

Abstract
Background and Purpose
The extent (number and diameter) of collateral vessels varies widely and is a major determinant, along with arteriogenesis (collateral remodeling), of variation in severity of tissue injury following large artery occlusion. Differences in genetic background underlie the majority of the variation in collateral extent in mice, through alterations in collaterogenesis (embryonic collateral formation). In brain and other tissues, ~80% of the variation in collateral extent among different mouse strains has been linked to a region on chromosome 7. We recently used congenic (CNG) fine-mapping of C57BL/6 (B6, high extent) and BALB/cBy (BC, low extent) mice to narrow the region to a 737 Kb locus, Dce1. Herein, we report the causal gene.
Methods
We used additional CNG mapping and knockout mice to narrow the number of candidate genes. Subsequent inspection identified a non-synonymous SNP between B6 and BC within Rabep2 (rs33080487). We then created B6 mice with the BC SNP at this locus plus three other lines for predicted alteration or knockout of Rabep2 using gene editing.
Results
The single amino acid change caused by rs33080487 accounted for the difference in collateral extent and infarct volume between B6 and BC mice attributable to Dce1. Mechanistically, variants of Rabep2 altered collaterogenesis during embryogenesis but had no effect on angiogenesis examined in vivo and in vitro. Rabep2 deficiency altered endosome trafficking known to be involved in VEGF-A→VEGFR2 signaling required for collaterogenesis.
Conclusions
Naturally occurring variants of Rabep2 are major determinants of variation in collateral extent and stroke severity in mice.
Keywords: collateral circulation, ischemic stroke, Rabep2, Rabaptin-5beta, Dce1
Introduction
Collaterals are interesting vessels. After sudden occlusion in brain, heart and other tissues, collateral-dependent blood flow can lessen or prevent tissue injury, depending primarily on collateral number and diameter at baseline (collateral extent) and their subsequent amount of anatomic lumen enlargement, termed collateral remodeling or arteriogenesis. Unfortunately, flow provided by the pial collaterals, ie, leptomeningeal arteriole-arteriole anastomoses interconnecting distal branches of the MCA, ACA and PCA trees, varies widely among patients immediately after acute ischemic stroke (AIS).1 Likewise, collateral flow also varies widely in heart and lower extremities of healthy individuals, ie, lacking detectable atherosclerosis.2,3 The cause of this variation is unknown. Evidence in humans and mice suggests that adverse effects of aging and other cardiovascular risk factors on collateral extent account for only a small portion of the variation.4,5 Recent studies have shown that differences in genetic background are major determinants in mice. Pial collateral extent varies 56 fold among 21 adult inbred mouse strains in brain and other tissues of the same individual.6 These differences reflect background-dependent variation in formation of pial collaterals (collaterogenesis) which occurs late in gestation.7 Collaterogenesis occurs via a unique sprouting-like mechanism.7 Endothelial tip and stalk cells branch from existing arterioles, migrate, and fuse with an arteriole in the adjacent tree. We recently reported that most of the genetic variation in murine collateral extent maps to a single quantitative trait locus (QTL) on chromosome 7 (Candq1)8 that was confirmed using SNP-association mapping among the above 21 strains.6 We subsequently constructed congenic (CNG) mice in which portions of Candq1 from a strain with abundant large-diameter collaterals (C57BL/6, B6) were introgressed into a strain with sparse small-diameter collaterals (BALB/cBy, BC).9 This led to the identification of a 737 Kb locus, Determinant of collateral extent-1 (Dce1), that when introgressed into the BC strain reversed 83 percent of the difference in collateral extent and, after permanent middle cerebral artery occlusion (pMCAO), increased cerebral blood flow to the area at risk and reduced infarct volume each by 85%.9
The Dce1 locus contains 28 protein-coding genes.9 The present study sought to identify the causative gene(s). We first verified that genetic background at Dce1 determines pial collateral extent and infarct volume by deriving an additional congenic line. This was followed by phenotyping collateral extent in mutant mice deficient at 13 of the above 28 genes, allowing their provisional elimination as candidates genes. Among the remaining 15 genes, we previously identified Rabep2 (Rabaptin-5β, FRA) as the highest-priority candidate.9 This was in part because it contains a SNP (rs33080487) that substitutes an adenine for a guanine in BC and almost all the other mouse strains with low collateral extent examined, resulting in an arginine-to-glutamine (R298Q) substitution predicted to be deleterious.9 We therefore hypothesized that variants of Rabep2 underlie the Dce1 locus, and used gene editing to create B6 mice with either the BC SNP at rs33080487 or deletion of Rabep2 altogether. The results identify Rabep2 as the causative allele at Dce1.
Rabep2 is a novel gene. Our results show that variants of it alter collaterogenesis in the embryo and thus collateral extent and infarct volume in the adult. It is possible that the same or a related gene(s) may contribute to the wide collateral variation in humans, opening the way for development of a biomarker for collateral extent, as well as a deeper understanding of why some patients with ischemic disease do better than others.
Methods
See the Online-only Data Supplement for details.
Mutant B6 mice were generated using CRISPR/Cas9 methodology: a guanine-to-adenine substitution (“SNP”), a 3-nucleotide deletion (“Del3”), a 1-guanine insertion (“InsG”) or a 1-guanine deletion (“DelG”) at rs33080487 (7:126,440,209) in Rabep2. Pial collaterals between the MCA and ACA trees were quantified in 3–4 month-old adults after maximal dilation, and in embryos by immunohistochemistry/fluorescence (IHC). Infarct volume was determined 24h after pMCAO. Endosomes were measured with IHC in embryonic pia. Neonatal retina, tumor and aortic ring angiogenic assays were standard, as were qPCR, immunoblot, immunoprecipitation, mass spectrometry and genotyping. This study conformed to the recommendations of STAIR (2012; see Supplement). Data are mean±SEM with significance (p<0.05) determined by ANOVA, Student’s and Bonferroni t-tests.
Results
A refined interval containing Dce1 narrows the candidate genes underlying collateral variation
During backcrossing CNG5 to BC mice we obtained a recombination that deleted 550 kb from CNG5 such that the introgressed region is 162 kb longer than the originally defined Dce1 (Figure I, Data Supplement). This CNG7B6 line has, on average 13.0 pial collaterals (MCA-ACA) per individual, compared to 0.7 in CNG7BC littermates and 19–20 in B6 (Figure 1). Thus the B6 allele of Dce1, when introgressed into BC, rescues 12–13 collaterals or ~65% of the BC deficit in collateral number. Similarly, pial collaterals in CNG7B6 are almost 2-fold larger in diameter and infarct volumes after pMCAO than in CNG7BC littermates (Figure 1). These values are similar to those reported previously for lines CNG3, CNG5 and CNG6 and their CNG-BC littermates (Figure 1).9 These data confirm our previous conclusion9 that a genetic element(s) in Dce1 is responsible for ~80% of the variation in collateral extent and infarct volume between B6 and BC mice. However, it should be pointed out that in mice, multiple branching of the MCA can also be associated with small infarction volumes in suture models.
Figure 1. The high-collateral allele of the congenic Dce1 locus rescues low collateral number and diameter and large infarct volume.

Collateral number in this and subsequent figures is the number of arteriole-to-arteriole anastomoses cross-connecting outer branches of the MCA and ACA trees. Introgression of the congenic (CNG) Dce1 locus-7 of C57BL/6 (B6) mice into BALB/cBy (BC) mice (CNG7BC, see Supplemental Figure I for genomic intervals of the 4 CNG lines shown) rescues 67% of the difference in number and 69% in average diameter of MCA-to-ACA collaterals and 93% in infarct volume between B6 and CNG7BC (essentially BC wildtype (WT)) mice. Lower right, representative images of TTC staining 24h after permanent MCAO) and collaterals (stars). Historic values9 for B6, BC and CNG3, 5 and 6 provided for comparison. For this and subsequent figures: congenic BC lines are controls for congenic derivation, thus essentially wildtype BC; number of animals given in columns; *p<0.05, **p<0.01, ***p<0.001; nsd, no significant difference.
We did not examine infarct volume in the CNG6 line previously characterized9 and since extinguished. This prevented us from addressing a recent report that a variant lying 3′ to Dce1 does not affect collateral extent but is protective against infarct volume when of B6 genotype.10 We were unable to confirm this conclusion: infarct volume did not decrease but instead trended greater between CNG7B6 that lack, and CNG5B6 and CNG3B6 that possess, B6 genome 3′ to Dce1 that includes the reported protective allele (Figure 1). One explanation is that the distal locus requires presence of additional genetic material not present in our congenic lines.
To narrow the 28 protein-coding gene candidates in Dce1,9 we phenotyped collateral extent in available knockout mice. Collateral extent was unaffected in mice deficient in Nfatc2ip, MapK3, Nupr1 or a 390 Kb region encompassing Slx1-to-Sept1b (Figures II,III). These findings when combined with absence of effect in Cln3−/− mice9 narrow the candidates to thirteen.
Variants of Rabep2 confer variation in collateral extent
Among the 28 genes in Dce1, a non-synonymous coding SNP (rs33080487, 7:126440209 (GRCm38), c.1258G>A in BC) causes the alteration R298Q in the Rabex-5 binding domain11 of Rabep2 (NP_085043.2), an alteration predicted to be deleterious by PolyPhen-2 (score=0.999). We thus edited this polymorphism into B6 mice (“SNP” line). Presumptive non-homologous end-joining gave 3 additional lines (Table I, Figure IV). Two lines, DelG and InsG, have a deletion and insertion, respectively, of one nucleotide leading to frame-shift, stop codons, and predicted non-sense mediated decay (NMD). In the third line, Del3, the codon for R298 was deleted, leading to expression of full-length Rabep2 minus R298. qPCR of DelG confirmed that Rabep2 message was reduced to 15% of wildtype, with the residual presumably reflecting message that had not yet undergone NMD.
Rabep2 expression was assessed (Figure 2A). Whole-brain and neocortex from wildtype and DelG mice probed with 2 specific rabbit sera (gifts of Dr. M Zerial) showed Rabep2 to be undetectable in DelG (A, upper panel). A commercial antibody (Proteintech™ 14625-AP) revealed several closely-spaced bands in WT, BC, SNP and Del3, while the lower band in DelG and InsG was absent. A pulldown from WT with this antibody yielded the lower band. Mass spectrometry (MS, MALDI and LTQ-Velos-Orbitrap) confirmed the product was Rabep2. We were unable to pull down enough of the upper 2 bands to get a definitive identification. However, MS analysis showed they are not Rabep2. Hence as predicted, both DelG and InsG mice are knockouts while expression of Rabep2 in SNP and Del3 appear unaffected. Rabep1 levels by qPCR and immunoblot are not altered in the edited mice (Figure VA,B). Each line reproduced at Mendelian ratios (Table II) and had normal growth. Founders were outcrossed to additional B6 for 2–5 generations, all wildtype were littermates, and their values of collateral number and diameter were very similar to our previous WT-B6,6–9 indicating no off-target effects.
Figure 2. A variant in Rabep2 accounts for the difference in collateral extent and infarct volume attributable to the Dce1 locus.
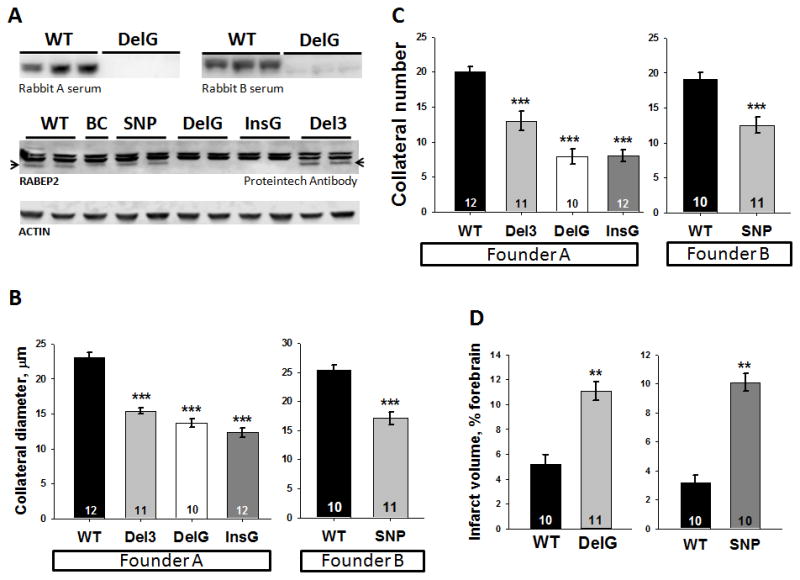
A, Immunoblots for Rabep2 in CRISPR/Cas9 adult brain (homozygous at the edited alleles in this and subsequent figures) and founder-derived WT-B6. Del3 and SNP have the coding SNP in Rabep2 deleted or altered in the 2 strains; DelG and InsG are different frameshift knockout edits of Rabep2. Upper panel, Anti-Rabep2 serum obtained from rabbits labels a 62 kDa protein in WT that is absent in DelG. Lower panel, Proteintech anti-Rabep2 labels 2 nonspecific upper bands and a lower band, Rabep2, that is absent in DelG and InsG (specificity determined by mass spectrometry). Collateral number (B) and diameter (C) were reduced in all lines compared to WT-B6. D, Compared to WT-B6, pMCAO in DelG and SNP gave increased infarct volumes similar to BC in Figure 1.
Collateral number was reduced to 8.0 collaterals in homozygous DelG and InsG mice, a deficit of 11–12 collaterals compared to wildtype littermates and WT-B6 (Figure 2C). Thus, the reduction in collateral number due to elimination of Rabep2 from B6 is virtually identical to the increase in collateral number caused by introduction of the B6 allele of Dce1 into BC, suggesting that variants of Rabep2 alone account for the entire effect of Dce1. Similarly, collateral diameter was reduced by 41% and 47% in DelG and InsG knockouts, representing 85 and 97 percent of the difference in diameter attributable to Dce1 (Figure 2B). This stronger effect on diameter than on number was previously noted for Dce19, where expression of the B6 allele in BC essentially restored diameter to the WT-B6 value.
Alterations at position 298 with retention of Rabep2 expression gave intermediate reductions in collateral number to 13.0 in Del3 (delR298) and 12.3 in SNP (R298Q). Thus, Rabep2 activity in these forms appears to be diminished, and variation at position 298 has less effect on the B6 background (reduction of 6–7 collaterals for R298Q) than on the BC background (rescue of 12–13 collaterals by R298Q). Diameter data yield similar conclusions (Figure 2B), although the difference between alteration and elimination of Rabep2 is less striking (Figure 2B). As expected, these changes in collateral extent have functional effects, since infarct volumes after pMCAO were increased 2–3-fold in DelG and SNP mice (Figure 2D). Heterozygous mice had intermediate phenotypes (Figure VI).
Rabep2 favors greater collateral extent and smaller infarcts in B6 females
Infarct volume was 35% smaller in female B6-WT; this difference was abolished in Rabep2−/− mice in association with a trend toward fewer collaterals of smaller diameter (Table III). Thus on the B6 background, the B6 allele of Rabep2 favors greater collateral extent and smaller infarcts in females. On BC-background Nfatc2ip−/− mice (discussed below) the B6 allele of Rabep2 does not confer protection against infarct volume.
Variants of Rabep2 alter collaterogenesis
Pial collateral extent in healthy adults is dependent on collateral formation in the embryo (ie, collaterogenesis).7 Consistent with this, collaterogenesis was inhibited at both E14.5 and E16.5 in Rabep2−/− mice, indicating that Rabep2 is required for robust collateral formation (Figure 3A). Likewise, CNG5B6 embryos formed more collaterals than CNG5BC, confirming that the B6 variant (of presumably Rabep2) increases collaterogenesis when on the BC background (Figure 3B). Consistent with diameter being larger and branch-density lower for pial capillary plexus vessels of BC versus B6 embryonic neocortex,7 diameter was greater and branch number lower in Rabep2−/− (Figure 3C), indicating that Rabep2 also impacts vascular patterning. Previous studies have observed these same differences in the early embryonic capillary plexus of BC versus B6 and Vegfa and Flk1 mutants and shown that the accompanying differences in collaterogenesis are not secondary to the differences in the plexus.7 and references therein
Figure 3. Rabep2 is a major gene in the collaterogenesis pathway that determines collateral extent in the adult.

Collateral formation is impaired in Rabep2−/− embryos (ie, DelG mice; E, embryonic day) versus WT (A), and enhanced in CNGB6 versus CNGBC (B), demonstrating that Rabep2 is a key gene in the collaterogenesis pathway. C, Rabep2 deletion also reduces branching and increases diameter of capillaries of embryonic pial plexus. D, Representative images of CD31-stained E14.5 pial vessels in WT and Rabep2−/−; arrowheads denote a newly-formed collateral; white box, region analyzed for plexus capillaries; asterisks, branch-points; lines, diameter.
Consistent with developmental and adult angiogenesis (sprouting of new capillary vessels from existing capillaries) being comparable in B6 and BC mice,see 7 for references angiogenesis in the newborn retina was similar in CNG5B6 and CNG5BC mice (Figure 4A). Likewise, orthotopic tumor growth and blood vessel infiltration (Figure 4B,C) and sprouting in aortic rings (Figure VII) were not different in CNG mice. Thus, Rabep2 is critical in the pathway that drives collaterogenesis but is not required for developmental or tumor angiogenesis.
Figure 4. Rabep2 variants do not alter angiogenesis.

A, Leading edge (top row) and trailing plexus (bottom row) of CD31-stained postnatal day-7 retina from CNGBC (BC-WT), CNGB6 and B6 mice. Introgression of the Dce1 into BC mice (CNGB6) does not confer greater number of tip cells, branch points and vascular density present in B6-WT. B,C, CNGBC and CNGB6 show comparable tumor growth after injection of 4T1 mammary tumor cells into breast pad of female CNG7 mice. C, Representative tumor and CD31-stained section (inset).
Rabep2 deficiency alters endosome trafficking
Little is known about the function of Rabep2 other than it binds activated Rabex5, Rab5 and Rab4 proteins during early endosomes (EEs) formation in cultured cells.12 Endosomes traffic receptors such as VEGFR2 to sites involved in downstream signaling, lysosomes, or back to the cell membrane.13–15 We thus examined EE in pial membrane of Rabep2−/− mice at E14.5 when collaterogenesis is underway. Endosome diameter was larger in Rabep2−/− while density was similar (Figure 5A–C). The requirement to examine EEs in situ precluded assessment of co-localization of VEGFR2 with EEA1. However, Vegfa, Vegfr2 and Vegfr1 expression was similar between groups (Figure 5D), suggesting that aberrant levels of these proteins do not underlie the reduced collaterogenesis in Rabep2−/− mice.
Figure 5. Rabep2 deletion increases early endosome diameter.

A, Early endosomes (EEs) in cells of E14.5 pia are larger diameter (4-parameter Weibull analysis (B)) in Rabep2−/− mice, suggesting an alteration in EE cycling. C, Representative images for data in A,B. D, Vegfa, Vegfr1 and Vegfr2 expression in embryonic brain is unaffected in Rabep2−/− mice.
Deficiency of Rabep2 had no effect on Rabex5 and Rab4 that Rabep2 is known to bind, nor on EEA1 or Rab7 which is present in late endosomes destined for recycling to the cell membrane (−). These proteins are in the VEGFA→VEGFR2→endosome→VEGFR2 recycling and signaling pathways (see Discussion). These findings, which are consistent with lack of effect on expression of VEGFA, VEGFR2and VEGFR1 (Figure 5D) strengthen the conclusion from Figure 5A–C that Rabep2 deficiency impairs endosome trafficking.
Capillary and microvessel densities in adult brain were not affected by Rabep2 deficiency (Figure 6B, VIII). This is consistent with data in Figures 4 and VII showing that angiogenesis was not altered in newborn retina, tumor growth, or aorta explants, reinforcing the conclusion that Rabep2 is important in the collaterogenic but not angiogenic pathways. Other genetic abnormalities have been associated with changes in capillary density, eg, Notch3 mutants with differences in pericyte coverage16,17 and aged mice harboring the human APP-Tg2576 transgene.18 However, expression of Notch3 was not different between CNG7BC and CNG7B6 mice (3 biological replicates done in triplicate and repeated 3 times, P > 0.8; data not shown).
Figure 6. Rabep2 deletion does not change expression of early endosome-associated proteins or alter capillary density.

A, Immunoblots in WT and Rabep2−/− (DelG) E16.6 mouse brain (3 of each; Figure VIII shows consistent beta-actin loading). Expression of Rabex5 and Rab4 by qPCR also did not differ; others above were not examined. B, Capillary density (2–10 μm diameter) in white and gray matter of M1 neocortex and underlying corpus callosum did not differ among WT, Rabep2−/− and SNP mice (p=0.97, p=0.42, ANOVA); the following also did not vary: microvessel density (2–40 μm diameter) capillary and microvessel pixel area(p=0.79, p=0.39, p=0.75, p=0.29). C, Schematic of Rabep2. Functional and structural domains of Rabep2 (554 amino acids; see scale bar) are identified by direct demonstration or prediction from homologous regions of Rabep1.12,13 and references therein We confirmed non-synonymous SNPs in strains LEWES/EiJ (P200R), A/J (R426H), SJL/J (R298Q) and SWR/J (R298Q). Ubiquination and phosphorylation sites are predicted (Phosphosite.org) and were confirmed for Lys92 and Ser180 by proteomics. CC, coiled-coil sequence.
The A-to-G SNP present in 129-background mice recapitulates the CNG phenotype
Before deriving Rabep2 edited mice, we examined Nfatc2ip knockouts (a gift from Dr. K Mowen).19 Nfatc2ip−/− mice were made using 129/SvJae embryonic stem cells and backcrossed 6–12 generations to BALB/c. Genotyping identified that a 129Sv flanking region was retained that included Rabep2 with the B6 allele at rs33080487 (Figure III). High-density SNP array analysis (GigaMUGA) confirmed the presence of approximately 3 Mb of 129-derived sequence around targeted Nfatc2ip (~7:125,221,537–128,681,374), extending well beyond Dce1 in both directions. Consistent with the presence of the B6 Rabep2 allele, Nfatc2ip−/− had 70% of collateral number and 93% of diameter of the difference attributable to the Dce1 locus evident in Figure 1. Likewise, they had the small infarct volume of B6-WT (Figures 1,2; infarct volumes were comparable, p=0.42; Figure IIIA–C). These values, which were reported previously9 and do not significantly differ from the CNG lines in Figure 1,9 confirm on a different background (BC) the major role of Rabep2 and SNP rs33080487 in collaterogenesis. These findings also show no contribution of Nfatc2ip.
Having identified Rabep2 as causal for Dce1, we combined new high-resolution SNP data (Sanger-1, Jackson Labs) with our own sequencing across Rabep2 to clarify uncertainties among 21 strains we previously phenotyped.6 Collateral phenotype now correlates almost exactly with genotype among 20 of 21 strains (Figure 6C, Table IV); we also identified three new SNPs in the lone SJL/J outlier strain that could reconcile its phenotype to Dce1 (see Table IV footnotes).
Discussion
Collateral status has been proposed to be important in defining the course after treatment in AIS.20,21 However, collateral flow varies widely among patients for largely unknown reasons.1–4 Recent studies in mice have provided a potential clue.5–9 Strains with differences in genetic background exhibit wide variation in collateral extent, approximately 80 percent of which depends on a polymorphic region on chromosome 7, Dce1.6,8,9 In the present study we demonstrate that a single gene within this region, Rabep2, is responsible for this variation. However, its contribution varies depending on genetic background. When the BC allele of Rabep2 is on the B6 background, it accounts for 36% and 72% of the differences in collateral number and diameter attributable to Dce1—as determined in the SNP and genetically comparable Del3 mice—and accounts for 64% and 89% of number and diameter when it is deleted as in the Rabep2−/− mice. By comparison, when the B6 allele is present on the BC background (CNG7B6 and Nfatc2ip−/− mice), it rescues 70% and 93% of number and diameter in WT-B6 mice. These values are identical to those reported previously9 and make clear that other genetic elements interact with Rabep2 or contribute independently to collateral variation.
Rabep2 is a ubiquitously expressed gene about which little is known. Studies in cultured cells show that Rabep2 participates in vesicular trafficking12 wherein cell surface receptors are internalized into vesicles that fuse into EEs in a Rab4 and Rab5-dependent manner.12–15 Membrane cargo is then returned to the plasma membrane via recycling endosomes (Rab4-dependent), concentrated in Golgi-associated stores, or transferred to lysosomal-bound late endosomes (Rab7- and Rab9-dependent).15,22–24 EE formation requires recruitment of Rab4, Rab5 and a number of Rab effectors. However, compared to the better known Rabep1 which when immuno-depleted in HUVEC results in severe disruption of EE fusion, depletion of Rabep2 has only a minor effect which may explain why no functional studies have appeared since its original description.12
Rabep2 shares 42% sequence identity and 53% similarity with Rabep1. Although they complex with some of the same proteins, there is no evidence that they directly interact.12 Similar to Rabep1, Rabep2 is recruited to EEs by Rab4 and Rab5 GTPases and Rabex5 (RabGEF1) where it complexes with other endosomal proteins.12,13,15 Redundancy exists for Rabep1 since overexpression of other binding partners can compensate for dysfunctional trafficking caused by deleting its binding domains.15 The B6-BC amino acid change (rs33080487) is in the coiled-coil domain of Rabep2 that enables binding to Rabex5 (CC2-1, Figure 6C). Deleting the homologous domain of Rabep1 prevented its recruitment to EEs.15 Accordingly, it is possible that the R298Q change alters binding of Rabep2 to Rabex5 and hinders Rab4 or Rab5 activation, and that deletion of Rabep2−/− further impairs activity. Consistent with this hypothesis, we observed larger endosomes in Rabep2−/− embryonic pia. This suggests that in Rabep2−/−, EEs initially form and fuse into larger vesicles (Rab5 function remains intact) but recycling is reduced because Rab4 action is hindered.
It is well established that VEGFR2 undergoes ligand-dependent and ligand-independent (constitutive) endocytosis,25–28 and that these signaling routes impact angiogenic sprouting and vascular development.eg, 29,30 Binding of VEGFA induces VEGFR2 trafficking through one of several pathways, including the Rab4-mediated “short-loop” fast-recycling route back to the cell membrane via EEs. Whether endocytosis enhances or dampens VEGFR2 signaling appears context dependent. For example, downstream VEGFR2 effectors MAPK and AKT required for VEGFA’s angiogenic actions show sustained activation in endothelial cells when EE formation is impaired,15 suggesting that endocytosis is required for VEGFR2 signaling. In contrast, ligand binding causes ubiquitination of VEGFR2 thus targeting for lysosomal degradation.26 Further complicating matters, presumed newly synthesized VEGFR2 stored in the Golgi can be, after VEGF-VEGFR2 binding to vesicles, quickly mobilized/targeted to either the cell membrane or lysosomes,31 and existing endosomes from plasma membrane can fuse with the Golgi.22 The signals that dictate where/when VEGFR2 is shuttled remain under investigation.
VEGFA→VEGFR2 signaling contributes importantly to embryonic collaterogenesis.7 Reduction of ligand or receptor during the narrow time-window of collaterogenesis reduces collateral formation thus collateral number and diameter in the adult.7 Herein we found that the embryonic pial plexus of Rabep2−/− mice had larger vessel diameters and less branching. This phenocopies previous findings in Vegfa-deficient and BC mice (compared to B6), the latter being consistent with reduced Vegfa expression in BC versus B6 mice.7 and references therein Since Vegfa, Vegfr2 and Vegfr1 transcripts were not decreased and EEs were larger in E14.5 Rabep2−/− mice, we hypothesize that when Rabep2 is lacking, deficient EE recycling sequesters inactive VEGFR2, effectively reducing the number available for signaling, with the result that collaterogenesis is reduced. Significant perinuclear VEGFR2 is evident in pial endothelial cells of E14.5 embryos (data not shown), thus transport from this location to the cell membrane may also be impaired.
Arteriogenesis, ie, anatomic enlargement of collateral lumen diameter that occurs after arterial obstruction (eg, pMCAO), is induced by shear stress.32 It is possible that the difference in arteriogenesis between these strains33 contributed to infarct volumes measured in the present study. However, we previously showed that this difference maps to a QTL on chromosome 11 different from Dce1/Rabep2 on chromosome 7.8 In agreement, introgression of Dce1 between the two strains had no effect on arteriogenesis of pial collaterals after pMCAO or hindlimb collaterals after femoral artery ligation.9 In addition, differences in infarct volume reported previously9 and in the current study closely follow the genetically engineered differences in native collateral extent.
This study has several limitations. First, determining how altered Rabep2 disrupts collaterogenesis will be challenging because there is no in vitro model of collaterogenesis, mice with transgenic, marker, and conditional alleles of Rabep2 will have to be constructed, and little is known about the functions of Rabep2. Second, although effective in our immunoblot and pulldown assays, available Rabep2 antibodies showed significant non-specific binding that precluded their use for IHC—findings we confirmed with mass spectrometry. Third, the importance of variants of Rabep2 in determining variation in collateral extent in mouse may not translate to human. However, since vascular developmental pathways are conserved among vertebrates, and mice and humans are 92% similar genetically, it is possible that polymorphisms at human RABEP2 are significant determinants. Other “collateral genes” are also likely important: Three additional QTL have been identified that account for almost all of the remaining variation in pial collaterals between B6 and BC mice beyond Rabep2,8 and several known angiogenic proteins significantly impact collaterogenesis thus collateral extent in pia and skeletal muscle (VEGFA, VEGFR2, Dll4, Notch, ADAM10, ADAM17, Gja4, Gja5) 7, 34–36 and possibly others (Egln1,37 NFkB138). Lastly, we have previously reported in mice that aging, impaired eNOS activity, hypertension, metabolic syndrome and other cardiovascular/stroke risk factors cause a reduction in pial collateral number and diameter, in association with increased infarct volume after pMCAO.5 and references therein However, these environmental effects on collateral extent are smaller than those attributable to differences in genetic background.
Summary/conclusion
Collateral-dependent blood flow following stroke is primarily determined by collateral extent at baseline and the amount of collateral remodeling/arteriogenesis following large artery occlusion. Herein, we show that Rabep2 is the causal gene at Dce1 responsible for most of the difference in native collateral extent in B6 and BC mice and, by inference based on previous SNP association studies8 and new SNPs we identified herein, in 18 of the 19 other strains examined. Whether this extends to humans is under investigation in a prospective study, GENEtic Determinants of Collateral Status in Stroke (GENEDCSS) (JEF pers comm). Confirmation that RABEP2 and other “collateral genes” links to variability in collateral status may lead to development of a test to identify individuals with poor collaterals before stroke or other occlusive disease occurs. In AIS patients, such a biomarker could also aid clinical decision-making and stratification into clinical studies; currently, there is no non-invasive method to assess collateral extent. In addition, the Rabep2 mutants described herein provide a set of mice that models individuals with poor, intermediate and good collaterals on an otherwise isogenic background for study of the collateral circulation.
Supplementary Material
Acknowledgments
The authors thank: Drs. F Pardo-Manuel de Villena, D Cowley, K Burridge for advice, D Miller for tissue and GigaMUGA analysis, and the Animal Models Core.
Sources of Funding
NIH-NHLBI HL111070 and NIH-NINDS NS083633 (JEF)
Footnotes
Disclosures
None.
References
- 1.Shuaib A, Butcher K, Mohammad AA, Saqqur M, Liebeskind DS, et al. Collateral blood vessels in acute ischaemic stroke: A potential therapeutic target. Lancet Neurol. 2011;10:909–921. doi: 10.1016/S1474-4422(11)70195-8. [DOI] [PubMed] [Google Scholar]
- 2.Meier P, Hemingway H, Lansky AJ, Knapp G, Pitt B, Seiler C. The impact of the coronary collateral circulation on mortality: A meta-analysis. Eur Heart J. 2012;33:614–621. doi: 10.1093/eurheartj/ehr308. [DOI] [PubMed] [Google Scholar]
- 3.Traupe T, Ortmann J, Stoller M, Baumgartner I, de Marchi SF, Seiler C. Direct quantitative assessment of the peripheral artery collateral circulation in patients undergoing angiography. Circulation. 2013;128:737–744. doi: 10.1161/CIRCULATIONAHA.112.000516. [DOI] [PubMed] [Google Scholar]
- 4.Menon BK, Smith EE, Coutts SB, Welsh DG, Faber JE, Goyal M, et al. Leptomeningeal collaterals are associated with modifiable metabolic risk factors. Ann Neurol. 2013;74:241–248. doi: 10.1002/ana.23906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore SM, Zhang H, Maeda N, Doerschuk CM, Faber JE. Cardiovascular risk factors cause premature rarefaction of the collateral circulation and greater ischemic tissue injury. Angiogenesis. 2015;18:265–281. doi: 10.1007/s10456-015-9465-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S, Zhang H, Wiltshire T, Sealock R, Faber JE. Genetic dissection of the canq1 locus governing variation in extent of the collateral circulation. PLoS One. 2012;7:e31910. doi: 10.1371/journal.pone.0031910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucitti JL, Mackey JK, Morrison JC, Haigh JJ, Adams RH, Faber JE. Formation of the collateral circulation is regulated by vascular endothelial growth factor-a and a disintegrin and metalloprotease family members 10 and 17. Circ Res. 2012;111:1539–1550. doi: 10.1161/CIRCRESAHA.112.279109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S, Zhang H, Dai X, Sealock R, Faber JE. Genetic architecture underlying variation in extent and remodeling of the collateral circulation. Circ Res. 2010;107:558–568. doi: 10.1161/CIRCRESAHA.110.224634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sealock R, Zhang H, Lucitti JL, Moore SM, Faber JE. Congenic fine-mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circ Res. 2014;114:660–671. doi: 10.1161/CIRCRESAHA.114.302931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keum S, Lee HK, Chu PL, Kan MJ, Huang MN, Gallione CJ, et al. Natural genetic variation of integrin alpha l (itgal) modulates ischemic brain injury in stroke. PLoS Genet. 2013;9:e1003807. doi: 10.1371/journal.pgen.1003807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Zhang T, Wang S, Gong Z, Tang C, Chen J, et al. Molecular mechanism for rabex-5 gef activation by rabaptin-5. Elife. 2014:3. doi: 10.7554/eLife.02687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gournier H, Stenmark H, Rybin V, Lippe R, Zerial M. Two distinct effectors of the small gtpase rab5 cooperate in endocytic membrane fusion. EMBO J. 1998;17:1930–1940. doi: 10.1093/emboj/17.7.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Renzis S, Sonnichsen B, Zerial M. Divalent rab effectors regulate the sub-compartmental organization and sorting of early endosomes. Nat Cell Biol. 2002;4:124–133. doi: 10.1038/ncb744. [DOI] [PubMed] [Google Scholar]
- 14.Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol. 2014;31:2–10. doi: 10.1016/j.semcdb.2014.03.034. [DOI] [PubMed] [Google Scholar]
- 15.Kalin S, Hirschmann DT, Buser DP, Spiess M. Rabaptin5 is recruited to endosomes by rab4 and rabex5 to regulate endosome maturation. J Cell Sci. 2015;128:4126–4137. doi: 10.1242/jcs.174664. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh M, Balbi M, Hellal F, Dichgans M, Lindauer U, Plesnila N. Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann Neurol. 2015;78:887–900. doi: 10.1002/ana.24512. [DOI] [PubMed] [Google Scholar]
- 17.Joutel A, Monet-Leprêtre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest. 2010;120:433–45. doi: 10.1172/JCI39733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuznetsova E, Schliebs R. β-Amyloid, cholinergic transmission, and cerebrovascular system – a developmental study in a mouse model of Alzheimer’s disease. Curr Pharm Des. 2013;19:6749–65. doi: 10.2174/13816128113199990711. [DOI] [PubMed] [Google Scholar]
- 19.Fathman JW, Gurish MF, Hemmers S, Bonham K, Friend DS, Grusby MJ, et al. Nip45 controls the magnitude of the type 2 t helper cell response. Proc Natl Acad Sci U S A. 2010;107:3663–3668. doi: 10.1073/pnas.0914700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang YH, Kang DH, Kim YW, Kim YS, Park SP, Liebeskind DS. Impact of time-to-reperfusion on outcome in patients with poor collaterals. AJNR Am J Neuroradiol. 2015;36:495–500. doi: 10.3174/ajnr.A4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mokin M, Rojas H, Levy EI. Randomized trials of endovascular therapy for stroke - impact on stroke care. Nat Rev Neurol. 2016;12:86–94. doi: 10.1038/nrneurol.2015.240. [DOI] [PubMed] [Google Scholar]
- 22.Barbero P, Bittova L, Pfeffer SR. Visualization of rab9-mediated vesicle transport from endosomes to the trans-golgi in living cells. J Cell Biol. 2002;156:511–518. doi: 10.1083/jcb.200109030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pagano A, Crottet P, Prescianotto-Baschong C, Spiess M. In vitro formation of recycling vesicles from endosomes requires adaptor protein-1/clathrin and is regulated by rab4 and the connector rabaptin-5. Mol Biol Cell. 2004;15:4990–5000. doi: 10.1091/mbc.E04-04-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 25.Ballmer-Hofer K, Andersson AE, Ratcliffe LE, Berger P. Neuropilin-1 promotes vegfr-2 trafficking through rab11 vesicles thereby specifying signal output. Blood. 2011;118:816. doi: 10.1182/blood-2011-01-328773. [DOI] [PubMed] [Google Scholar]
- 26.Ewan LC, Jopling HM, Jia H, Mittar S, Bagherzadeh A, Howell GJ, et al. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic. 2006;7:1270–1282. doi: 10.1111/j.1600-0854.2006.00462.x. [DOI] [PubMed] [Google Scholar]
- 27.Gampel A, Moss L, Jones MC, Brunton V, Norman JC, Mellor H. Vegf regulates the mobilization of vegfr2/kdr from an intracellular endothelial storage compartment. Blood. 2006;108:2624–2631. doi: 10.1182/blood-2005-12-007484. [DOI] [PubMed] [Google Scholar]
- 28.Jopling HM, Odell AF, Hooper NM, Zachary IC, Walker JH, Ponnambalam S. Rab gtpase regulation of vegfr2 trafficking and signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:1119–1124. doi: 10.1161/ATVBAHA.109.186239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, et al. Vegf receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18:713–724. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakayama M, Nakayama A, van Lessen M, Yamamoto H, Hoffmann S, Drexler HC, et al. Spatial regulation of vegf receptor endocytosis in angiogenesis. Nat Cell Biol. 2013;15:249–260. doi: 10.1038/ncb2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manickam V, Tiwari A, Jung JJ, Bhattacharya R, Goel A, Mukhopadhyay D, et al. Regulation of vascular endothelial growth factor receptor 2 trafficking and angiogenesis by golgi localized t-snare syntaxin 6. Blood. 2011;117:1425–1435. doi: 10.1182/blood-2010-06-291690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Royen N, Piek JJ, Schaper W, Fulton WF. A critical review of clinical arteriogenesis research. J Am Coll Cardiol. 2009;55:17–25. doi: 10.1016/j.jacc.2009.06.058. [DOI] [PubMed] [Google Scholar]
- 33.Chalothorn D, Clayton JA, Zhang H, Pomp D, Faber JE. Collateral density, remodeling, and vegf-a expression differ widely between mouse strains. Physiol Genomics. 2007;30:179–191. doi: 10.1152/physiolgenomics.00047.2007. [DOI] [PubMed] [Google Scholar]
- 34.Cristofaro B, Shi Y, Faria M, Suchting S, Leroyer AS, Trindade A, et al. Dll4-notch signaling determines the formation of native arterial collateral networks and arterial function in mouse ischemia models. Development. 2013;140:1720–1729. doi: 10.1242/dev.092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buschmann I, Pries A, Styp-Rekowska B, Hillmeister P, Loufrani L, Henrion D, et al. Pulsatile shear and gja5 modulate arterial identity and remodeling events during flow-driven arteriogenesis. Development. 2010;137:2187–2196. doi: 10.1242/dev.045351. [DOI] [PubMed] [Google Scholar]
- 36.Fang JS, Angelov SN, Simon AM, Burt JM. Cx37 deletion enhances vascular growth and facilitates ischemic limb recovery. Am J Physiol Heart Circ Physiol. 2012;301:H1872–1881. doi: 10.1152/ajpheart.00683.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R. Nature. 2011;479:122–126. doi: 10.1038/nature10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tirziu D, Jaba IM, Yu P, Larrivee B, Coon BG, Cristofaro B, et al. Endothelial nuclear factor-kappab-dependent regulation of arteriogenesis and branching. Circulation. 2012;126:2589–2600. doi: 10.1161/CIRCULATIONAHA.112.119321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.