

The RUNX family of genes are well established as transcription factors essential for differentiation and development in higher organisms. The RUNX family is profoundly implicated in cancer: dysregulation of RUNX genes has been linked to initiation as well progression of diverse cancer types.1 RUNX proteins derived their name from the Runt domain, an evolutionarily conserved 128-amino acid region, which endows RUNX proteins with sequence-specific DNA binding. Interestingly, unlike many transcription factors in mitosis, RUNX proteins are found on key mitotic structures such as the centrosome, spindle and midbody2 – raising the intriguing possibility that RUNX proteins play active roles in mitosis, a cell cycle phase traditionally associated with major cessation of transcription.
One of the most obvious changes to the RUNX proteins during mitosis is the massive phosphorylation of all 3 members of the human RUNX family. And yet, the underlying reason remains obscure. Recently, we found that 2 key residues at the N-terminus domain of RUNX3 – threonine 14 (T14) and threonine 173 (T173) – are critical for mitotic-specific hyperphosphorylation of RUNX3.3 The crystal structure of the Runt domain showed that the peptide comprising T173 and its flanking amino acid residues constitutes a key structure necessary for the Runt-DNA interaction; moreover, T173 contacts the phosphate backbone of the DNA double helix via polar interaction.4 Mitosis-specific phosphorylation of T173 regulates RUNX3 subcellular localization through inhibition of the DNA-binding function of the Runt domain – RUNX proteins are detached from the DNA and redistributed to the cytoplasm and mitotic structures such as the centrosome and midbody, which are necessary for spindle formation and cytokinesis, respectively. Aurora kinases are master regulators of mitosis with similar localization patterns as RUNX3. The identification of RUNX3 as a novel substrate of Aurora kinases, which induces T173 phosphorylation, therefore suggests non-canonical roles for RUNX3 at specific stages of mitosis. We showed that RUNX3 knockdown results in delayed mitotic entry while RUNX3 phosphorylation is a likely regulatory element for mitotic entry.
The strong conservation of T173 and its flanking residues in divergent organisms, from the unicellular holozoan Capsaspora owczarzaki to human, indicates that T173 phosphorylation evolved to regulate the primordial role of RUNX and perhaps, tailor it to mitosis. Mutation of the T173 residue and equivalents – all to isoleucine – were discovered in all RUNX family members in human diseases. In particular, RUNX3 (T173I) and RUNX1 (equivalent, T196I) mutations were cancer associated (http://cancer.sanger.ac.uk). Unlike wild-type RUNX3, T173 mutants promoted colony formation on soft agar, suggesting that mutation or phosphorylation of T173 is associated with loss of growth inhibition. Our work suggests that T173 phosphorylation normally acts to regulate mitosis but with the accompanying caveat: the risk of cancer formation if T173 phosphorylation is not tightly confined to mitosis. Since the tumor suppressor property of RUNX3 mainly resides in its ability to regulate transcription of genes that induce cell cycle arrest, apoptosis and senescence,1 the frequent upregulation of Aurora kinases in cancer5 could reasonably be expected to attenuate RUNX3 transcription activity, and therefore its tumor suppressor function, during non-mitotic phases.
We had earlier reported an acetylation site at lysine 171 (K171), which is important for complex formation between RUNX3 and bromodomain-containing protein BRD2 in a K-Ras- dependent manner. 6 The RUNX3-BRD2 complex mediates transcription of growth inhibitory and tumor suppressor genes p21WAF1(CIP1 and p14ARF respectively to protect cells from oncogenic signals.6 Aurora kinase B preferentially phosphorylates threonine/serine residues with upstream basic residues. K171, at the –2 position, conforms to an Aurora kinase B target motif (Fig. 1). Acetylation of K171 might therefore interfere with phosphorylation of T173. It is conceivable that the conservation of the phospho-site T173 and acetyl-site K171 is due to selective pressure for cross-check between modifications – as a safeguard against tumorigenesis.
Figure 1.
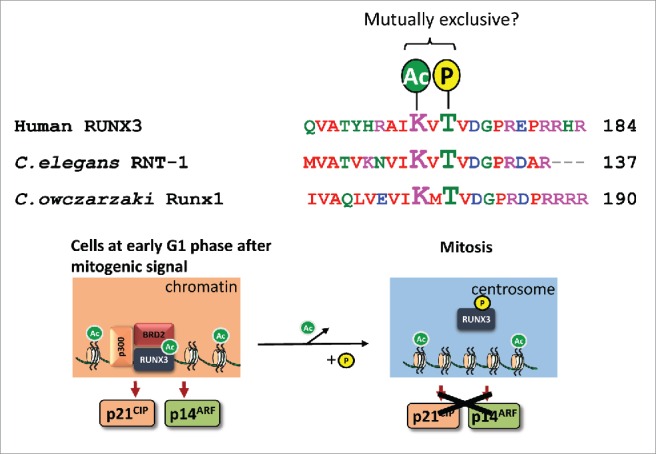
Proposed model for crosstalk between post-translational modifications of the highly conserved T173 and K171 residues Top, alignment of human RUNX3 with RUNX proteins from Caenorhabditis elegans and Capsaspora owczarzaki. Bottom, upon mitogenic stimulation, p300 mediates acetylation of RUNX3; acetylated RUNX3 binds to BRD2 to activate p21WAF1(CIP and p14ARF transcription during early stages of G1 phase – this is a key cellular mechanism to safeguard against persistent mitogenic signals6. As the cell cycle progresses, we propose that deacetylation of K171 results in cessation of p21WAF1(CIP and p14ARF transcription in S phase, and permits T173 phosphorylation during G2/M transition – the phosphorylated RUNX3 is released from DNA and subsequently localizes to the centrosome to license mitotic entry.
Finally, our work reinforced the exciting notion that all RUNX proteins possess non-transcriptional roles. We propose that RUNX proteins have transcriptional and non-transcriptional roles that function in a complementary manner to maintain cell identity. Earlier, we reported that RUNX1 and RUNX3 are integral components of the Fanconi anemia DNA repair pathway.7 Both RUNX proteins, independent of their transcriptional regulation roles, recruit FANCI/FANCD2 to damaged sites for effective repair. Moreover, Runx3-deficient mice showed accelerated gastric tumor development when challenged with mutagen.1 Taken together with our findings on RUNX in mitosis, we hypothesize that RUNX proteins ensure cellular identity through 2 main mechanisms: (1) maintain cell phenotype through its transcriptional programs; (2) safeguard genome integrity through its non-transcriptional regulation of mitosis and DNA repair. Conceptually, the transcriptional and non-transcriptional roles of RUNX proteins are mutually exclusive functions that represent 2 sides of the same coin. It allows for coordinated and sequential actions of RUNX when responding to oncogenic cues. It also explains why tight regulation of RUNX dosage is important for normal cell growth and why RUNX haploinsufficiency and overexpression are frequently associated with cancer development.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Ito Y, Bae S-C, Chuang LSH. The RUNX family: developmental regulators in cancer.. Nat Rev Cancer 2015; 15:81-95; PMID:25592647; http://dx.doi.org/ 10.1038/nrc3877 [DOI] [PubMed] [Google Scholar]
- [2].Chuang LSH, Lai SK, Murata-Hori M, Yamada A, Li H-Y, Gunaratne J, Ito Y. RUNX3 interactome reveals novel centrosomal targeting of RUNX family of transcription factors.. Cell Cycle 2012; 11:1938-47; PMID:22544322; http://dx.doi.org/ 10.4161/cc.20278 [DOI] [PubMed] [Google Scholar]
- [3].Chuang LSH, Khor JM, Lai SK, Garg S, Krishnan V, Koh C-G, Lee SH, Ito Y. Aurora kinase-induced phosphorylation excludes transcription factor RUNX from the chromatin to facilitate proper mitotic progression.. Proc Natl Acad Sci U S A 2016; 113:6490-5; PMID:27217562; http://dx.doi.org/ 10.1073/pnas.1523157113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bravo J, Li Z, Speck NA, Warren AJ. The leukemia-associated AML1 (Runx1)–CBFβ complex functions as a DNA-induced molecular clamp.. Nat Struct Biol 2001; 8:371-8; PMID:11276260; http://dx.doi.org/ 10.1038/86264 [DOI] [PubMed] [Google Scholar]
- [5].Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma . Nature 2014; 513:202-9; PMID:25079317; http://dx.doi.org/ 10.1038/nature13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee Y-S, Lee J-W, Jang J-W, Chi X-Z, Kim J-H, Li Y-H, Kim M-K, Kim D-M, Choi B-S, Kim E-G, et al. Runx3 inactivation is a crucial early event in the development of lung Adenocarcinoma.. Cancer Cell 2013; 24:603-16; PMID:24229708; http://dx.doi.org/ 10.1016/j.ccr.2013.10.003 [DOI] [PubMed] [Google Scholar]
- [7].Wang CQ, Krishnan V, Tay LS, Chin DWL, Koh CP, Chooi JY, Nah GSS, Du L, Jacob B, Yamashita N, et al. Disruption of Runx1 and Runx3 leads to bone marrow failure and Leukemia predisposition due to transcriptional and DNA repair defects.. Cell Rep 2014; 8:767-82; PMID:25066130; http://dx.doi.org/ 10.1016/j.celrep.2014.06.046 [DOI] [PubMed] [Google Scholar]