

Despite over 25 y of accumulating evidence, the transition of the major human tumor suppressor – TP53 – into a potent oncogene is still controversial. Recent studies involving combinations of “omics” methods and a parallel use of multiple experimental models revealed that missense mutant p53 proteins profoundly modify cell's global molecular homeostasis. These results will hopefully contribute to lifting the remaining doubts and establishing mutant TP53 as a critical oncogene in human cancer.
In recent years, genome-wide, multi-cancer studies have confirmed a long standing assumption that TP53 is the most frequently mutated gene in human neoplasias (with a frequency of 30–40% overall). Its primary role is tumor suppression and the mutations inactivate the anti-cancer abilities of p53 proteins. However, the mode of its inactivation is exceptional among tumor suppressors, as in 70–80% of cases TP53 undergoes missense point mutations, and the resulting p53 proteins are stabilized in tumor or metastasis microenvironments. Since the 1990s scientists have observed that these “p53 mutants” may transform human cells, findings that have gradually lead to the discovery of multiple cancer-relevant pathways controlled by mutant p53. The oncogenic gain-of-function (GOF) of mutant p53 includes inactivation of p63/p73 proteins, cyclin upregulation, integrin recycling, steroid synthesis, nucleotide metabolism or Warburg effect. We have recently reviewed these results elsewhere.1
This portrait is now being extended by the use of genomic, transcriptomic and proteomic methods in parallel cancer models. Our group used multi-omics to answer several fundamental questions on mutant p53 GOF2: Is mutant p53 primarily an oncogenic transcription co-factor, as suggested earlier in numerous studies? Is the mutant p53 oncogenic program mostly shared between different missense mutants or is it specific to mutant variants and/or cellular backgrounds? How can different p53 mutants be efficiently targeted by a simple therapeutic protocol?
First, by integrated DNA-interactomic (ChIP-sequencing), transcriptomic and proteomic data from a single mutant p53 triple negative breast cancer (TNBC) cell line, our study revealed that, albeit binding of mutant p53 in proximity of gene promoters leads in the majority of cases to modulation of the corresponding transcripts, on the protein level the relation is more complex. The majority of proteins significantly upregulated by mutant p53 was accompanied by transcript upregulation, while downregulation of proteins turned out to be mostly independent of transcripts.
Strikingly, the subsequent overlap of 5 distinct mutant p53 transcriptomic programs in TNBC cell lines revealed that in the common, “core” transcriptional program the most enriched group of mutant p53 targets are Nrf2-controlled 20S/26S protesome and immunoproteasome subunits. Hence, mutant p53 strongly upregulates proteasome activity in several cancer models and in TNBC patients. It became apparent that the mutant p53-dependent upregulation of cell's protein levels is indeed directly linked to the mutant p53's role as a potent transcriptional activator, while the mutant p53-dependent downregulation of proteins is mostly controlled post-transcriptionally, through the proteasome machinery.2
Of note, the downstream effects of the mutant p53-Nrf2-proteasome axis include destabilization of the antioncogenic KSRP protein – a component of Dicer and Drosha miRNA processing complexes, responsible for maturation of a subset of oncosuppressive miRNAs.2 To our knowledge, this is the first indication of a direct link between elevated proteasome activity and alteration of miRNA homeostasis relevant for carcinogenesis. Together with a recent study by Garibaldi and coworkers showing that mutant p53 inhibits Drosha/Microprocessor complex,3 and earlier studies on mutant p53 influence on Dicer and Drosha,4,5 our results demonstrate that mutant p53 has a profound global effect on cellular miRNA processing via several overlapping routes.
Apart from the core mutant p53 program we also looked at the cell line-specific mutant p53 transcriptional signatures in the 5 TNBC cell lines. Much to our surprise these signatures contained relatively few significantly modulated pro-oncogenic transcripts and performed weakly in the association with breast cancer patient poor prognosis. In contrast, the common and the proteasome subunit gene signatures, containing transcripts shared by the mutant p53 program in 5 cell lines, prognosed the bad outcome more efficiently.2 This indicates that missense p53 mutants largely share the most significant downstream oncogenic program.
A concordant conclusion was obtained by Shelley Berger's group, who compared the DNA-binding patterns, obtained by ChIP-sequencing, of 3 mutant p53 variants from breast cancer cells against 2 wild-type p53 DNA-interactomes.6 Strikingly, in the common mutant p53 DNA-interactome Zhu and coworkers found other broad process effectors – methyltranferases (MLL1, MLL2) and an acetyltranferse (MOZ), whose Ets2-mediated activation by mutant p53 leads to epigenetic reprogramming and increased cancer growth.6
Thus, epigenetic modification, proteome reshaping and miRNA modulation mechanisms joined the earlier-discovered mutant p53-dependent genomic instability 7 in the set of mechanisms that globally drive the molecular landscape of the cell toward transformation (Fig. 1).
Figure 1.
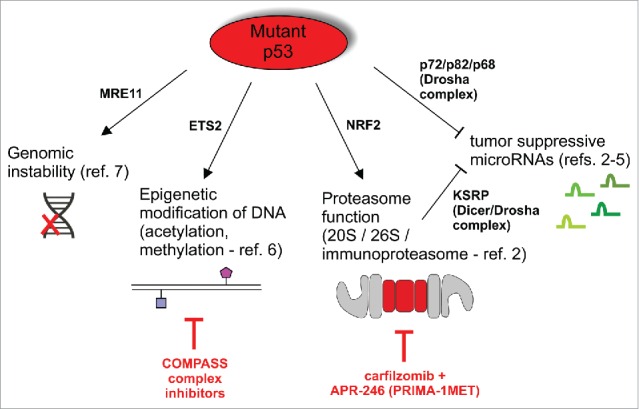
A scheme of global influences exerted by mutant p53 on cell's molecular homeostasis to drive tumorigenesis, and proposed treatment solutions.
The important issue to address in the near future is how to take advantage of these broad processes in clinical cancer treatment. Zhu et al. noticed that mutant p53 cells are sensitized to COMPASS methyltransferase complex inhibitors in vitro,6 while we found that mutant p53 induces resistance to proteasome inhibitors (such as carfilzomib), which can be overcome in TNBC xenografts by targeting of mutant p53 by APR-246 (PRIMA-1MET).2 Now these approaches, that could provide simple therapeutic strategies to target a large number of tumors bearing different mutant p53 proteins, need to be progressed toward the clinical tests.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Walerych D, Lisek K, Del Sal G. Mutant p53: One, No One, and One Hundred Thousand. Front Oncol 2015; 5:289; PMID:26734571; http://dx.doi.org/ 10.3389/fonc.2015.00289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Walerych D, Lisek K, Sommaggio R, Piazza S, Ciani Y, Dalla E, Rajkowska K, Gaweda-Walerych K, Ingallina E, Tonelli C, et al.. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol 2016; 18:897–909; PMID:27347849; http://dx.doi.org/ 10.1038/ncb3380 [DOI] [PubMed] [Google Scholar]
- [3].Garibaldi F, Falcone E, Trisciuoglio D, Colombo T, Lisek K, Walerych D, Del Sal G, Paci P, Bossi G, Piaggio G, et al.. Mutant p53 inhibits miRNA biogenesis by interfering with the microprocessor complex. Oncogene 2016; 35:3760–70; PMID:26996669; http://dx.doi.org/ 10.1038/onc.2016.51 [DOI] [PubMed] [Google Scholar]
- [4].Muller PA, Trinidad AG, Caswell PT, Norman JC, Vousden KH. Mutant p53 regulates Dicer through p63-dependent and -independent mechanisms to promote an invasive phenotype. J Biol Chem 2014; 289:122–32 ; PMID:24220032; http://dx.doi.org/ 10.1074/jbc.M113.502138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature 2009; 460:529–33; PMID:19626115; http://dx.doi.org/ 10.1038/nature08199 [DOI] [PubMed] [Google Scholar]
- [6].Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, et al.. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015; 525:206–11; PMID:26331536; http://dx.doi.org/ 10.1038/nature15251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol 2007; 9:573–80; PMID:17417627; http://dx.doi.org/ 10.1038/ncb1571 [DOI] [PubMed] [Google Scholar]