

The accurate repair of DNA double-strand breaks (DSBs) is essential for cell survival and maintenance of genome integrity. In most cases, cells counteract DSBs by employing 2 highly conserved repair pathways: non-homologous end-joining (NHEJ) and homologous recombination (HR). When these pathways are impaired due to a lack of homologous donor sequence or conditions that block end-to-end ligation, alternative repair occurs, such as break-induced replication (BIR) or else imprecise or microhomology-mediated end-joining. Since these alternative pathways tend to be highly mutagenic, the choice of the repair pathway is controlled on at least 3 levels: by cell cycle stage, the chromatin context of the damage and the subnuclear position of the breaks.
In budding yeast, persistent DSBs are recruited to the nuclear periphery and associate with nuclear pores through the Nup84 subcomplex or with an inner nuclear membrane SUN domain protein called Mps3.1 The cell cycle stage influences target site choice: pores are used in both G1 and S/G2-phases of cell cycle, while Mps3 binding only occurs in S/G2-phase cells. In S-phase cells, collapsed or stalled replication forks at extended triplet repeats, as well as eroded telomeres, were shown to shift to nuclear pores.1 Importantly, these 2 perinuclear binding sites differentially affected the repair outcome. Mps3 appears to sequester resected DSBs and thereby inhibits aberrant recombination events, whereas nuclear pores are implicated in the non-canonical repair pathways such as BIR and imprecise end-joining (see reviews1-3 and Horigome et al4,5). There is, however, some cross-talk between the sites, as Mps3 may contribute to proper pore assembly, complicating the interpretation of repair data based on mps3 mutants.
Several recent papers highlight the importance of SUMO (small ubiquitin-like modifier) as a driver for perinuclear anchoring of DSBs.5-7 The nuclear pore harbors the SUMO protease Ulp1 and Slx5/Slx8 SUMO-targeted ubiquitin ligase (STUbL) and an earlier genetic study revealed that nuclear pores, Slx5/Slx8, and the proteasome act on the same pathway in DNA repair. Extensive SUMOylation events occur in response to DNA breaks in multiple species, such that factors involved in various pathways of repair become modified. Intriguingly, Horigome et al showed that the target of DSB relocation at the nuclear envelope depends on the nature of SUMOylation mediated by the E3 ligases Siz2 and Mms21.5 In G1- and S-phase cells, a polySUMOylation chain deposited coordinately by Mms21 and Siz2 recruits the Slx5/Slx8 STUbL to persistent breaks. Then Slx5 mediates binding to the nuclear pore subcomplex, Nup84. Slx5 alone can shift DNA to pores when it is targeted to a tagged locus through a DNA binding domain, even in the absence of damage. This artificial targeting of Slx5 bypasses the need for polySUMOylation for relocation. Nonetheless, at endogenous breaks5 and shortened telomeres,6 both SUMOylation and Slx8 are needed to stabilize Slx5 binding and allow the damaged site to shift to the Nup84.
In S-phase cells, monoSUMOylation mediated by the SMC5/6-Mms21 E3 complex correlated with the association of resected DSBs with the SUN domain protein, Mps3, and this can occur in the absence of Slx5. Moreover, the targeted binding of a polymer of SUMO residues (4 head-to-tail linked Smt3 residues) to an undamaged chromatin locus, allowed it to bind to pores, while the targeting of a single Smt3 residue (mono-SUMO), shifted the same locus to Mps3. Importantly, the polySUMO-dependent relocation to pores still required Slx5, arguing that this STUbL and its SUMO interacting motifs must recognize a polySUMO chain to mediate relocation (Fig. 1).
Figure 1.
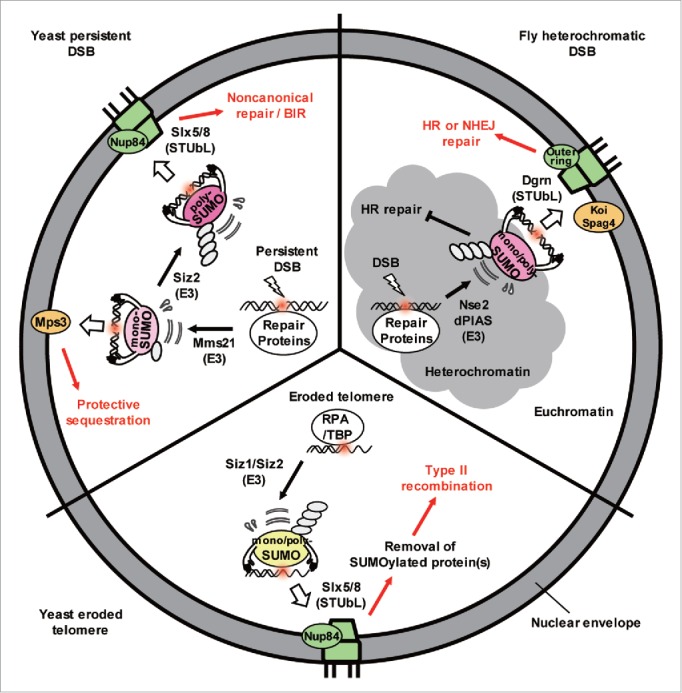
The extent of SUMO chain formation affects spatial sequestration of damage and the repair pathway choice. Repair proteins bind to DSBs and eroded telomeres in yeast and become modified by Mms21 and/or Siz1/Siz2 SUMO ligases. If monoSUMOylated, the DSBs shift to Mps3 where aberrant recombination is inhibited. If Siz2 adds a polySUMO chain, it is recognized by Slx5/Slx8, a STUbL enzyme that shifts the damage to nuclear pores. At the pore, ubiquitination of the polySUMOylated substrates and proteasome degradation facilitate alternative repair pathways. Similar events happen to DSBs in heterochromatin in flies (see text).
The question arises as to whether one or multiple SUMOylation targets are crucial for the relocation. This may well depend on the type of damage. At eroded telomeres RPA was shown to be a SUMOylation target. Since it recruits Slx5/Slx8, it was proposed to be involved in targeting the telomere to nuclear pores for an alternative pathway of repair.
In Drosophila, Ryu et al. showed that DSBs in heterochromatin shift away from the compacted chromatin domain and bind to either the nuclear pore (Nup107 or Nup160) and/or the SUN domain proteins (Koi or Spag4) in a SUMOylation- and STUbL (Dgrn)-dependent manner.7 As in yeast, both nuclear pores and the SUN domain proteins work in concert with Smc5/6 and its targeted SUMO ligase Mms21 (Nse2), yet the 2 perinuclear binding sites act independently from each other. The recruitment of the fly Slx5/Slx8 homolog (Dgrn) requires SUMO ligases Nse2 and dPIAS, which modifies multiple repair factors at the site of damage. SUMOylation appears to trigger TopBP1/ATRIP displacement to allow Rad51 binding and HR.
While problematic breaks in yeast may resort to more than one alternative pathway of repair, it seems that the association of eroded telomeres with pores facilitates a single BIR-type repair which generates type II survivors: amplification of TG repeats copied from other telomeres is a mechanism dependent on Mre11, Rad50, and Xrs2, the yeast Rad51 homolog Rad59, and a RecQ helicase, Sgs1. Triggering this recombination event at TG repeats may not be unlike the repair event that occurs in repetitive satellite DNA of metazoans like flies. For DSBs without TG sequences, BIR is also pore-enhanced, and for triplet repeat expansions, fork recovery through breaks that can occur at expanded triplet repeats also are favored by a transient shift to pores in late S-phase. These findings confirm that the pore relocation is likely to be functionally relevant for certain types of repair.
In conclusion, distinct perinuclear subcompartments act as healing hubs for eukaryotic DNA damage. How the translocation occurs is still largely unclear, and whether the outcome of relocation is always a ubiquitination and degradation event, is also unknown. Nonetheless, it is tempting to hypothesize that it may be important to clear away proteins that might mediate end-joining, to allow an alternative repair pathway to initiate. Such alternative repair mechanisms may be crucial in regions rich in repeats, such as heterochromatin at centromeres or telomeres. Future studies will no doubt reveal if the “SUMO struggle” at the edge of the nuclear ring, ends up in the destruction of one or another repair factor, yielding an ultimate winner of the match.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Geli V, Lisby M. Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. Bioessays 2015; 37:1287-92; PMID:26422820; http://dx.doi.org/ 10.1002/bies.201500084 [DOI] [PubMed] [Google Scholar]
- [2].Ptak C, Wozniak RW. Nucleoporins and chromatin metabolism. Curr Opin Cell Biol 2016; 40:153-60.; PMID:27085162; http://dx.doi.org/ 10.1016/j.ceb.2016.03.024 [DOI] [PubMed] [Google Scholar]
- [3].Kalousi A, Soutoglou E. Nuclear compartmentalization of DNA repair. Curr OpinGenet Dev 2016; 37:148-57.; PMID:27266837; http://dx.doi.org/ 10.1016/j.gde.2016.05.013 [DOI] [PubMed] [Google Scholar]
- [4].Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM. SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell 2014; 55:626-39.; PMID:25066231; http://dx.doi.org/ 10.1016/j.molcel.2014.06.027 [DOI] [PubMed] [Google Scholar]
- [5].Horigome C, Bustard DE, Marcomini I, Delgoshaie N, Tsai-Pflugfelder M, Cobb JA, Gasser SM. PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev 2016; 30(8):931-45 ; PMID:27056668; http://dx.doi.org/ 10.1101/gad.277665.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Churikov D, Charifi F, Eckert-Boulet N, Silva S, Simon MN, Lisby M, Geli V. SUMO-Dependent Relocalization of Eroded Telomeres to Nuclear Pore Complexes Controls Telomere Recombination. Cell Rep 2016; 15(6):1242-53; PMID:27134164; http://dx.doi.org/ 10.1016/j.celrep.2016.04.008 [DOI] [PubMed] [Google Scholar]
- [7].Ryu T, Spatola B, Delabaere L, Bowlin K, Hopp H, Kunitake R, Karpen GH, Chiolo I. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat Cell Biol 2015; 17:1401-11; PMID:26502056; http://dx.doi.org/ 10.1038/ncb3258 [DOI] [PMC free article] [PubMed] [Google Scholar]