

The discovery of DNA damage-tolerant and error-prone Y-family Trans-Lesion Synthesis (TLS) polymerases in eukaryotes almost 2 decades ago provided a molecular basis for mutagenesis and chemical carcinogenesis. A recent report identifies a new biochemical mechanism by which many cancer cells aberrantly activate TLS. Pathological activation of TLS represents a new way in which neoplastic cells might acquire some of the “emerging hallmarks and enabling characteristics” of cancer.
Collectively the 4 Y-family TLS DNA polymerases Polη, Polκ, Polι and REV1 enable replicative bypass of diverse DNA lesions and confer viability in the face of genotoxic exposures.1 However, TLS is error-prone and can cause mutations. A link between TLS and cancer was firmly established by the seminal finding that Polη (which performs efficient and error-free bypass of UV-induced cyclobutane pyrimidine dimers or CPD), is functionally inactivated in skin cancer-prone xeroderma pigmentosum-Variant (XPV) patients.2 The hypermutability of UV-irradiated XPV cells is due to compensatory and error-prone TLS of CPD by the “wrong” DNA polymerases when Polη is absent. However, with the exception of XPV, TLS polymerases are not generally known to be dysfunctional in cancer, and the extent to which TLS polymerase imbalance contributes to the mutational landscape of tumor cells is unknown.
Gao et al. recently identified a new molecular mechanism by which an apical component of the TLS pathway, the E3 ubiquitin ligase RAD18, is inappropriately activated in cancer cells.3 In response to replication fork stalling RAD18 mono-ubiquitinates PCNA. Y-family DNA polymerases preferentially associate with PCNA in its ubiquitinated form. Thus RAD18 triggers a DNA damage-tolerant mode of synthesis that averts replication fork collapse and prevents Double Strand Break (DSB) formation (Fig. 1).4 However, owing to its error-propensity, TLS carries the risk of mutagenesis and must be used sparingly. Slight increases in RAD18 expression can activate TLS, even in the absence of DNA damage. Therefore, restrained use of RAD18 is important to prevent mutagenesis.
Figure 1.
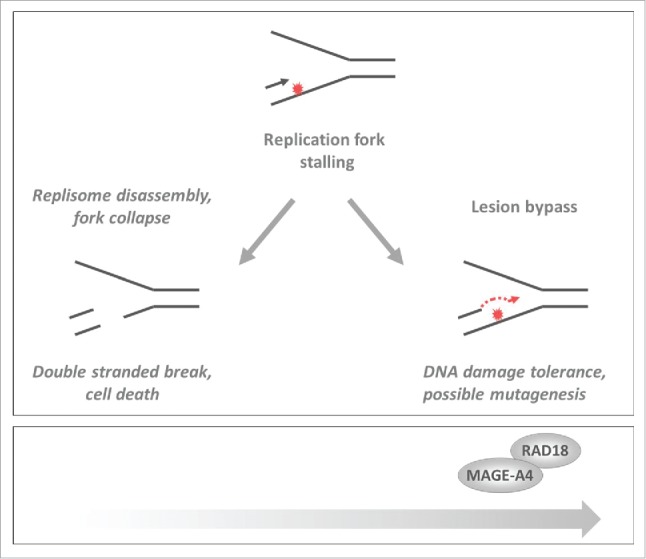
Effect of MAGE-A4 on responses to DNA damage and replication stress. In cancer cells MAGE-A4-RAD18 promotes TLS thereby averting replication fork collapse and conferring DNA damage tolerance while increasing the risk of mutagenesis. The black and red arrows (and) indicate leading strand 5′–3′ DNA synthesis and the red “explosion” indicates a fork-stalling DNA lesion.
Gao and colleagues identified Melanoma Antigen-A4 (MAGE-A4) as a major binding partner and stabilizer of RAD18 in lung adenocarcinoma cells. MAGE-A4 is a “Cancer/Testes Antigen” (CTA) that is ordinarily germline-restricted and absent from normal somatic cells but aberrantly overexpressed in many tumors.5 Owing to their tumor-specific expression, CTAs have been considered as targets for cancer immune therapy. Recently there have been tantalizing hints that CTAs might play active roles in carcinogenesis. In a landmark study, Potts identified several of the ∼45 MAGE-family proteins as activating binding partners of specific E3 RING ubiquitin ligases.6 Therefore, MAGEs may reprogram ubiquitin signaling networks in cancer cells. It is important to define the full repertoire of MAGE-E3 ligase complexes, identify their effector pathways and test their roles in cancer.
In contrast with other known MAGE/E3 ligase complexes, MAGE-A4 is not an allosteric activator of RAD18 catalytic activity. Instead, MAGE-A4 protects RAD18 from ubiquitin-mediated degradation. However, a conserved di-lysine motif that mediates interactions of other MAGEs with their E3 ligase partners is also necessary for MAGE-A4-dependent RAD18 stability. Remarkably, some cancer cell lines have developed reliance upon MAGE-A4 for maintaining RAD18 levels and sustaining DNA damage-tolerant DNA synthesis.
The finding that RAD18 regulation is fundamentally different between normal and cancer cells challenges the general assumption that TLS is merely a “housekeeping” mechanism in all cells. Our understanding of RAD18 signaling stems largely from studies conducted with highly transformed cancer cell lines in which the TLS pathway may already be “re-wired”. It will be interesting to determine whether additional mechanisms serve to activate TLS in cancer cells.
The demonstration that cancer cells “hijack” MAGE-A4 to reprogram TLS clearly suggests a new way for tumors to achieve both DNA damage tolerance and mutability. However many questions remain. For example the mechanism by which MAGE-A4 is upregulated during carcinogenesis is unknown. It is important to test the prediction that MAGE-A4-dependent TLS confers a selective advantage that favors carcinogenesis. Neoplastic cells often exist in unfavorable environments and experience DNA replication stress from metabolic, onocgenic and pharmaceutical sources. It is possible that MAGE-A4-RAD18 facilitates tolerance of stresses experienced during tumorigenesis.
Mutagenesis occurs when TLS polymerases inaccurately replicate undamaged DNA or DNA templates harboring non-cognate lesions. The four Y-family DNA polymerases are differentially dependent on PCNA-ubiquitination for recruitment to replication forks.7 MAGE-A4-induced RAD18 expression and PCNA mono-ubiquitination might over-ride normal constraints over TLS polymerase activity. Therefore, it will be interesting to determine how MAGE-A4-RAD18 influences the fidelity of different Y-family polymerases when replicating undamaged DNA, and templates containing cognate or non-cognate lesions. RAD18 activates several additional genome maintenance mechanisms including the Fanconi Anemia pathway and Homologous Recombination.4 Therefore, MAGE-A4 has potential to promote repair of diverse DNA lesions that are relevant to cancer etiology and therapy.
It is tempting to speculate that CTA-induced genome maintenance will emerge as a broad new paradigm for pathological DNA damage tolerance and genome instability. Neoplastic cells depend heavily on DNA damage tolerance and mutagenesis to survive, adapt, and resist therapy. These dependencies on DNA damage tolerance and mutagenesis are vulnerabilities that could be exploited to sensitize cancer cells to intrinsic or therapeutic stresses. CTA-dependent genome maintenance pathways represent appealing new targets for therapies that would be harmless to normal cells.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: Specificity of struction and function.. Annu Rev Biochem 2005; 74:317-53; PMID:15952890; http://dx.doi.org/ 10.1146/annurev.biochem.74.082803.133250 [DOI] [PubMed] [Google Scholar]
- [2].Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase.. Nature 1999; 399:700-4; PMID:10385124; http://dx.doi.org/ 10.1038/21447 [DOI] [PubMed] [Google Scholar]
- [3].Gao Y, Mutter-Rottmayer E, Greenwalt AM, Goldfarb D, Yan F, Yang Y, Martinez-Chacin RC, Pearce KH, Tateishi S, Major MB, et al. A neomorphic cancer cell-specific role of MAGE-A4 in trans-lesion synthesis.. Nature commun 2016; 7:12105; PMID:27377895; http://dx.doi.org/ 10.1038/ncomms12105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hedglin M, Benkovic SJ. Regulation of Rad6/Rad18 activity during DNA damage tolerance. Annu rev biophys 2015; 44:207-28; PMID:26098514; http://dx.doi.org/ 10.1146/annurev-biophys-060414-033841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Simpson AJ, Caballero OL, Jungbluth A, Chen Y-T, Old LJ. Cancer/testis antigens, gametogenesis and cancer.. Nat Rev Cancer 2005; 5:615-25; PMID:16034368; http://dx.doi.org/ 10.1038/nrc1669 [DOI] [PubMed] [Google Scholar]
- [6].Doyle JM, Gao J, Wang J, Yang M, Potts PR. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases.. Mol Cell 2010; 39:963-74; PMID:20864041; http://dx.doi.org/ 10.1016/j.molcel.2010.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Durando M, Tateishi S, Vaziri C. A non-catalytic role of DNA polymerase η in recruiting Rad18 and promoting PCNA monoubiquitination at stalled replication forks.. Nucleic Acids Res 2013; 41:3079-93; PMID:23345618; http://dx.doi.org/ 10.1093/nar/gkt016 [DOI] [PMC free article] [PubMed] [Google Scholar]