ensure reproducibility in our discipline.
Defining and quantifying substrates in aLQTS
In the aLQTS the primary substrate is abnormal electrical function precipitated by the block of cardiac ion channels (primarily hERG) as a side‐effect of both cardiac and non‐cardiac drugs. Specifically, the disruption of the repolarisation reserve is thought to amplify transmural spatial dispersion of repolarisation, establishing voltage gradients that can act as a substrate for re‐entry (Antzelevitch, 2007), while other factors such as temporal dispersion of repolarisation (i.e. beat to beat instability) are also thought to contribute (Frommeyer & Eckardt, 2015). The challenge of quantifying the substrate therefore lies in accurately defining and modelling the pharmacology of drug–channel interactions in the context of the cardiac action potential. To do this requires that we revisit the question of what degree of complexity these models must encapsulate to make in silico prediction of their proarrhythmic propensity a reality?
Measuring and modelling hERG block
Whilst it is certainly true that potency of block is not a good indication of proarrhythmia (Redfern et al. 2003; Di Veroli et al. 2013 a), a key outstanding question in determining the extent to which it is necessary to develop biophysically accurate models of drug binding is: are the kinetics of drug interaction with hERG, and indeed other channels, important in determining proarrhythmic risk? In support of this concept, Di Veroli et al. proposed that kinetics, rather than multichannel block profiles, might underlay the difference in proarrhythmic propensity between the drugs verapamil and bepridil (Di Veroli et al. 2013 a). More recently, Lee et al. demonstrated in silico how the kinetics of drug binding can modify both the degree of prolongation observed with an IC50 concentration of drug well as the Action Potential (AP) morphology, in terms of the degree of triangulation for example (Lee et al. 2015), that reflect likely differences in the associated risk profile (Hondeghem & Carlsson, 2001). This study, however, was completely theoretical, and it remains to be seen where real drugs fall into this theoretical landscape, and whether the range of kinetics observed in vitro allows for these complex dependencies.
When generating models of the kinetics of drug binding to ion channels, many of the same issues that arise in relation to modelling ion channel function also come to light. Multiple published models exist whose structures vary according to considerations such as to which states does a compound bind/unbind (Di Veroli et al. 2013 a); is binding voltage dependent (Moreno et al. 2011); do drugs get trapped behind a closed activation gate (Mitcheson et al. 2000); or does drug binding alter the intrinsic gating behaviour of the channel (Lee et al. 2015)? The key to addressing these issues in silico lies with accurately measuring and modelling the kinetics of drug binding to hERG (as well as other channels). Much of the complexity in achieving this stems from the fact that drugs can differentially bind to the open and inactive states of the hERG channel (Suessbrich et al. 1997; Walker et al. 1999; Weerapura et al. 2002; Perrin et al. 2008 a). Most studies assessing binding kinetics depend on complex voltage protocols that result in cycling of the channel between open and inactive states in a voltage dependent manner (Vandenberg et al. 2012). As a result, it is difficult to confidently deconvolute drug binding kinetics from voltage dependent channel gating. One way to tackle this problem is to directly monitor the drug–channel interaction at a single voltage (Milnes et al. 2010; Hill et al. 2014). Using similar approaches with other drugs presents us with the opportunity to accurately model the mechanism by which drugs block the hERG channel, and interrogate in detail questions relating to drug binding kinetics. For example, how do the kinetics of binding and unbinding vary with temperature? This is a question that is critical to answer if we want to extrapolate from data acquired in high throughout systems at room temperature to physiological temperatures in silico. How do the kinetic properties of the drug correlate with the observance of proarrhythmic markers such as triangulation, reverse use dependence, instability and dispersion (TRIaD criteria) and indeed do any specific kinetic properties correlate with risk?
With a view to answering these questions, a central tenet of CiPA is to include kinetics of block, rather than just potency, in their in silico models to estimate risk. However, CiPA is bound by practicalities of industrial screening in the extent to which kinetics can be measured. For example, most high‐throughput patch clamp systems are not able to switch perfusion solutions fast enough to allow direct, accurate measurement of drug binding and unbinding. Furthermore, the extent to which the kinetics of drug binding are actually important in determining risk is not yet fully understood. As a result, first‐pass measures of drug‐binding kinetics deemed appropriate for CiPA may not necessarily equate to the more academically attractive endpoint of fully characterising the kinetics of drug binding in order to constrain mechanistically accurate models of the interaction. However, it is clear the most effective and efficient approach to tackle this problem in the coming years is to leverage the resources and expertise available across the range of stakeholders from academia, the pharmaceutical industry and regulatory bodies to ensure that we have the right predictive models that will benefit patients.
Modelling multichannel block
Alongside hERG‐specific drug interactions, it is increasingly clear that a more complete pharmacological profile of individual compounds needs to be considered in order to properly assign risk (Mirams et al. 2011; Kramer et al. 2013), and therefore needs to be described in silico as we work towards holistic models of drug effects. Even a small degree of mixed channel block can shorten or prolong the QT interval by a few milliseconds, enough to be measured in a clinical ECG study. However, it is not known what (if any) risk is associated with these changes. It is therefore important to understand whether small degrees of block of multiple cardiac ion channels can combine to give rise to increased proarrhythmia, or alternatively perhaps confer some safety margin by compensating for block that would ordinarily be proarrhythmic. Such effects are certainly observed in the clinic, with drugs such as verapamil and amiodarone for example, whose multichannel pharmacology, especially block of cardiac calcium channels, appear to significantly abrogate the risk profile that would be associated with their block of hERG. Mathematical models offer an intuitive and biophysically based way to combine the effects of multiple ion channel block in order to tackle this question. In silico approaches based on concentration–effect data derived from high‐throughput drug screens on multiple ion channels have been examined in this regard and have shown good qualitative and quantitative agreement with experimental results from rabbit ventricular wedge preparations (Beattie et al. 2013) but less good prediction of results from thorough QT studies (Mirams et al. 2014), suggesting there is still work to be done in this area. A limitation of these studies is that simple concentration–effect data do not take into account the full complexity of drug–channel interactions, such as descriptions of binding and unbinding kinetics as described above for hERG, meaning this is an obvious area to address in seeking to improve in silico models of multichannel block. It should be noted, however, that development of Markov‐type descriptions of channel gating and pharmacology for multiple ion channels represents a major undertaking, so once again warrants careful consideration of the depth of characterisation that is deemed necessary.
A second aspect to consider in relation to multichannel block pertains to polypharmacy. Many, if not most, adverse proarrhythmic effects are seen in patients who are taking multiple drugs (‘torsade coadministration’ has > 16,700 hits on Google Scholar as of October 2015, see e.g. (Chézalviel‐Guilbert et al. 1995; Atar et al. 2003; Kounas et al. 2005; Kaźmierczak et al. 2007). With the correct application of in silico modelling of multichannel pharmacology, predictions could be used to avoid co‐administration of particularly dangerous compounds. This advance will require risk information to be disseminated to the doctors in the clinic differently: risk is no longer associated with the ‘black box’ for a particular drug, but a new ‘black box’ would be generated for the patient's particular prescription. However, in this scenario of combinations of compounds, each with multi‐target pharmacology, it is likely that historically trained statistical models will not be capable of accurate predictions – there is quickly a combinatorial explosion with many risk factors; so no training procedure will be able to assemble enough cases to cover ‘all the options’ we may wish to consider. For this reason, we consider that biophysically based mechanistic models will continue to have a larger predictive advantage over purely statistical models in future.
Patient specific factors influencing the substrate in aLQTS
The major focus of current regulatory guidelines is the identification of compounds that carry risk of TdP. However, even in the context of ‘dangerous drugs’, the incidence of TdP is relatively low. For example, the incidence of TdP in randomised trials of dofetilde is reported to be 0.8–3.3% (Torp‐Pedersen et al. 1999; Singh et al. 2000). This raises the question of what makes these subsets of patients vulnerable to drug‐induced TdP? If we were able to tackle this problem, it may soon be plausible to move risk stratification from the pill to the patient, taking into account the sub‐groups that a patient falls into, in terms of age, sex, genetic background and disease states. All of these are well‐recognised risk factors for proarrhythmia (Zeltser et al. 2003; Frommeyer & Eckardt, 2015), and are known to influence the electrophysiology of the cell in ways that can be encapsulated in mathematical model parameters (Yang & Clancy, 2012).
Genetic background
The concept of ‘repolarisation reserve’ has provided the conceptual framework to understand why a person's genetic background may alter their response to QT prolonging drugs (Roden, 2006). The central tenet of this concept is that normal repolarisation is a function of several redundant mechanisms, where the overall repolarisation potential is determined by the level of expression and function of multiple cardiac ion channels, which in turn are coded for by an individual's genetic background. In this scenario, the effect of QT prolonging drugs occurs in the context of different electrical environments from person to person, meaning the level of risk is specific to the individual.
Our ability to characterise this electrical background, at least at the genetic level, has increased exponentially with the advent of next generation sequencing. In this regard, the early tranche of studies identified both cardiac (Weeke et al. 2014) and non‐cardiac (Jamshidi et al. 2012) genes involved in risk determination in aLQTS. In this time of precision medicine (Collins & Varmus, 2015), in silico approaches hold the promise of (1) allowing us to interpret this wealth of available data; (2) quantitatively analyse the effect of variable genetic backgrounds; (3) identify potentially protective and/or risky backgrounds, and most importantly (4) make predictions about how an individual or group of individuals may respond to a QT prolonging drug. Early efforts to tackle these questions have involved using multivariate analysis to examine how variable genetic background altered action potential duration (APD) prolongation in response to I Kr block (Sarkar & Sobie, 2011), and provide an excellent platform for further in silico study of this area. At the cellular level this approach could be extended to better proarrhythmic markers such as triangulation or early after depolarisations (EADs), while simulation of organ‐level effects will allow us to take into account tissue properties relevant to proarrhythmia such as transmural dispersion (Akar & Rosenbaum, 2003), alternative ECG markers associated with risk (discussed below) and more complex rate dependent effects. In this regard, the potential to extend sensitivity analysis to the organ level now exists (Sadrieh et al. 2014), and presents an opportunity to tackle this problem in the coming years.
Tissue structure
A second factor to consider in identifying patients at risk is the tissue structure of the myocardium in each individual. A confounding effect in the current approach is that early phase clinical trials are carried out in cohorts of healthy individuals, and consequently do not take into account potential patient specific co‐morbidities such as tissue structure alterations that can influence the level of arrhythmia risk associated with a drug. The patients at greatest risk of cardiac adverse effects are likely to be those with pre‐existing heart disease and associated structural remodelling. For example, the patchy fibrosis characteristic of ageing, hypertensive heart disease and heart failure gives rise to structural discontinuities across a range of scales that can contribute to initiation of arrhythmia (Tanaka et al. 2007; Tusscher et al. 2007). Furthermore, the more compact fibrosis of healed myocardial infarcts can also provide a focused substrate for circus re‐entry (Jalife, 2000). It is likely that such structural abnormalities could help explain the variability in risk profiles observed with drugs that prolong the QT interval. This concept is supported by early clinical trials of antiarrhythmic drugs such as the Cardiac Arrhythmia Suppression Trial (CAST) that was designed to test the hypothesis that suppression of premature ventricular complexes with class I antiarrhythmic agents would reduce mortality after a myocardial infarction, but instead found that the tested drugs actually decreased survival (The Cardiac Arrhythmia Suppression Trial (CAST) Investigators, 1989; Echt et al. 1991).
From the perspective of computational physiology, multiple in silico approaches have already demonstrated the role of structural discontinuity in arrhythmogenesis (Caldwell et al. 2009; Engelman et al. 2010; Rutherford et al. 2012). Specifically, it has been shown that patchy fibrosis can give rise to re‐entrant activation as well as dynamic functional substrates that maintain re‐entrant circuits (Engelman et al. 2010). It is a short conceptual leap to suppose that these structurally defined substrates and triggers will amplify the specific proarrhythmic properties of drugs in aLQTS. Of particular interest in this regard will be an evaluation of rate dependent effects. For example, drugs that interact with the hERG channel with different kinetics and/or state dependence may well differentially affect the rate dependent instability that arises from structural defects such as fibrosis. A further factor to consider regarding the role of structural alteration in modifying risk is that the repolarisation heterogeneity, which is thought to contribute to initiation of arrhythmias in aLQTS, is amplified by structural remodelling (Akar & Rosenbaum, 2003). In the structurally normal heart, this heterogeneous repolarisation dispersion is modulated by electrotonic coupling (Nattel et al. 2011) that one could argue is reduced in structural heart disease, providing further amplification of proarrhythmic effects. Such problems, which are likely to include identifying precise mechanistic features of drug binding that contribute to structural‐dependent risk modification, are very difficult to tackle either in vivo or in vitro. However, in silico approaches offer the flexibility to quantitatively address these issues.
To achieve this, however, in addition to appropriate molecular models of drug–channel interaction, will also require high resolution structural imaging to provide accurate and detailed meshes for in silico approaches. Over the years imaging approaches have revealed detailed laminar architecture of the heart including collagen organisation and myofibre orientation that are critical in defining electrical propagation. Similar approaches have also been extended to imaging of infarct and peri‐infarct zones in animal models allowing in silico evaluation of how electrical and structural disruptions interact to precipitate potentially arrhythmic phenotypes (Rutherford et al. 2012; Deng et al. 2015). A critical area for development in this field, as we work towards clinical integration, will be the continued growth of imaged‐based analysis in human subjects. Imaged‐based models, that include details of structural discontinuities exist for both human atria (Zhao et al. 2013; McDowell et al. 2015) and ventricles (Wallman et al. 2014; Ukwatta et al. 2015), while recent developments have focused on techniques and pipelines for extracting useful structural meshes of human hearts and infarct scars from standard medical imaging approaches (Crozier et al. 2015; Ukwatta et al. 2015). These pipelines are steps toward structure‐based personalisation of in silico approaches that will, over time, also establish populations of cardiac structures. In the context of aLQTS, for example, this will allow us to consider more broadly how electrical disturbances that occur as adverse effects of drugs are influenced by the range of structural discontinuities present in human subjects, to provide a more nuanced consideration of risk stratification in this area.
In silico driven risk stratification for aLQTS
Tackling the range of issues outlined above will provide the necessary inputs to facilitate organ‐level simulations aimed at identifying ECG biomarkers that are more predictive of TdP than the current standard of using the QT interval. Resolving this problem in silico embodies the motivation of the Physiome project. Many of the issues faced in risk prediction in aLQTS are related to the fact that it is almost impossible to predict the path from molecular event – drug binding to an ion channel in this case – to emergent properties of the cardiac electrical system that might manifest on the ECG as a useful biomarker for proarrhythmic risk. The ultimate goal for in silico risk prediction is therefore to incorporate the factors discussed above, including details of molecular interactions, cellular biophysics, tissue structure and geometry of the heart, into multiscale, organ level simulations that can predict these emergent outputs, which can then be correlated with the degree of risk associated with known compounds, validated against clinical ECG data, and used for future risk prediction.
Organ level simulations for identification of new ECG biomarkers of risk
To date, in silico studies of the effects of drug binding to hERG and other channels have largely been focused at the cellular (Mirams et al. 2011; Beattie et al. 2013; Romero et al. 2014) and pseudo‐tissue level (Roden & Viswanathan, 2005; Brennan et al. 2009), and these studies have certainly highlight the potential of the approach. The extension of this analysis to the level of the organ is less developed, perhaps because of the computational demands, or simply an acceptance that simpler in silico approximations are, on balance, a reasonable compromise given the shortcomings of current molecular and cellular level models that we have highlighted above. However, the feasibility of the approach has been demonstrated in several recent studies. Zemzemi et al. reported simulations from human torso and derived a 12 lead ECG, including a basic representation of hERG block (Zemzemi et al. 2013). More recently, quantitative analysis of the molecular basis of the T wave based on highly parallelised simulations of many thousands of heartbeats, from hearts with different genetic and molecular backgrounds, highlighted how the subtleties of the T wave shape are related to underlying ion channel conductances (Sadrieh et al. 2014). This parallelised approach should prove especially useful when applied to identification of new ECG biomarkers of risk. An efficient approach might be to simulate many thousands of ECGs from hearts with variable tissue structure and genetic backgrounds in the presence of drugs with different kinetic properties and pharmacological profiles to create a library of simulated ECGs that can be screened for ECG biomarkers.
This of course begs the question of what specific properties of the ECG waveform should be considered in the search for these markers of risk? In this regard, there is a growing appreciation that the morphology of the T wave can help paint a much more complete picture of cardiac electrophysiology (Vaglio et al. 2008; Johannesen et al. 2014; Sadrieh et al. 2014; Vicente et al. 2015). This idea is supported by clinical data that illustrate how the morphology of the T wave is informative about the range of ion channels blocked by a drug (Vicente et al. 2015). More recently research from the Mayo clinic has also demonstrated that specific aspects of T wave morphology – specifically the slope of the descending limb – are correlated with risk of TdP (Sugrue et al. 2015). It therefore seems likely that features of the morphology of the T wave on the ECG should be a primary target in our search for new biomarkers. In addition to this, dynamic features of the ECG (such as temporal changes in QT interval and other morphological features of the signal) are a relatively neglected aspect of the ECG that may contribute to better understanding the various arrhythmogenic mechanisms associated with single‐ and multi‐channel drugs (Page et al. 2015). The volume of data that can be generated representing a range of pharmacological, genetic and environmental backgrounds using parallelised in silico cardiac models presents an opportunity to study these dynamic features in detail and develop them as a complementary facet to T‐wave morphology as ECG biomarkers for drug safety and efficacy. Furthermore, the flexibility of simulation relative to more conventional in vitro or in vivo assays offers the potential to interrogate a range of rate dependence and complex stimulation protocols, and to correlate the emergence specific ECG parameters with the onset of arrhythmogenic properties (such as cellular EADS) and even the initiation of TdP.
The future: moving computational cardiology into the clinic
There is still a huge amount of work to be done to achieve the goal embodied in the human Physiome project, i.e. being able to develop individualised in silico models, in a realistic timeframe, that can help determine the risk of cardiac arrhythmias in patients. Success will require input from all sectors of the biomedical research enterprise, spanning the academic, industrial and clinical spheres and must be truly multi‐disciplinary. An integrative approach will involve extensive detailed genotyping, comprehensive in vitro and in vivo phenotyping of multiple systems including electrical, calcium handling, imaging, contractile and metabolic systems, and ultimately high performance computer modelling to integrate the in vitro and in vivo results to develop quantitative insights into the nature of arrhythmogenic substrates and triggers. This approach, combined with a ‘big data’ analysis of continuous ECG traces will allow the identification of novel ECG biomarkers that have much greater power to discriminate between patients at high risk of proarrhythmic events and those at low risk. Drawing from the experience in the field of aLQTS discussed above, and the gaps in our knowledge that it highlights, we submit that the following specific goals should be pursued as a matter of priority to accelerate the convergence of computational and clinical cardiology:
-
1.
A model of calcium cycling scalable to tissue and organ level simulation. Modelling intracellular calcium, even at the single cell level, is currently a bottleneck for in silico studies of arrhythmia. Studies using many‐core accelerators such as Intel's Xeon Phi or NVIDIA GPU have mainly focused on increasing the spatial resolution of simulating subcellular calcium down to the nanometer. While these approaches tackle the programming and performance challenges associated with these architectures, our focus should be more on numerical methods to work towards a multiscale model of calcium cycling that can be scaled up to the tissue level, albeit with necessary simplifications.
-
2.
A consensus model of adrenergic effects on cardiac electrophysiology. While adrenergic stimulation, either as fight or flight surge, or increased basal tone in the context of ischaemia has been implicated in arrhythmogenesis, this factor is largely absent from models of human ventricular electrophysiology. Particular focus needs to be placed on developing models of the effects of adrenergic stimulation on the major components of repolarisation reserve (I Ks, I CaL), as well as intracellular calcium signalling in human cells.
-
3.
Development of IPSC derived cardiomyocytes for model validation. Current uptake suggests IPSC derived cardiomyocytes are set to play a major role as a source of human tissue for validation of computer models of cellular electrophysiology and pharmacology as well as providing data to constraint models of particular disease phenotypes. While this goal is outside the immediate computational domain, development of maturation strategies for these cells, such that data can be obtained that is equivalent to adult human tissue, is critical. Furthermore, since industry as well is aggressively pursuing this goal, primarily as part of the CiPA initiative, ongoing collaboration and engagement with industry in this sector is the likely the most productive and efficient approach.
-
4.
Continuation of efforts to build central repositories and tools for sharing and validation of models. The CellML language and repository already provide a centralised source for model sharing, while the recently published EP Web Lab allows standardised testing of these models. This resource should be extended to provide a centralised repository for published data, protocols, structural meshes and associated validation datasets such as in vitro electrophysiology and clinical ECG data. The Physiome community should explore working groups around facilitating such resources.
-
5.
Establishment of deep ECG phenotype resources. Accurate phenotyping of ECG datasets is critical for the iterative improvement of whole heart simulations as well as identification and validation of novel ECG biomarkers of risk identified in silico. Such analysis is likely to involve detailed characterisation of ECG wave morphology, as well as the dynamic changes that are associated with emergence of ectopic triggers. These types of analysis will necessitate use of data from Holter and/or implantable loop recorder ECGs to characterise how the heart responds to a wide range of physiological stimuli as well as to capture relatively infrequent, but information rich regions preceding and following ectopic beats.
If such goals can be met, and can be demonstrated to work in a relatively ‘simple’ case such as aLQTS, this will provide an impetus to pursue similar studies in other genetic arrhythmia syndromes (Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia (CPVT), hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy (ARVC)) and ultimately in the more complex scenario of SCD complicating ischaemic heart disease which involves electrical, calcium, metabolic and structural remodelling.
Additional information
Competing interests
None declared.
Funding
A.P.H. is funded by an Australian Research Council Future Fellowship FT110100075. J.I.V. is funded by a National Health and Medical Research Council fellowship 1019693. G.M. is funded by a joint Wellcome Trust/Royal Society fellowship 101222/Z/13/Z.
References
- Adeniran I, McPate MJ, Witchel HJ, Hancox JC & Zhang H (2011). Increased vulnerability of human ventricle to re‐entrant excitation in hERG‐linked variant 1 short QT syndrome. PLoS Comput Biol 7, e1002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akar FG & Rosenbaum DS (2003). Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res 93, 638–645. [DOI] [PubMed] [Google Scholar]
- Alliance G (2014). Creating a global alliance to enable responsible sharing of genomic and clinical data. Global Alliance for Genomics and Health.
- Antzelevitch C (2007). Ionic, molecular, and cellular bases of QT‐interval prolongation and torsade de pointes. Europace 9 (Suppl. 4), iv4–iv15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C & Burashnikov A (2011). Overview of basic mechanisms of cardiac arrhythmia. Card Electrophysiol Clin 3, 23–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atar S, Freedberg NA, Antonelli D & Rosenfeld T (2003). Torsades de pointes and QT prolongation due to a combination of loratadine and amiodarone. Pacing Clin Electrophysiol 26, 785–786. [DOI] [PubMed] [Google Scholar]
- Beattie KA, Luscombe C, Williams G, Munoz‐Muriedas J, Gavaghan DJ, Cui Y & Mirams GR (2013). Evaluation of an in silico cardiac safety assay: using ion channel screening data to predict QT interval changes in the rabbit ventricular wedge. J Pharmacol Toxicol Methods 68, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler GW & Reuter H (1977). Reconstruction of the action potential of ventricular myocardial fibres. J Physiol 268, 177–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers D (2001). Excitation‐contraction coupling and cardiac contractile force, 2nd ed. Springer Netherlands.
- Bers DM (2002). Cardiac excitation‐contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Bett GCL, Zhou Q & Rasmusson RL (2011). Models of HERG gating. Biophys J 101, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Box G (1976). Science and statistics. Journal of the American Statistical Association 71, 791–799. [Google Scholar]
- Brennan T, Fink M & Rodriguez B (2009). Multiscale modelling of drug‐induced effects on cardiac electrophysiological activity. Eur J Pharm Sci 36, 62–77. [DOI] [PubMed] [Google Scholar]
- Caldwell BJ, Trew ML, Sands GB, Hooks DA, LeGrice I & Smaill BH (2009). Three distinct directions of intramural activation reveal nonuniform side‐to‐side electrical coupling of ventricular myocytes. Circ Arrhythm Electrophysiol 2, 433–440. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Kong CHT, Imtiaz MS & Laver D (2013). Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys J 104, 2149–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal‐Vergara X Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, Ge Y, Cohen N, Edelmann LJ, Chang B, Waghray A, Su J, Pardo S, Lichtenbelt KD, Tartaglia M, Gelb BD, Lemischka IR. (2010). Patient‐specific induced pluripotent stem‐cell‐based models of LEOPARD syndrome. Nature 465, 808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai J, Hake J, Wu N, Wen M & Cai X (2013). Towards simulation of subcellular calcium dynamics at nanometre resolution. International Journal of High Performance Computing Applications DOI: 10.1177/1094342013514465. [Google Scholar]
- Chézalviel‐Guilbert F, Davy JM, Poirier JM & Weissenburger J (1995). Mexiletine antagonizes effects of sotalol on QT interval duration and its proarrhythmic effects in a canine model of torsade de pointes. J Am Coll Cardiol 26, 787–792. [DOI] [PubMed] [Google Scholar]
- Collins FS & Varmus H (2015). A new initiative on precision medicine. N Engl J Med 372, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper J, Scharm M & Mirams GR (2016). The Cardiac Electrophysiology Web Lab. Biophys J 110, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier A, Augustin CM, Neic A, Prassl AJ, Holler M, Fastl TE, Hennemuth A, Bredies K, Kuehne T, Bishop MJ, Niederer SA & Plank G (2015). Image‐based personalization of cardiac anatomy for coupled electromechanical modeling. Ann Biomed Eng 44, 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins MA, Dalal PJ, Bugana M, Severi S & Sobie EA (2014). Comprehensive analyses of ventricular myocyte models identify targets exhibiting favorable rate dependence. PLoS Comput Biol 10, e1003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vreede‐Swagemakers JJ, Gorgels AP, Dubois‐Arbouw WI, van Ree JW, Daemen MJ, Houben LG & Wellens HJ (1997). Out‐of‐hospital cardiac arrest in the 1990’s: a population‐based study in the Maastricht area on incidence, characteristics and survival. J Am Col Cardiol 30, 1500–1505. [DOI] [PubMed] [Google Scholar]
- Deng D, Arevalo H, Pashakhanloo F, Prakosa A, Ashikaga H, McVeigh E, Halperin H & Trayanova N (2015). Accuracy of prediction of infarct‐related arrhythmic circuits from image‐based models reconstructed from low and high resolution MRI. Front Physiol 6, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo R & Albert CM (2012). Epidemiology and genetics of sudden cardiac death. Circulation 125, 620–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Veroli GY, Davies MR, Zhang H, Abi‐Gerges N & Boyett MR (2013. a). hERG inhibitors with similar potency but different binding kinetics do not pose the same proarrhythmic risk: implications for drug safety assessment. J Cardiovasc Electrophysiol 25, 197–207. [DOI] [PubMed] [Google Scholar]
- Di Veroli GY, Davies MR, Zhang H, Abi‐Gerges N & Boyett MR (2013. b). High‐throughput screening of drug‐binding dynamics to HERG improves early drug safety assessment. Am J Physiol Heart Circ Physiol 304, H104–H117. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D & Noble D (1985). A model of cardiac electrical activity incorporating ionic pumps and concentration changes. Philos Trans R Soc Lond B Biol Sci 307, 353–398. [DOI] [PubMed] [Google Scholar]
- Doherty JU, Kienzle MG & Waxman HL (1983). Programmed ventricular stimulation at a second right ventricular site: an analysis of 100 patients, with special reference to sensitivity, specificity and characteristics of patients with induced ventricular tachycardia. Am J Cardiol 52, 1184–1189. [DOI] [PubMed] [Google Scholar]
- Dokos S & Lovell NH (2004). Parameter estimation in cardiac ionic models. Prog Biophys Mol Biol 85, 407–431. [DOI] [PubMed] [Google Scholar]
- Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias‐Manno D, Barker AH, Arensberg D, Baker A, Friedman L & Greene HL (1991). Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 324, 781–788. [DOI] [PubMed] [Google Scholar]
- Eng G, Lee BW, Protas L, Gagliardi M, Brown K, Kass RS, Keller G, Robinson RB & Vunjak‐Novakovic G (2016). Autonomous beating rate adaptation in human stem cell‐based cardiomyocytes. Nat Commun 7, 10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman ZJ, Trew ML & Smaill BH (2010). Structural heterogeneity alone is a sufficient substrate for dynamic instability and altered restitution. Circ Arrhythm Electrophysiol 3, 195–203. [DOI] [PubMed] [Google Scholar]
- Fermini B Hancox JC, Abi‐Gerges N, Bridgland‐Taylor M, Chaudhary KW, Colatsky T, Correll K, Crumb W, Damiano B, Erdemli G, Gintant G, Imredy J, Koerner J, Kramer J, Levesque P, Li Z, Lindqvist A, Obejero‐Paz CA, Rampe D, Sawada K, Strauss DG, Vandenberg JI (2016). A new perspective in the field of cardiac safety testing through the comprehensive in vitro proarrhythmia assay paradigm. J Biomol Screen 21, 1–11. [DOI] [PubMed] [Google Scholar]
- Fink M, Niederer SA, Cherry EM, Fenton FH, Koivumäki JT, Seemann G, Thul R, Zhang H, Sachse FB, Beard D, Crampin EJ & Smith NP (2011). Cardiac cell modelling: observations from the heart of the cardiac physiome project. Prog Biophys Mol Biol 104, 2–21. [DOI] [PubMed] [Google Scholar]
- Fink M & Noble D (2009). Markov models for ion channels: versatility versus identifiability and speed. Philos Transact A Math Phys Eng Sci 367, 2161–2179. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration, HHS (2005. a). International Conference on Harmonisation; guidance on S7B nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals; availability. Notice. Fed Regist 70, 61133–61134. [PubMed] [Google Scholar]
- Food and Drug Administration, HHS (2005. b). International Conference on Harmonisation; guidance on E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs; availability. Notice. Fed Regist 70, 61134–61135. [PubMed] [Google Scholar]
- Frommeyer G & Eckardt L (2015). Drug‐induced proarrhythmia: risk factors and electrophysiological mechanisms. Nat Rev Cardiol 13, 36–37. [DOI] [PubMed] [Google Scholar]
- Gardner RT, Wang L, Lang BT, Cregg JM, Dunbar CL, Woodward WR, Silver J, Ripplinger CM & Habecker BA (2015). Targeting protein tyrosine phosphatase σ after myocardial infarction restores cardiac sympathetic innervation and prevents arrhythmias. Nat Commun 6, 6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur N & Rudy Y (2011). Multiscale modeling of calcium cycling in cardiac ventricular myocyte: macroscopic consequences of microscopic dyadic function. Biophys J 100, 2904–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AL (2013). Molecular and genetic basis of sudden cardiac death. J Clin Invest 123, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi E, Pasqualini FS & Bers DM (2010). A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol 48, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenendaal W, Ortega FA, Kherlopian AR, Zygmunt AC, Krogh‐Madsen T & Christini DJ (2015). Cell‐specific cardiac electrophysiology models. PLoS Comput Biol 11, e1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins ER, Goel P, Puglisi JL, Bers DM, Cannell M & Sneyd J (2007). Modelling calcium microdomains using homogenisation. J Theor Biol 247, 623–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AP, Perrin MJ, Heide J, Campbell TJ, Mann SA & Vandenberg JI (2014). Kinetics of drug interaction with the Kv11.1 potassium channel. Mol Pharmacol 85, 769–776. [DOI] [PubMed] [Google Scholar]
- Hondeghem L & Carlsson L (2001). Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation 103, 2004. [DOI] [PubMed] [Google Scholar]
- Hummel JD, Strickberger SA, Daoud E, Niebauer M, Bakr O, Man KC, Williamson BD & Morady F (1994). Results and efficiency of programmed ventricular stimulation with four extrastimuli compared with one, two, and three extrastimuli. Circulation 90, 2827–2832. [DOI] [PubMed] [Google Scholar]
- Hunter PJ & Borg TK (2003). Integration from proteins to organs: the Physiome Project. Nat Rev Mol Cell Biol 4, 237–243. [DOI] [PubMed] [Google Scholar]
- Hunter P, Robbins P & Noble D (2002). The IUPS human Physiome Project. Pflugers Arch 445, 1–9. [DOI] [PubMed] [Google Scholar]
- Iribe G, Kohl P & Noble D (2006). Modulatory effect of calmodulin‐dependent kinase II (CaMKII) on sarcoplasmic reticulum Ca2+ handling and interval‐force relations: a modelling study. Philos Transact A Math Phys Eng Sci 364, 1107–1133. [DOI] [PubMed] [Google Scholar]
- Jafri MS, Rice JJ & Winslow RL (1998). Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys J 74, 1149–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalife J (2000). Ventricular fibrillation: mechanisms of initiation and maintenance. Annu Rev Physiol 62, 25–50. [DOI] [PubMed] [Google Scholar]
- Jamshidi Y, Nolte IM, Dalageorgou C, Zheng D, Johnson T, Bastiaenen R, Ruddy S, Talbott D, Norris KJ, Sneider H, George AL, Marshall V, Shakir S, Kannankeril PJ, Munroe PB, Camm AJ, Jeffery S, Roden DM & Behr ER (2012). Common variation in the NOS1AP gene is associated with drug‐induced QT prolongation and ventricular arrhythmia. J Am Coll Cardiol 60, 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- January CT & Riddle JM (1989). Early afterdepolarizations: mechanism of induction and block. A role for L‐type Ca2+ current. Circ Res 64, 977–990. [DOI] [PubMed] [Google Scholar]
- Johannesen L, Vicente J, Mason JW, Sanabria C, Waite‐Labott K, Hong M, Guo P, Lin J, Sørensen JS, Galeotti L, Florian J, Ugander M, Stockbridge N & Strauss DG (2014). Differentiating drug‐induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin Pharmacol Ther 96, 549–558. [DOI] [PubMed] [Google Scholar]
- Johnstone RH, Chang ETY, Bardenet R, de Boer TP, Gavaghan DJ, Pathmanathan P, Clayton RH & Mirams GR (2015). Uncertainty and variability in models of the cardiac action potential: Can we build trustworthy models? J Mol Cell Cardiol (in press; DOI: 10.1016/j.yjmcc.2015.11.018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin A, Usher‐Smith J, Jones VJ, Huang CL‐H & Sabir IN (2010). Cardiac arrhythmia: a simple conceptual framework. Trends Cardiovasc Med 20, 103–107. [DOI] [PubMed] [Google Scholar]
- Kannankeril P, Roden DM & Darbar D (2010). Drug‐induced long QT syndrome. Pharmacol Rev 62, 760–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaźmierczak J, Peregud‐Pogorzelska M & Rzeuski R (2007). QT Interval prolongation and torsades de pointes due to a coadministration of ciprofloxacin and azimilide in a patient with implantable cardioverter‐defibrillator. Pacing Clin Electrophysiol 30, 1043–1046. [DOI] [PubMed] [Google Scholar]
- Keating MT & Sanguinetti MC (2001). Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104, 569–580. [DOI] [PubMed] [Google Scholar]
- Kim C, Majdi M, Xia P, Wei KA, Talantova M, Spiering S, Nelson B, Mercola M & Chen H‐SV (2010). Non‐cardiomyocytes influence the electrophysiological maturation of human embryonic stem cell‐based cardiomyocytes during differentiation. Stem Cells Dev 19, 783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounas SP, Letsas KP, Sideris A, Efraimidis M & Kardaras F (2005). QT interval prolongation and torsades de pointes due to a coadministration of metronidazole and amiodarone. Pacing Clin Electrophysiol 28, 472–473. [DOI] [PubMed] [Google Scholar]
- Kramer J, Obejero‐Paz CA, Myatt G, Kuryshev YA, Bruening‐Wright A, Verducci JS & Brown AM (2013). MICE models: superior to the HERG model in predicting Torsade de Pointes. Sci Rep 3, 2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme MA & Murry CE (2011). Heart regeneration. Nature 473, 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Mann SA, Windley MJ, Imtiaz MS, Vandenberg JI & Hill AP (2015). In silico assessment of kinetics and state dependent binding properties of drugs causing acquired LQTS. Prog Biophys Mol Biol 120, 89–99. [DOI] [PubMed] [Google Scholar]
- Li W, Knowlton D, Van Winkle DM & Habecker BA (2004). Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Heart Circ Physiol 286, H2229–H2236. [DOI] [PubMed] [Google Scholar]
- Lloyd CM, Lawson JR, Hunter PJ & Nielsen PF (2008). The CellML Model Repository. Bioinformatics 24, 2122–2123. [DOI] [PubMed] [Google Scholar]
- Lu Y, Mahaut‐Smith MP, Varghese A, Huang CL, Kemp PR & Vandenberg JI (2001). Effects of premature stimulation on HERG K+ channels. J Physiol 537, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo CH & Rudy Y (1994). A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res 74, 1071–1096. [DOI] [PubMed] [Google Scholar]
- McDowell KS, Zahid S, Vadakkumpadan F, Blauer J, MacLeod RS & Trayanova NA (2015). Virtual electrophysiological study of atrial fibrillation in fibrotic remodeling. PLoS One 10, e0117110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihic A, Li J, Miyagi Y, Gagliardi M, Li S‐H, Zu J, Weisel RD, Keller G & Li R‐K (2014). The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell‐based cardiomyocytes. Biomaterials 35, 2798–2808. [DOI] [PubMed] [Google Scholar]
- Milnes JT, Witchel HJ, Leaney JL, Leishman DJ & Hancox JC (2010). Investigating dynamic protocol‐dependence of hERG potassium channel inhibition at 37 degrees C: Cisapride versus dofetilide. J Pharmacol Toxicol Methods 61, 178–191. [DOI] [PubMed] [Google Scholar]
- Mines GR (1914). On circulating excitations in heart muscles and their possible relation to tachycardia and fibrillation. Transactions of the Royal Society of Canada 8, 43–52. [Google Scholar]
- Mirams GR, Cui Y, Sher A, Fink M, Cooper J, Heath BM, McMahon NC, Gavaghan DJ & Noble D (2011). Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovasc Res 91, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirams GR, Davies MR, Brough SJ, Bridgland‐Taylor MH, Cui Y, Gavaghan DJ & Abi‐Gerges N (2014). Prediction of thorough QT study results using action potential simulations based on ion channel screens. J Pharmacol Toxicol Methods 70, 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirams GR, Pathmanathan P, Gray RA, Challenor P & Clayton RH. (2016). Uncertainty and variability in computational and mathematical models of cardiac physiology. J Physiol 594, 6833–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J & Sanguinetti MC (2000). Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J Gen Physiol 115, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JD, Zhu ZI, Yang P‐C, Bankston JR, Jeng M‐T, Kang C, Wang L, Bayer JD, Christini DJ, Trayanova NA, Ripplinger CM, Kass RS & Clancy CE (2011). A computational model to predict the effects of class I anti‐arrhythmic drugs on ventricular rhythms. Sci Transl Med 3, 98ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott‐Flügel L, Dorn T, Goedel A, Höhnke C, Hofmann F, Seyfarth M, Sinnecker D, Schömig A & Laugwitz K‐L (2010). Patient‐specific induced pluripotent stem‐cell models for long‐QT syndrome. N Engl J Med 363, 1397–1409. [DOI] [PubMed] [Google Scholar]
- Nagahara D, Nakata T, Hashimoto A, Wakabayashi T, Kyuma M, Noda R, Shimoshige S, Uno K, Tsuchihashi K & Shimamoto K (2008). Predicting the need for an implantable cardioverter defibrillator using cardiac metaiodobenzylguanidine activity together with plasma natriuretic peptide concentration or left ventricular function. J Nucl Med 49, 225–233. [DOI] [PubMed] [Google Scholar]
- Narsinh K, Narsinh KH & Wu JC (2011). Derivation of human induced pluripotent stem cells for cardiovascular disease modeling. Circ Res 108, 1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattel S, Antzelevitch C & Noble D (2011). Resolving the M‐cell debate: why and how. Heart Rhythm 8, 1293–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivala M, de Lange E, Rovetti R & Qu Z (2012). Computational modeling and numerical methods for spatiotemporal calcium cycling in ventricular myocytes. Front Physiol 3, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivala M, Song Z, Weiss JN & Qu Z (2015). T‐tubule disruption promotes calcium alternans in failing ventricular myocytes: mechanistic insights from computational modeling. J Mol Cell Cardiol 79, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D (1962). A modification of the Hodgkin–Huxley equations applicable to Purkinje fibre action and pace‐maker potentials. J Physio 160, 317–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D, Varghese A, Kohl P & Noble P (1998). Improved guinea‐pig ventricular cell model incorporating a diadic space, IKr and IKs, and length‐ and tension‐dependent processes. Can J Cardiol 14, 123–134. [PubMed] [Google Scholar]
- Nunes SS, Miklas JW, Liu J, Aschar‐Sobbi R, Xiao Y, Zhang B, Jiang J, S Massé, Gagliardi M, Hsieh A, Thavandiran N, Laflamme MA, Nanthakumar K, Gross GJ, Backx PH, Keller G & Radisic M (2013). Biowire: a platform for maturation of human pluripotent stem cell‐based cardiomyocytes. Nat Methods 10, 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'hara T, Virág L, Varro A & Rudy Y (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7, e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olde Nordkamp LRA, Postema PG, Knops RE, van Dijk N, Limpens J, Wilde AAM & de Groot JR (2015). Implantable cardioverter‐defibrillator harm in young patients with inherited arrhythmia syndromes: A systematic review and meta‐analysis of inappropriate shocks and complications. Heart Rhythm 13, 443–454. [DOI] [PubMed] [Google Scholar]
- Otsuji TG, Minami I, Kurose Y, Yamauchi K, Tada M & Nakatsuji N (2010). Progressive maturation in contracting cardiomyocytes derived from human embryonic stem cells: Qualitative effects on electrophysiological responses to drugs. Stem Cell Res 4, 201–213. [DOI] [PubMed] [Google Scholar]
- Page A, Aktas MK, Soyata T, Zareba W & Couderc J‐P (2015). “QT clock” to improve detection of QT prolongation in long QT syndrome patients. Heart Rhythm 13, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin MJ, Kuchel PW, Campbell TJ & Vandenberg JI (2008. a). Drug binding to the inactivated state is necessary but not sufficient for high‐affinity binding to human ether‐à‐go‐go‐related gene channels. Mol Pharmacol 74, 1443–1452. [DOI] [PubMed] [Google Scholar]
- Perrin MJ, Subbiah RN, Vandenberg JI & Hill AP (2008. b). Human ether‐a‐go‐go related gene (hERG) K+ channels: Function and dysfunction. Prog Biophys Mol Biol 1–12. [DOI] [PubMed] [Google Scholar]
- Piper DR, Varghese A, Sanguinetti MC & Tristani‐Firouzi M (2003). Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc Natl Acad Sci USA 100, 10534–10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z & Weiss JN (2015). Mechanisms of ventricular arrhythmias: from molecular fluctuations to electrical turbulence. Annu Rev Physiol 77, 29–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao C, Prodromakis T, Kolker L, Chaudhry UAR, Trantidou T, Sridhar A, Weekes C, Camelliti P, Harding SE, Darzi A, Yacoub MH, Athanasiou T & Terracciano CM (2013). The effect of microgrooved culture substrates on calcium cycling of cardiac myocytes derived from human induced pluripotent stem cells. Biomaterials 34, 2399–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PKS, Strang I, Sullivan AT, Wallis R, Camm AJ & Hammond TG (2003). Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 58, 32–45. [DOI] [PubMed] [Google Scholar]
- Restrepo JG, Weiss JN & Karma A (2008). Calsequestrin‐mediated mechanism for cellular calcium transient alternans. Biophys J 95, 3767–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM (2006). Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med 259, 59–69. [DOI] [PubMed] [Google Scholar]
- Roden DM & Viswanathan PC (2005). Genetics of acquired long QT syndrome. J Clin Invest 115, 2025–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero L, Trenor B, Yang P‐C, Saiz J & Clancy CE (2014). In silico screening of the impact of hERG channel kinetic abnormalities on channel block and susceptibility to acquired long QT syndrome. J Mol Cell Cardiol 72C, 126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenqvist M, Beyer T, Block M, Dulk den K, Minten J & Lindemans F (1998). Adverse events with transvenous implantable cardioverter‐defibrillators: a prospective multicenter study. European 7219 Jewel ICD investigators. Circulation 98, 663–670. [DOI] [PubMed] [Google Scholar]
- Rovetti R, Cui X, Garfinkel A, Weiss JN & Qu Z (2010). Spark‐induced sparks as a mechanism of intracellular calcium alternans in cardiac myocytes. Circ Res 106, 1582–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford SL, Trew ML, Sands GB, LeGrice I & Smaill BH (2012). High‐resolution 3‐dimensional reconstruction of the infarct border zone: impact of structural remodeling on electrical activation. Circ Res 111, 301–311. [DOI] [PubMed] [Google Scholar]
- Sadrieh A, Domanski L, Pitt‐Francis J, Mann SA, Hodkinson EC, Ng CA, Perry MD, Taylor JA, Gavaghan D, Subbiah RN, Vandenberg JI & Hill AP (2014). Multiscale cardiac modelling reveals the origins of notched T waves in long QT syndrome type 2. Nat Commun 5, 5069. [DOI] [PubMed] [Google Scholar]
- Sager PT, Gintant G, Turner JR, Pettit S & Stockbridge N (2014). Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J 167, 292–300. [DOI] [PubMed] [Google Scholar]
- Saini H, Navaei A, Van Putten A & Nikkhah M (2015). 3D cardiac microtissues encapsulated with the co‐culture of cardiomyocytes and cardiac fibroblasts. Adv Healthc Mater 4, 1961–1971. [DOI] [PubMed] [Google Scholar]
- Sarkar AX & Sobie EA (2011). Quantification of repolarization reserve to understand interpatient variability in the response to proarrhythmic drugs: A computational analysis. Heart Rhythm 8, 1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schendel T & Falcke M (2010). Efficient and detailed model of the local Ca2+ release unit in the ventricular cardiac myocyte. Genome Inform 22, 142–155. [PubMed] [Google Scholar]
- Schendel T, Thul R, Sneyd J & Falcke M (2011). How does the ryanodine receptor in the ventricular myocyte wake up: by a single or by multiple open L‐type Ca2+ channels? Eur Biophys J 41, 27–39. [DOI] [PubMed] [Google Scholar]
- Schimpf R, Wolpert C, Bianchi F, Giustetto C, Gaita F, Bauersfeld U & Borggrefe M (2003). Congenital short QT syndrome and implantable cardioverter defibrillator treatment:. J Cardiovasc Electrophysiol 14, 1273–1277. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Wang F, Puglisi J, Weber C & Bers DM (2004). A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J 87, 3351–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Zoble RG, Yellen L, Brodsky MA, Feld GK, Berk M & Billing CB (2000). Efficacy and safety of oral dofetilide in converting to and maintaining sinus rhythm in patients with chronic atrial fibrillation or atrial flutter: the symptomatic atrial fibrillation investigative research on dofetilide (SAFIRE‐D) study. Circulation 102, 2385–2390. [DOI] [PubMed] [Google Scholar]
- Sinnecker D, Laugwitz K‐L & Moretti A (2014). Induced pluripotent stem cell‐based cardiomyocytes for drug development and toxicity testing. Pharmacol Ther 143, 246–252. [DOI] [PubMed] [Google Scholar]
- Stern MD, Ríos E & Maltsev VA (2013). Life and death of a cardiac calcium spark. J Gen Physiol 142, 257–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson WG & Epstein LM (2003). Predicting sudden death risk for heart failure patients in the implantable cardioverter‐defibrillator age. Circulation 107, 514–516. [DOI] [PubMed] [Google Scholar]
- Suessbrich H, Schönherr R, Heinemann SH, Attali B, Lang F & Busch AE (1997). The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in Xenopus oocytes. Br J Pharmacol 120, 968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugrue A, Kremen V, Qiang B, Sheldon SH, DeSimone CV, Sapir Y, Striemer BL, Brady P, Asirvatham SJ, Ackerman MJ, Friedman P & Noseworthy PA (2015). Electrocardiographic predictors of torsadogenic risk during dofetilide or sotalol initiation: utility of a novel T wave analysis program. Cardiovasc Drugs Ther 29, 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K & Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Zlochiver S, Vikstrom KL, Yamazaki M, Moreno J, Klos M, Zaitsev AV, Vaidyanathan R, Auerbach DS, Landas S, Guiraudon G, Jalife J, Berenfeld O & Kalifa J (2007). Spatial distribution of fibrosis governs fibrillation wave dynamics in the posterior left atrium during heart failure. Circ Res 101, 839–847. [DOI] [PubMed] [Google Scholar]
- The Cardiac Arrhythmia Suppression Trial (CAST) Investigators (1989). Preliminary Report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med 321, 406–412. [DOI] [PubMed] [Google Scholar]
- Torp‐Pedersen C, Møller M, Bloch‐Thomsen PE, Køber L, Sandøe E, Egstrup K, Agner E, Carlsen J, Videbaek J, Marchant B & Camm AJ (1999). Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med 341, 857–865. [DOI] [PubMed] [Google Scholar]
- Tuan H‐TM, Williams GSB, Chikando AC, Sobie EA, Lederer WJ & Jafri MS (2011). Stochastic simulation of cardiac ventricular myocyte calcium dynamics and waves. Conf Proc IEEE Eng Med Biol Soc 2011, 4677–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusscher Ten KHWJ, Hren R & Panfilov AV (2007). Organization of ventricular fibrillation in the human heart. Circ Res 100, e87–e101. [DOI] [PubMed] [Google Scholar]
- Ukwatta E, Arevalo H, Rajchl M, White J, Pashakhanloo F, Prakosa A, Herzka DA, McVeigh E, Lardo AC, Trayanova NA & Vadakkumpadan F (2015). Image‐based reconstruction of three‐dimensional myocardial infarct geometry for patient‐specific modeling of cardiac electrophysiology. Med Phys 42, 4579–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaglio M, Couderc J‐P, McNitt S, Xia X, Moss AJ & Zareba W (2008). A quantitative assessment of T‐wave morphology in LQT1, LQT2, and healthy individuals based on Holter recording technology. Heart Rhythm 5, 11–18. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y & Hill AP (2012). hERG K+ channels: structure, function, and clinical significance. Physiol Rev 92, 1393–1478. [DOI] [PubMed] [Google Scholar]
- Vicente J, Johannesen L, Mason JW, Crumb WJ, Pueyo E, Stockbridge N & Strauss DG (2015). Comprehensive T wave morphology assessment in a randomized clinical study of dofetilide, quinidine, ranolazine, and verapamil. J Am Heart Assoc 4, e001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierheller J, Neubert W, Falcke M, Gilbert SH & Chamakuri N (2015). A multiscale computational model of spatially resolved calcium cycling in cardiac myocytes: from detailed cleft dynamics to the whole cell concentration profiles. Front Physiol 6, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BD, Singleton CB, Bursill JA, Wyse KR, Valenzuela SM, Qiu MR, Breit SN & Campbell TJ (1999). Inhibition of the human ether‐a‐go‐go‐related gene (HERG) potassium channel by cisapride: affinity for open and inactivated states. Br J Pharmacol 128, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallman M, Smith NP & Rodríguez B (2014). Computational methods to reduce uncertainty in the estimation of cardiac conduction properties from electroanatomical recordings. Med Image Anal 18, 228–240. [DOI] [PubMed] [Google Scholar]
- Weeke P, Mosley JD, Hanna D, Delaney JT, Shaffer C, Wells QS, Van Driest S, Karnes JH, Ingram C, Guo Y, Shyr Y, Norris K, Kannankeril PJ, Ramirez AH, Smith JD, Mardis ER, Nickerson D, George AL & Roden DM (2014). Exome sequencing implicates an increased burden of rare potassium channel variants in the risk of drug‐induced long QT interval syndrome. J Am Coll Cardiol 63, 1430–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapura M, Hébert TE & Nattel S (2002). Dofetilide block involves interactions with open and inactivated states of HERG channels. Pflugers Arch 443, 520–531. [DOI] [PubMed] [Google Scholar]
- Wood AJ & Roden DM (2004). Drug‐induced prolongation of the QT interval. N Engl J Med 350, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Yang P‐C & Clancy CE (2012). In silico prediction of sex‐based differences in human susceptibility to cardiac ventricular tachyarrhythmias. Front Physiol; 3, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltser D, Justo D, Halkin A, Prokhorov V & Heller K (2003). Torsade de pointes due to noncardiac drugs: most patients have easily identifiable risk factors. Medicine 82, 282–290. [DOI] [PubMed] [Google Scholar]
- Zemzemi N, Bernabeu MO, Saiz J, Cooper J, Pathmanathan P, Mirams GR, Pitt‐Francis J & Rodríguez B (2013). Computational assessment of drug‐induced effects on the electrocardiogram: from ion channel to body surface potentials. Br J Pharmacol 168, 718–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Butters TD, Zhang H, LeGrice I, Sands GB & Smaill BH (2013). Image‐based model of atrial anatomy and electrical activation: a computational platform for investigating atrial arrhythmia. IEEE Trans Med Imaging 32, 18–27. [DOI] [PubMed] [Google Scholar]