

Abstract
Human centromeres are defined by chromatin containing the histone H3 variant CENP-A assembled onto repetitive alphoid DNA sequences. By inducing rapid, complete degradation of endogenous CENP-A, we now demonstrate that once the first steps of centromere assembly have been completed in G1/S, continued CENP-A binding is not required for maintaining kinetochore attachment to centromeres or for centromere function in the next mitosis. Degradation of CENP-A prior to kinetochore assembly is found to block deposition of CENP-C and CENP-N, but not CENP-T, thereby producing defective kinetochores and failure of chromosome segregation. Without the continuing presence of CENP-A, CENP-B binding to alphoid DNA sequences becomes essential to preserve anchoring of CENP-C and the kinetochore to each centromere. Thus, there is a reciprocal interdependency of CENP-A chromatin and the underlying repetitive centromere DNA sequences bound by CENP-B in the maintenance of human chromosome segregation.
Graphical Abstract
INTRODUCTION
A correct balance of chromosome distribution following cell division is a pre-requisite for normal development. Indeed, whole-chromosome aneuploidy is responsible for many human genetic diseases and cancer. Centromeres are fundamental for chromosome inheritance serving as the unique chromosomal locus for the assembly of the kinetochore, a multi-subunit structure that attaches to spindle microtubules, and for centromeric cohesion prior to sister chromatid separation (Fukagawa & Earnshaw 2014).
From fission yeast to humans, centromeres are epigenetically identified by chromatin assembled with the histone H3 variant CENP-A, a key component of all centromeres (McKinley & Cheeseman 2015). CENP-A is required and essential to preserve centromere position (Fachinetti et al., 2013) by directing its self-replication at mitotic exit of every cell cycle (Jansen et al. 2007; Shelby et al. 1997) through the histone chaperone HJURP (Dunleavy et al. 2009; Foltz et al. 2009). Key determinants of this function are the CENP-A targeting domain (CATD)(Black et al. 2004) together with the amino- and carboxy-terminal tails that mediate kinetochore assembly (Fachinetti et al. 2013; Logsdon et al. 2015). Indeed, CENP-A has been reported to directly interact with several subunits of the Constitutive Centromere-Associated Network (CCAN) onto which the kinetochore is formed (Hori et al. 2008; Foltz et al. 2006; Anon 2008; Okada et al. 2006; Cheeseman & Desai 2008; Izuta et al. 2006; Carroll et al. 2009; Carroll et al. 2010; Fachinetti et al. 2013; Kato et al., 2013; Guse et al. 2011). However, whether CENP-A is necessary for maintaining the kinetochore and, consequently, required for proper chromosome segregation is unclear. Efforts to either reduce CENP-A levels via siRNA-mediated silencing over a two-day period or eliminate new CENP-A synthesis by gene disruption (seven days to achieve complete depletion) suggested that both the CCAN and the entire kinetochore complex would rapidly disassemble upon loss of anchoring to CENP-A (Régnier et al. 2005; Liu et al. 2006). Consequently, the pathways for kinetochore assembly were studied in cells by artificially tethering centromeric components to LacO arrays to bypass the CENP-A requirement (Gascoigne et al. 2011; Logsdon et al. 2015), which was thought to act as an essential connection between chromatin and kinetochore.
Since CENP-A is a long-lived protein (Smoak et al. 2016; Fachinetti et al. 2013) and is essential for maintenance of centromere identity, without ability to induce rapid depletion of it at known points in the cell cycle, effects on centromere maintenance and kinetochore function following CENP-A loss cannot be separated from CENP-A’s known role in specifying centromere position. Consequently, the importance of centromeric chromatin containing CENP-A in kinetochore maintenance and chromosome segregation has remained untested. In this manuscript, we now develop an approach to allow rapid (<1 hour), inducible degradation of the endogenous CENP-A at every stage of the cell cycle. While overall CENP-A is essential for centromere function, induced degradation of CENP-A demonstrates that after initial centromere assembly continued CENP-A binding to centromeric chromatin is dispensable for preserving centromere-bound kinetochores. In CENP-A-depleted centromeres maintenance of kinetochore-centromere attachment and high fidelity chromosome segregation in the next mitosis is dependent on CENP-B.
RESULTS
Rapid and complete depletion of the endogenous CENP-A in human cells
To produce cells whose centromeres were supported by an inducible degradable CENP-A, genome editing was used to add amino-terminal EYFP and auxin inducible degron (AID) tags to endogenous CENP-A to facilitate its rapid and complete depletion in human cells (Nishimura et al. 2009; Holland et al. 2012). We did this by 1) modifying both endogenous CENP-A alleles with amino-terminal EA (EYFP-AID) tags in a diploid, non-transformed human RPE-1 cell line or 2) tagging one allele and inactivating the other in a pseudo-diploid colorectal human cancer DLD-1 cell line (Fig. 1A and Supplementary Fig. S1A–B). Additionally, in both CENP-AEA/EA and CENP-A−/EA cell lines, a gene encoding the plant E3 ubiquitin ligase osTIR1 was stably integrated to enable rapid degradation of AID-CENP-A upon addition of the synthetic auxin indole-3-acetic acid (IAA). Fusion of the AID and EYFP tags to one or both CENP-A allele(s) did not interfere with CENP-A or centromere function, as long-term maintenance of cell viability was found in CENP-A-deficient RPE-1 (CENP-A−/−) cells when rescued with a EYFP-AIDCENP-A transgene (Supplementary Fig. S1C–E).
Figure 1. Complete and rapid removal of the centromere epigenetic mark CENP-A in human cells.

(A) Schematic of the TALEN-mediated genome editing to endogenously tag CENP-A with AID (A) and EYFP (E) in the indicated cell lines. Position of exons, introns and start/stop codons are indicated. (B) Immuno-blot of RPE-1 cells following treatment with Auxin (IAA) at the indicated time points (hours). α-tubulin was used as a loading control. A schematic of the soluble CENP-A-associated factors is also shown. (C) Representative immunofluorescence images on DLD-1 cells showing CENP-A depletion after 24 hours treatment with IAA. CENP-B was used to mark centromere position. (D) Quantification of the experiment in C by using an antibody against CENP-A or by monitoring EYFP intensity. Unpaired t test: *** p < 0.0001. (E) Degradation kinetics of CENP-A in RPE-1 cells with or without (+/-) IAA treatment measured by EYFP intensity during live cell imaging. IAA was added at the microscope stage. Σn = 10 cells (F) Representative images show immuno-fluorescence on RPE-1 cells expressing the cell cycle indicator FUCCI. Red (G1), yellow (S) or green (S/G2) circles indicate the cell cycle position +/− IAA treatment. A schematic of the experimental designed is also shown. (G) Images of representative crystal violet–stained colonies from the colony formation assay +/− IAA treatment in RPE-1 cells. (H) Cell counting experiment on DLD-1 CENP-A−/EA cells +/− IAA treatment. IAA was added at day 0 and kept for a maximum of 7 days. Error bars represent the SEM of five independent experiments. Unpaired t test: ** p = 0.0026. See also Supplementary Fig. S1. Scale bars = 5 um.
Auxin addition led to complete depletion of all endogenous CENP-A protein, measured using three complementary approaches: immunoblotting (using anti - CENP-A and -GFP antibodies; Fig. 1B and Supplementary Fig. S1F), immunofluorescence (using anti-GFP and an anti-CENP-A antibody - Fig. 1C, D and Supplementary Fig. S1G) and immunoprecipitation (following chromatin precipitation with an anti-GFP antibody; Supplementary Fig. S1H–J). Loss of CENP-A appeared complete at individual centromeres [we have previously demonstrated that our immunofluorescence approach could identify as little as 1 molecule (Fachinetti et al., 2013), starting from the initial 400 molecules per human centromere (Bodor et al. 2014)]. Importantly, complete degradation of CENP-A was also observed (by immunofluorescence using a CENP-A antibody recognizing a.a. 3–19) on C-terminal-tagged CENP-A (CENP-A3HA-AID), demonstrating that the entire protein was removed from centromeres (Supplementary Fig. S1G).
CENP-A depletion occurred rapidly (with first-order kinetics) and to completion within 50 min, with a half-life of 9 min; Fig. 1E and Supplementary Fig. S1K and Movie S1) in all examined cell lines and at every stage of the cell cycle [with cell cycle position visualized using the fluorescence cell cycle indicator (FUCCI) reporter system (Sakaue-Sawano et al. 2008) or by arresting cells in mitosis with a microtubule depolymerizing drug; Fig. 1F and Supplementary Fig. S1L). CENP-A degradation also triggered rapid, partial degradation of its pre-nucleosomal assembly factor HJURP (Foltz et al. 2009; Dunleavy et al. 2009) (Fig. 1B), in agreement with the reciprocal stabilization of the CENP-ACID/HJURPCAL1 complex observed in flies (Bade et al. 2014). As expected, CENP-A depletion led to cell lethality in both RPE-1 and DLD-1 cells as observed by a colony forming assay (a two weeks growth assay; Fig. 1G and Supplementary Fig. S1M). However, surprisingly, cell death was not immediate, but initiated after the second cell cycle, presumably from rampant chromosome mis-segregation (Fig. 1 H).
CENP-A is dispensable for kinetochore maintenance and chromosome segregation during mitosis, but essential for overall centromere function
To address if CENP-A-depleted centromeres retained their function in mediating chromosome attachment to mitotic spindle microtubules, we followed chromosome segregation by live cell imaging (after stable insertion of mCherry-H2B) in cells that underwent a first or second round of mitosis without CENP-A (2 hours or 24 hours of IAA treatment, respectively; Fig. 2A). Remarkably, no significant increase in chromosome segregation defects was observed in RPE-1 CENP-AEA/EA cells with auxin-induced degradation of CENP-A in the 2 hours preceding the first mitosis (2h IAA) (Fig. 2B). On the other hand, after 24 hours (about one full cell cycle) without CENP-A, cells that entered mitosis had severe accumulation of mis-aligned chromosomes, extended mitotic duration, and subsequent formation of micronuclei at mitotic exit (Fig. 2B, C). Importantly, the frequency of mis-segregated chromosomes was dependent on when in the cycle CENP-A was depleted. CENP-A depletion during G1 produced the highest error frequency in the first mitosis after CENP-A degradation, with a much lower error rate when CENP-A was depleted from S-phase cells and an even lower rate when depleted in G2 (Fig. 2D and Supplementary Fig. S2A, B).
Figure 2. CENP-A is dispensable for centromere/kinetochore maintenance and chromosome segregation once centromere is assembled.

(A) Schematic of the experiments shown in B–C. (B) Bar graph shows the percentage of chromosome mis-segregation events by live cell imaging in non-treated conditions or following IAA treatment for 2 or 24 hours, respectively. Error bars represent the SEM of three independent experiments. Individual Σn = ~35 cells. Unpaired t test: ** p = 0.0099 and 0.0065. (C) Each individual point represents a single cell. Time in mitosis was defined as the period from NEBD to chromosome decondensation. Error bars represent the SEM of three independent experiments. Unpaired t test: **** p < 0.0001. (D) Bar graph shows the frequency of chromosome mis-segregation (micronuclei and mis-aligned chromosomes, 2 hour IAA treatment condition) versus the cell cycle phase in which CENP-A was depleted (determined by measuring the time required for the cells to enter into mitosis in asynchronous population, using cell co-expressing PCNAGFP as an indicator of the cell cycle or by synchronizing cell in G1 by adding IAA either at t= 0 hr or at t = 9 hr) in DLD-1 cells. Unpaired t test: * p = 0.0142, ** p = 0.0024. (E) Schematic of the experiments shown in F–G. (F) Bar graphs showing centromere intensities of CENP-A, CENP-B, CENP-C, CENP-I and CENP-T for the indicated cell line following IAA treatment. Values represent the mean of six independent experiments combining analysis performed in RPE-1 and DLD-1 cells. Individual Σn = ~30 cells, Σn = 25 centromeres per cell. Error bars represent the SEM (standard error of the mean). Unpaired t test: *** p < 0.0001, ** p < 0.07, * p < 0.05. (G) Box & whisker plots of Dsn1 or Hec1 intensities at the centromere measured on metaphase spreads. Unpaired t test: * p = 0.017, *** p = 0.0005, **** p < 0.0001. See also Supplementary Fig S2
We then investigated the consequences of CENP-A depletion on binding of centromere/kinetochore components and on the kinetochore capture of spindle microtubules. We measured the binding stability of several components of the CCAN (including CENP-B, CENP-C, CENP-T and CENP-I) in interphase or of the mature kinetochore in mitosis (including Dsn1, a subunit of the Mis12 complex, and Hec1, a subunit of the Ndc80 complex) following CENP-A depletion for either 2 or 4 hours or for an entire cell cycle (24 hours) (Fig. 2E–G). CENP-A depletion for 4 hours resulted in only a minor reduction in interphase on all measured centromere-bound components except CENP-C (Fig. 2F), which underwent an immediate, substantial (~70%) reduction. CENP-T and CENP-I levels at centromeres significantly decreased in cells only following an entire cell cycle without CENP-A (Fig. 2F, G). Centromere-bound CENP-B also slightly diminished following CENP-A depletion (Fig. 2F), consistent with direct stabilization of CENP-B via the CENP-A amino terminal tail (Fachinetti et al. 2013; Fachinetti et al. 2015).
Similarly, degradation of CENP-A within 2 hours of mitotic entry left binding of components of the mature mitotic kinetochore, Dsn1 and Hec1, almost unchanged. In cells entering mitosis 24 hours (i.e., one cell cycle) after CENP-A depletion both Dsn1 and Hec1 were reduced (by ~50% - Fig. 2G). Microtubules were still stably bound to kinetochores in the absence of CENP-A (4 hr or 24 hr after addition of IAA), as observed by immunofluorescence following cold treatment to disassemble all but kinetochore microtubules (Supplementary Fig. S2C).
Altogether, these findings revealed a dual response to CENP-A depletion immediately prior cell division: it is dispensable for kinetochore maintenance and chromosome segregation in the first mitosis, but essential to preserve centromere function in a subsequent cell cycle. The reduced binding stability of centromere/kinetochore components following CENP-A depletion for 24 hours is likely to be the primary deficits that lead to severe chromosome mis-segregation in CENP-A-depleted centromeres.
CENP-A is critical for initial assembly of centromeric components CENP-C and CENP-N, but not CENP-T
We next tested if the severe chromosome segregation defects in mitosis following an entire cell cycle without CENP-A (24 hours treatment) could be the result of failure of centromere assembly. Firstly, we tested the dependency of loading of new molecules of CENP-C — known to directly interact with CENP-A (Carroll et al. 2010; Kato et al., 2013; Falk et al. 2015) and to be recruited at ectopic sites by CENP-A/HJURP [(Logsdon et al. 2015; Tachiwana et al. 2015)] — on the continued presence of CENP-A at native centromeres. For this, we tagged a single CENP-C allele with RFP-AID to follow its localization at CENP-A-containing or CENP-A-depleted centromeres (Fig. 3A–C). After IAA treatment to induce CENP-C or both CENP-C/CENP-A degradation (red bars, Fig. 3C) and IAA removal to permit re-accumulation of CENP-C and/or CENP-A, respectively (see immunoblot, Supplementary Fig. S2D), new CENP-CmRFP-AID deposition at centromeres was measured in cells released from G1 cell cycle arrest. Despite continued presence of CENP-B at centromeric loci following CENP-A degradation, CENP-CmRFP-AID failed to re-localize to centromeres in CENP-A-depleted cells following IAA removal (plain versus striped light blue bars, Fig. 3B, C and Supplementary Fig. S2E). Taken together, these findings reveal a requirement for CENP-A for new CENP-C loading (Fig. 2F) at native centromeres.
Figure 3. CENP-A is required to regulate early steps of centromere assembly, such as for CENP-C and CENP-N, but not CENP-T.

(A) Schematic of the experiments shown in B and C. (B) Representative immunofluorescence images show de novo CENP-CAID-mRFP loading at centromeres. (C) Bar graphs showing centromere intensities of CENP-CAID-mRFP in the indicated cell lines. Error bars represent the SEM of three independent experiments. Individual Σn = ~30 cells, Σn = 25 centromeres for cell. Unpaired t test: *** p < 0.0001. (D) Schematics of the experiments shown in E–G. (E) Representative immunofluorescence images show de novo CENP-N3HA-SNAP loading at centromeres. HA antibody was used to identify unlabeled CENP-N. (F–G) Bar graphs showing centromere intensities of CENP-N3HA-SNAP or CENP-T3HA-SNAP respectively in the indicated cell lines. Values represent the mean of three independent experiments. Error bars represent the SEM. Individual Σn = ~30 cells, Σn = 25 centromeres for cell. Unpaired t test: * p = 0.01, ** p = 0.0028, *** p = 0.0003. See also Supplementary Fig S2. Scale bars = 5 um.
Moreover, we found that deposition of new CENP-C occurred within a window of a few hours after mitotic exit [determined using live cell imaging on CENP-C+/AE cells released from mitotic block (nocodazole) or S-phase block (thymidine), or in untreated conditions (Supplementary Fig. S2F–H)]. Therefore, we conclude that CENP-C loading takes place only a few hours after CENP-A deposition (Supplementary Fig. S2I) and is dependent on CENP-A.
We then measured CENP-N and CENP-T stability and their deposition in CENP-A-depleted cells using the SNAP-tag method (Bodor et al. 2012) (Fig. 3D–G). Both CENP-N and CENP-T are essential components for centromere function (Foltz et al. 2006; McKinley et al. 2015), and their depletion causes a strong mitotic arrest within 2 hours of IAA addition (Supplementary Fig. S2J and Wood et al., 2016). Surprisingly, following CENP-A depletion and release into S-phase, pre-deposited CENP-N3HA-SNAP (labeled by covalent linkage to TMR) remained stably bound to centromeres (Fig. 3E, F). In contrast, new CENP-N failed to deposit at CENP-A-depleted centromeres (Fig. 3E, F) as observed by initial addition of BTP (a non-fluorescent substrate used to block subsequent visualization of all pre-deposited SNAP-tagged molecules) followed by new protein synthesis labeled by the SNAP substrate TMR. Pre-deposited centromeric CENP-T was reduced following CENP-A depletion, but its deposition was not prevented in the absence of CENP-A (Fig. 3G). Taken together, these results show that CENP-A itself is required for new assembly of CENP-C and CENP-N, but not of CENP-T. However, CENP-A is not necessary in the short-term for CENP-N maintenance once already assembled at centromeres.
CENP-B binding to alphoid DNA is necessary and sufficient for kinetochore anchoring via CENP-C on CENP-A-depleted centromeres
Previously, we demonstrated that the Y chromosome recruits a reduced level of CENP-C and mis-segregates at a higher rate compared to the X chromosome or to any of the autosomes (Fachinetti et al. 2015). We suggested that this inherent instability could be due to the complete absence of the Y chromosome’s centromeric sequences of CENP-B binding boxes (Earnshaw et al. 1989; Miga et al. 2014), which represent the sites for the centromeric DNA binding protein CENP-B. Indeed, alone among the human chromosomes, the Y does not have the CENP-B-mediated backup pathway for maintenance of CENP-C at its centromere (Fachinetti et al. 2015).
Consistent with this, the first mitosis following CENP-A depletion in male DLD-1 cells was accompanied by an overall increase in chromosome segregation defects compared to RPE-1 cells (Supplementary Fig. S3A). Detailed analysis revealed that, on average, in most cells in the first mitosis after CENP-A degradation one chromosome failed (or delayed) in alignment to metaphase, while two or more were mis-aligned in the second mitosis after removal of CENP-A (Supplementary Fig. S3B). FISH analysis revealed that depletion of CENP-A resulted in a significant accumulation of micronuclei (MN) carrying the Y chromosome (~36% of MN+Y; 1 cell out of 7 cells carried Y chromosome in MN; Fig. 4A) accompanied by the loss of centromere-bound CENP-C as observed by both IF-FISH (Fig. 4B) and Chromatin Immuno-Precipitation (ChIP) analysis on CENP-C (Supplementary Fig. S3C). [In agreement with our previous findings (Fachinetti et al. 2015), even in cells with normal CENP-A levels there was already a selective reduction of CENP-C binding to the Y centromere compared to other centromeric region.] These findings suggest that the majority of single mis-aligned chromosomes in the first mitosis following CENP-A depletion are the Y chromosome.
Figure 4. CENP-B is sufficient and essential to maintain kinetochore assembly and consequently faithful chromosome segregation in the absence of CENP-A.

(A) (left) Representative images show a micronucleus containing the Y chromosome by dual FISH analysis. (Right) Graphs show the frequency of micronuclei formation (X axis) versus the frequency of a micronucleus containing the chromosome Y or chromosome 4 +/− IAA treatment for 24 hours. n = ~400 cells. Unpaired t test: * p = 0.01, ** p = 0.0094 (B) Representative images of an immuno-fluorescence coupled with FISH showed CENP-C binding to centromere on the Y or X chromosome +/− IAA treatment for 24 hours. Scale bar = 5 μm. (C) Schematic of the experimental design shown in D-G. (D) Immuno-blot shows depletion of endogenous CENP-B using the CRISPR technology. α-tubulin was used as a loading control. (E) Cell counting experiment on RPE-1 +/− IAA treatment and/or CENP-B gene. IAA was added at day 0 and kept for a maximum of 7 days. Error bars represent the SEM of four independent experiments. Unpaired t test: ** p = 0.0043; 0.0016. (F) Representative immuno-fluorescence FISH to measure CENP-C levels following CENP-A depletion (by IAA) in CENP-B depleted cells. A FISH probe against CENP-B boxes was used to mark centromere position. (G) Quantification of the experiment show in D. Each dot represents an average of 25 centromeres in a single cell. Unpaired t test: *** p < 0.0001 (H) Schematic of the experiments shown in I–K. (I) Bar graph shows the percentage of chromosome mis-segregation events observed by live cell imaging following siRNA depletion of GAPDH or CENP-B and IAA treatment for 2 hours, respectively. Error bars represent the SEM of three independent experiments. Individual Σn = ~60 cells. Unpaired t test: * p = 0.02, ** p = 0.0068 (J) Bar graph shows the number (1 or >2) of mis-aligned chromosomes in percentage from analysis in E. Error bars represent the SEM of three independent experiments. Unpaired t test: ** p = 0.0093. (K) Scatter plot graph shows the time in mitosis (from NEBD to chromosome decondensation). Each individual point represents a single cell. Error bars represent the SEM of three independent experiments. Unpaired t test: *** p < 0.0001. (L) Box & whisker plots of Dsn1 and Hec1 intensities at the centromere measured on metaphase spreads. Unpaired t test: ** p = 0.002, *** p = 0.0005. See also Supplementary Fig S3 and S4. Scale bars = 5 μm.
The preceding results imply that CENP-C-mediated kinetochore assembly stabilized by its binding to CENP-B at centromeric sequences is sufficient to mediate continued kinetochore function and chromosome segregation in the complete absence of CENP-A. To further test this hypothesis, both CENP-B alleles in the RPE-1 CENP-AAE/AE cell line were disrupted using CRISPR/Cas9-mediated genome editing (Fig. 4C, D). Without CENP-B, cell lethality was drastically accelerated upon depletion of CENP-A (compare triangle-red to square-green lines, Fig. 4E). Co-depletion of CENP-A and CENP-B abolished centromeric localization of CENP-C and CENP-T (Fig. 4F, G and Supplementary Fig. S3D). Accordingly, normal CENP-C levels could be restored by expression of a siRNA-resistant, full-length CENP-B rescue construct, but not a mutant lacking its DNA binding domain (Yoda et al. 1992) and therefore its centromeric localization (Supplementary Fig. S3E–G). Short-term reduction of CENP-B levels in CENP-A-/EA DLD-1 (by siRNA - Supplementary Fig. S3E) caused severe chromosome segregation errors beginning in the first mitosis following CENP-A depletion, with cells accumulating significantly more than 1 mis-aligned chromosome, developing micronuclei, and displaying an extended mitotic duration (Fig. 4H–K and Supplementary Movie S2 and S3).
Chromosome segregation failure following CENP-A and CENP-B depletion correlated with rapid dissociation of the kinetochore complex Mis12 (measured by Dsn1) and the Ndc80 complex (measured by Hec1 binding; Fig. 4L and Supplementary Fig. S3H, I). Altogether, these findings indicate that once the first steps of kinetochore assembly have been completed, centromere and kinetochore components of all chromosomes except the Y are (at least partially) stably maintained in the first mitosis following CENP-A depletion. Furthermore, binding of CENP-B to centromeric DNA is necessary for preserving centromeric CENP-C and faithful chromosome segregation.
Since it has been previously reported that CENP-C is essential for maintaining centromere function (Fukagawa et al. 1999; McKinley et al. 2015) and co-depletion of CENP-A and CENP-B led to CENP-C loss and immediate chromosome mis-segregation (Fig. 4), we next tested whether CENP-C was required for retention of mitotic centromere function on pre-assembled kinetochores. In order to obtain the precise temporal control of inducible CENP-C degradation [missing in previously reported systems (Falk et al. 2015; McKinley et al. 2015)], one or both alleles of CENP-C were tagged at the C-terminus with AID-EYFP in wild-type DLD-1 cells. Addition of IAA rapidly depleted CENP-C (Supplementary Fig. S4A–C), suppressed long-term viability (Supplementary Fig. S4D), and induced rapid cell death (Supplementary Fig. S4E). In contrast to CENP-A degradation alone but similar to co-depletion of both CENP-A and CENP-B, depletion of CENP-C resulted in immediate chromosome segregation failure during the first mitosis (Supplementary Fig. S4F–H) and rapid de-stabilization of CENP-T and the kinetochore complexes Mis12 and (partially) Ndc80 (Supplementary Fig. S4I). This latter finding extends the previous report of a direct stabilization of CENP-T via CENP-C (Klare et al. 2015). Altogether, these results demonstrated that CENP-C is a key component for direct maintenance of kinetochore architecture and faithful chromosome segregation.
DISCUSSION
We propose a model (Fig. 5) in which centromere identity is initially maintained via the CENP-ACATD/HJURP interaction at mitotic exit (Black et al. 2007; Fachinetti et al. 2013; Foltz et al. 2009; Dunleavy et al. 2009). CENP-A deposition is essential to recruit CENP-C in mid G1 and CENP-N in S-phase (Fig. 3A–F and Supplementary Fig. S2F–I; Hellwig et al. 2011). Accordingly, removal of CENP-A in G1 led to an increase in the frequency of chromosome mis-segregation beginning in the next mitosis (Fig. 2D). Further, CENP-B binding to α-satellite DNAs in the proximity of CENP-A chromatin enhanced centromeric CENP-C stability (Fig. 4 and Supplementary Fig. S3G). The CENP-A/CENP-C complex is then required to sustain centromeric CENP-T assembly [which normally occurs in late S-phase - (Prendergast et al. 2011)] through interdependent interactions among the CCAN subunits (McKinley et al. 2015; Weir et al. 2016). Our data also suggest that CENP-N and CENP-T, both immediately required for cell division (Wood et al. 2016) (Supplementary Fig. S2J), have to cooperate with CENP-C (or at least with its CENP-B-bound fraction) to nucleate a functional kinetochore, in agreement with previous reports (McKinley et al. 2015). Nevertheless, once these centromeric components are assembled, CENP-A is no longer essential for kinetochore tethering to the centromere nor its function in spindle microtubule capture, with the CCAN/kinetochore retained at each centromere (Fig. 2 and Supplementary Fig. S2C). Thus, while CENP-A depletion after CENP-C/N recruitment alters kinetochore composition in the proximal mitosis, it does not disrupt centromere/kinetochore function in chromosome segregation in that mitosis (Fig. 2F, G).
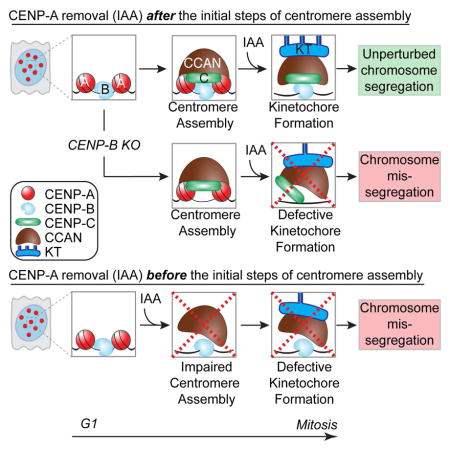
Figure 5. Model of centromere function mediated by centromeric chromatin and DNA sequences.

At exit of mitosis, centromeric chromatin replication and identity is mediated by CENP-A (in red) deposition via interaction with HJURP. CENP-A then mediates the assembly of CENP-C (in green) in mid-G1 followed by CENP-N/L (in orange) during S-phase. These steps might be interconnected. At this point, CENP-A becomes dispensable for mitotic centromere function as long as CENP-B (in light blue) is stably bound to centromeric sequences to support CENP-C binding. Assembly of the other subunits of the CCAN, such as CENP-T/W (in yellow) and HIKM (in brown), allows the full recruitment of the kinetochore complex (in grey) required to mediate centromere function. In summary, we propose that the kinetochore is tethered to the centromere through a dual linkage of CENP-A chromatin and CENP-B-bound DNA sequences, as the two major links from the DNA to the kinetochore to mediate successful chromosome segregation.
These results support an essential role for CENP-A before mitosis in mediating the initial steps of centromere assembly, but in contrast to CENP-C/-T/-N, does not play an active role in centromere-dependent chromosome movement (Fig. 5). In support of this model, CENP-C and CENP-T have been reported to be sufficient for kinetochore assembly at ectopic loci (Gascoigne et al. 2011). Furthermore, we now provide evidence that CENP-B binding to α-satellite DNA, previously only proposed to support centromere function (Fachinetti et al. 2015), is indeed sufficient (and essential) for maintenance of a pre-assembled kinetochore and to support chromosome segregation through stabilization of CENP-C (Fig. 4).
These findings demonstrate a reciprocal, but non-exclusive, interdependency on centromeric chromatin (marked by CENP-A) and specific centromeric sequences (bound by CENP-B) for tethering the kinetochore complex to centromeres via CENP-C stabilization throughout mitosis. They also implicate CENP-B as an important contributor of the CCAN complex to modulate centromere function and strength, which may have implications for karyotypic evolution (Chmátal et al. 2014) due to the variations in the frequency of CENP-B boxes between the centromeres of each mammalian chromosome.
EXPERIMENTAL PROCEDURES
Constructs
osTIR19myc, H2BmRFP, CENP-N3HA-SNAP, CENP-T3HA-SNAP and PCNAGFP were cloned into a pBabe-based vector for retrovirus generation. mCherryH2B was cloned into a pSMPUW-based vector for lentivirus generation The FUCCI system was integrated by lentiviral insertion and clones were selected by FACS. For tetracycline-inducible expression, siRNA resistant CENP-B-GFP or ΔNH2CENP-B-GFP was cloned into a pcDNA5/FRT/TO-based vector (Invitrogen).
Cell culture conditions
Cells were maintained at 37°C in a 5% CO2 atmosphere. Flp-In TRex-DLD-1 were grown in Dulbecco's modified medium (DMEM) containing 10% tetracycline free fetal bovine serum (GE Healthcare), 100 U/ml penicillin, 100 U/ml streptomycin and 2 mM L-glutamine, while hTERT RPE-1 cells were maintained in DMEM:F12 medium containing 10% tetracycline free fetal bovine serum (Pan Biotech), 0.348% sodium bicarbonate, 100 U/ml penicillin, 100 U/ml streptomycin and 2 mM L-glutamine. IAA (SIGMA, I5148) was used at 500 uM, colcemid (Roche) and Nocodazole (SIGMA) was used at 0.1 mg/ml, thymidine at 2 mM, Doxycycline (SIGMA) at 1 mg/ml and Palbociclib at 1 uM. Cold treatment experiment to determine kinetochore/microtubules stability was performed for 15’ on ice.
Generation of stable cell lines
Stable, isogenic cell lines expressing CENP-B-GFP FL or ΔN were generated using the FRT/Flp-mediated recombination system as described previously (Fachinetti et al. 2013). The different transgenes used in this study were introduced by retroviral delivery as described previously (Fachinetti et al. 2013). Stable integration was selected with 5 ug/ml puromycin or 10 ug/ml blasticidin S and single clones isolated using fluorescence activated cell sorting (FACS Vantage; Becton Dickinson, Franklin Lakes, NJ).
siRNA, SNAP-tagging, clonogenic colony assay and cell counting experiments
siRNAs were introduced using Lipofectamine RNAiMax (Invitrogen). A pool of four siRNAs directed against CENP-B and GAPDH (Fachinetti et al. 2013) was purchased from Dharmacon. SNAP labeling was conducted as described previously (Jansen et al. 2007). Clonogenic colony assays were done as described (Fachinetti et al. 2013). For the counting experiment, cells were plated at 1*105 cells/ml in a 6 well plate. After 24 hours Auxin was added to the medium. Cells were then counted and divided every other day for 7 days.
Gene targeting
TALENs were assembled using the Golden Gate cloning strategy and library as described previously (Fachinetti et al. 2015) and cloned into a modified version of pcDNA3.1 (Invitrogen) containing also the Fok I endonuclease domain as previously described (Fachinetti et al. 2015). TALENs were designed to the N-terminal region of CENP-A gene: GTCATGGGCCCGCGCC and GGCCCCGAGGAGGCGCA. RPE-1 and DLD-1 cells were co-transfected with the TALEN expression vectors and the donor cassette (containing the two homology arms for CENP-A N-terminal region and the AID and EYFP gene) by nucleofection (Lonza) and positive clones were selected by FACS. CENP-C targeting was done as previously described (Fachinetti et al. 2015). CENP-B gene was deleted as described before (Fachinetti et al. 2015).
Surveyor Nuclease Assay
A surveyor nuclease assay was performed as previously described (Fachinetti et al. 2015). Briefly, 1*106 U2OS cells were transfected with 1000 ng of the sgRNA/Cas9 expression vector by nucleofection (Lonza) using program X-001 and a nucleofection buffer (100 mM KH2PO4, 15 mM NaHCO3, 12 mM MgCl2, 6 H20, 8 mM ATP, 2 mM glucose, pH 7.4). 48 hours following transfection, genomic DNA was isolated using the quick g-DNA miniprep isolation kit (Zymo) and PCR was performed using Q5 polymerase (NEB) with CENP-A specific primers sitting just outside of the target sequence (forward primer: 5’ GACTTCTGCCAAGCACCG 3’; reverse primer: 5’ GCCTCGGTTTTCTCCTCTTC 3’). PCR products were denatured, annealed, treated with the surveyor nuclease (Transgenomics), separated on a 10% TBE polyacrylamide gel and visualized by ethidium bromide staining.
Immunoblotting
For immunoblot analysis protein samples were separated by SDS-PAGE, transferred onto nitrocellulose membranes (BioRad) and then probed with the following antibodies: DM1A (α-tubulin, 1:5000), CENP-A (Cell Signaling, 1:1000), GFP (cell signaling, 1:1000), HJURP [Covance, 1:1000(Foltz et al. 2009)], CENP-B (Abcam and Upstate, 1:1000), GAPDH (Abcam, 1:10000), CENP-C (a gift from Iain Cheeseman, MIT, Boston and Ben Black, UPENN, Philadelphia), c-Myc (SIGMA, 1:1000) and H4 (Abcam, 1:250).
Immunofluorescence, chromosome spreads, live-cell microscopy and IF-FISH
Cells were fixed in 4% formaldehyde at room temperature or in methanol at −20°C for 10 min. Incubations with primary antibodies were conducted in blocking buffer for 1 hr at room temperature using the following antibodies: CENP-A (Abcam, 1:1500), CENP-C (MBL, 1:1000), CENP-B (Abcam, 1:1000), ACA (Antibodies Inc, 1:500), Hec1 (Abcam, 1:1000), Dsn1 (1:1000, a gift from A. Desai, Ludwig, San Diego), CENP-I (a gift from Song-Tao Liu, University of Toledo, OH), DM1A (α-tubulin, 1:2000), CENP-T (Covance, 1:5000) and HA-11 (Covance, 1:1000). Immunofluorescence on chromosome spreads was done as described previously (Fachinetti et al. 2015). Immunofluorescence images were collected using a Deltavision Core system (Applied Precision). For live cell imaging, cells were plated on high optical quality plastic slides (IBIDI) and imaged using a Deltavision Core system (Applied Precision) or spinning disk with deconvolution and denoising (Nikon). For IF-FISH we follow the IF protocol followed by the FISH protocol (see below).
FISH experiment
Cells were fixed in Carnoy’s Fixative (Meth-Acetic Acid 3:1) for 15 minutes at room temperature, rinsed in 80% ethanol and air dried for 5 minutes. Probe mixtures (MetaSystems) were applied and sealed with a coverslip. Slides were denatured at 75°C for 2 minutes and incubated at 37°C overnight in a humidified chamber. Slides were washed with 0.4X SCC at 72°C for 2 minutes, 4X SCC, 0.1% Tween-20 at room temperature for 30 sec, and rinsed with PBS. Slides were incubated with DAPI solution for 10 minutes before mounting in anti-fade reagent.
Centromere quantification
Interphase centromere quantifications: quantification of centromere signal intensity on interphase cells was done manually as described (Fachinetti et al. 2013) or using an automated system (Fachinetti et al. 2015). Briefly for the manual quantification, un-deconvolved 2D maximum intensity projections were saved as un-scaled 16-bit TIFF images and signal intensities determined using MetaMorph (Molecular Devices). A 15 x 15 pixel circle was drawn around a centromere (marked by ACA or CENP-B staining) and an identical circle drawn adjacent to the structure (background). The integrated signal intensity of each individual centromere was calculated by subtracting the fluorescence intensity of the background from the intensity of the adjacent centromere. 25 centromeres were averaged to provide the average fluorescence intensity for each individual cell and more than 30 cells were quantified per experiment.
Chromatin extraction and affinity purification
Nuclei from 1*109 DLD-1 cells were prepared as previously described (Foltz et al. 2006), except for reducing the NaCl to 150 mM in the wash buffer. Chromatin was digested at room temperature using 140 units/ml of micrococcal nuclease (cat# 10107921001; Roche, Indianapolis, IN) for 20 minutes to produce mononucleosomes and short oligonucleosomes of up to three nucleosomes or for 35 minutes to produce a pool of mono-nucleosomes. Following micrococcal nuclease treatment, extracts were supplemented with 5 mM EGTA and 0.05% NP40 and centrifuged at 10,000g for 15 min at 4 °C. For affinity purification, GFP-tagged chromatin was immunoprecipitated using mouse anti-GFP antibody (clones 19C8 and 19F7, Monoclonal Antibody Core Facility at Memorial Sloan-Kettering Cancer Center, New York) coupled to Dynabeads M-270 Epoxy (cat# 14301, Life Technologies, Grand Island, NY). Chromatin extracts were incubated with antibody-bound beads for 16 h at 4 °C. Bound complexes were washed once in buffer A (20 mM HEPES at pH 7.7, 20 mM KCl, 0.4 mM EDTA and 0.4 mM DTT), once in buffer A with 300 mM KCl and finally twice in buffer A with 300 mM KCl, 1 mM DTT and 0.1% Tween 20.
DNA extraction
Following elution of the chromatin from the beads, Proteinase K (100 μg/ml) was added and samples were incubated for 2 h at 55 °C. DNA was purified from proteinase K treated samples using a DNA purification kit following the manufacturer instructions (Promega, Madison, USA) and was subsequently analyzed by running a 2% low melting agarose (APEX) gel.
Chromatin Immuno-precipitation and qPCR analysis
Cells were crosslinked in 0,75% formaldehyde for 10 minutes at room temperature. The reaction was stopped by adding 125mM glycine for 5 minutes at room temperature. Chromatin was fragmented by sonication in a ChIP buffer (50mM Hepes-KOH pH 7.5, 140mM NaCl, 1mM EDTA pH 8, 1% Triton X-100, 0,1% Sodium Deoxycholate, 0,1% SDS). The soluble chromatin was diluted 1:10 with RIPA buffer (50mM TRIS HCl pH 7.6, 150mM NaCl, 1mM EDTA, 1% NP-40, 1% Sodium Deoxycholate, 0,1% SDS, 1% Protease Inhibitor), pre-cleared with Dynabeads Protein G (Thermo Fisher Scientific) and immunoprecipitated overnight at 4°C with anti-CENP-C (MBL). Chromatin was then washed once in Low Salt Wash Buffer (0,1% SDS, 1% Triton X-100, 2mM EDTA, 50mM Tris HCl pH 7.6), once in High Salt Wash Buffer (0,1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris HCl pH 7.6, 500mM NaCl) and once in LiCl Wash Buffer (0,25M LiCl, 1% NP-40, 1% Sodium Deoxycholate, 1mM EDTA, 10mM Tris HCl). Samples were eluted with Elution Buffer at 30°C for 15 minutes and then incubated at 65°C overnight with 5M NaCl. Then samples were incubated with 10mg/ml RNase A and 20mg/ml Proteinase K for 1 hour at 45°C and DNA was purified by Phenol-Chloroform. The recovered DNA and the soluble chromatin (input) were quantified by qPCR using the Light Cycler 480 (Roche). The following primers were used to amplify Y centromere (Fw: TCCTTTTCCACAATAGACGTCA; Rev: GGAAGTATCTTCCCTTAAAAGCTATG), Telomere (Fw: ACACTAAGGTTTGGGTTTGGGTTTGGGTTTGGGTTAGTGT; Rev: TGTTAGGTATCCCTATCCCTATCCCTATCCCTATCCCTAACA), satellite 2 (Fw: CTGCACTACCTGAAGAGGAC; Rev: GATGGTTCAACACTCTTACA), 17 centromere (Fw: CAACTCCCAGAGTTTCACATTGC; Rev: GGAAACTGCTCTTTGAAAAGGAACC), alpha satellite (Fw: TCATTCCCACAAACTGCGTTG; Rev: TCCAACGAAGGCCACAAGA).
Statistical methods
Statistical analysis of all the graphs was performed using the Unpaired t test in Prism 6 in which the follow parameters were consider: P value, P value summary, significant difference (P < 0.05), two-tailed P value and “t, df” values.
Supplementary Material
Acknowledgments
The authors would like to thank B.E. Black (University of Pennsylvania, Philadelphia), C. Bartocci (Institut Curie, Paris), Dong Hyun Kim (Ludwig, La Jolla), Amira Abdullah (Ludwig, La Jolla) and Vincent Fraiser (Institut Curie, Paris) for helpful suggestions and technical help. K.McKinley and I. Cheeseman (MIT, Boston), A. Desai (Ludwig, La Jolla), G. Orsi and G. Almouzni (I. Curie, Paris), I. Draskovic and A. Londono (I. Curie, Paris), Song-Tao Liu (University of Toledo, OH), A. Miyawaki (Hirosawa, Japan) and B.E. Black (University of Pennsylvania, Philadelphia) for providing reagents. We also thank the FACS facility in the Sanford Consortium for Regenerative Medicine (La Jolla, CA), the microscopy platform at Institut Curie supported by the French National Research Agency through the « Investments for the Future » program (France-BioImaging, ANR-10-INSB-04), the Fondation pour la Recherche Médicale (FRM N° DGE20111123020) and the Canceropôle-IdF (n°2012-2-EML-04-IC-1 & n° 2011-1-LABEL-IC-4). D.W.C. has received support from R01 GM074150 from the NIH. D.W.C. receives salary support from the Ludwig Institute for Cancer Research. D.F. receives salary support from the CNRS. D.F. has received support by Labex « CelTisPhyBio », the Institut Curie and the ATIP-Avenir 2015 program. This work has also received support under the program «Investissements d’Avenir » launched by the French Government and implemented by ANR with the references ANR-10-LABX-XXX and ANR-10-IDEX-0001-02 PSL.
Footnotes
AUTHOR CONTRIBUTION
D.F. and M.A.M. performed gene-targeting of CENP-A and CENP-C. Y.NA. performed affinity purification experiments. PL performed the micronuclei experiment on the Y and contributed to text editing. V.B. performed CENP-A depletion on synchronized cells. V.B. and S.E. carried out ChIP analysis of CENP-C. S.H., M.D. and D.F. performed and analyzed all the remaining experiments. D.F. and D.W.C. conceived the experimental design and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anonymous. CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. 2008;135(6):1039–1052. doi: 10.1016/j.cell.2008.10.019. [DOI] [PubMed] [Google Scholar]
- Bade D, et al. The E3 Ligase CUL3/RDX Controls Centromere Maintenance by Ubiquitylating and Stabilizing CENP-A in a CAL1-Dependent Manner. Developmental Cell. 2014;28(5):508–519. doi: 10.1016/j.devcel.2014.01.031. [DOI] [PubMed] [Google Scholar]
- Black BE, et al. Centromere Identity Maintained by Nucleosomes Assembled with Histone H3 Containing the CENP-A Targeting Domain. Molecular Cell. 2007;25(2):309–322. doi: 10.1016/j.molcel.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Black BE, et al. Structural determinants for generating centromeric chromatin. Nature. 2004;430(6999):578–582. doi: 10.1038/nature02766. [DOI] [PubMed] [Google Scholar]
- Bodor DL, et al. Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. In: Bonifacino Juan S, et al., editors. Current protocols in cell biology / editorial board. Unit8.8. Chapter 8. 2012. [DOI] [PubMed] [Google Scholar]
- Bodor DL, et al. The quantitative architecture of centromeric chromatin. eLife. 2014;3:e02137. doi: 10.7554/eLife.02137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, et al. Centromere assembly requires the direct recognition of CENP-A nucleosomes by CENP-N. Nature Cell Biology. 2009;11(7):896–902. doi: 10.1038/ncb1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Milks KJ, Straight AF. Dual recognition of CENP-A nucleosomes is required for centromere assembly. The Journal of Cell Biology. 2010;189(7):1143–1155. doi: 10.1083/jcb.201001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. 2008;9(1):33–46. doi: 10.1038/nrm2310. [DOI] [PubMed] [Google Scholar]
- Chmátal L, et al. Centromere strength provides the cell biological basis for meiotic drive and karyotype evolution in mice. Current biology : CB. 2014;24(19):2295–2300. doi: 10.1016/j.cub.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy EM, et al. HJURP Is a Cell-Cycle-Dependent Maintenance and Deposition Factor of CENP-A at Centromeres. Cell. 2009;137(3):485–497. doi: 10.1016/j.cell.2009.02.040. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Ratrie H, Stetten G. Visualization of centromere proteins CENP-B and CENP-C on a stable dicentric chromosome in cytological spreads. Chromosoma. 1989;98(1):1–12. doi: 10.1007/BF00293329. [DOI] [PubMed] [Google Scholar]
- Fachinetti D, et al. A two-step mechanism for epigenetic specification of centromere identity and function. Nature Cell Biology. 2013;15(8):1–13. doi: 10.1038/ncb2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinetti D, et al. DNA Sequence-Specific Binding of CENP-B Enhances the Fidelity of Human Centromere Function. Developmental Cell. 2015;33(3):314–327. doi: 10.1016/j.devcel.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk SJ, et al. Chromosomes. CENP-C reshapes and stabilizes CENP-A nucleosomes at the centromere. Science. 2015;348(6235):699–703. doi: 10.1126/science.1259308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, et al. The human CENP-A centromeric nucleosome-associated complex. Nature Cell Biology. 2006;8(5):458–469. doi: 10.1038/ncb1397. [DOI] [PubMed] [Google Scholar]
- Foltz DR, et al. Centromere-Specific Assembly of CENP-A Nucleosomes Is Mediated by HJURP. Cell. 2009;137(3):472–484. doi: 10.1016/j.cell.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukagawa T, et al. CENP-C is necessary but not sufficient to induce formation of a functional centromere. The EMBO Journal. 1999;18(15):4196–4209. doi: 10.1093/emboj/18.15.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukagawa T, Earnshaw WC. The centromere: chromatin foundation for the kinetochore machinery. Developmental Cell. 2014;30(5):496–508. doi: 10.1016/j.devcel.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, et al. Induced Ectopic Kinetochore Assembly Bypasses the Requirementfor CENP-A Nucleosomes. Cell. 2011;145(3):410–422. doi: 10.1016/j.cell.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse A, et al. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature. 2011;477(7364):354–358. doi: 10.1038/nature10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, et al. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(49):E3350–7. doi: 10.1073/pnas.1216880109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, et al. CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell. 2008;135(6):1039–1052. doi: 10.1016/j.cell.2008.10.019. [DOI] [PubMed] [Google Scholar]
- Izuta H, et al. Comprehensive analysis of the ICEN (Interphase Centromere Complex) components enriched in the CENP-A chromatin of human cells. Genes to Cells. 2006;11(6):673–684. doi: 10.1111/j.1365-2443.2006.00969.x. [DOI] [PubMed] [Google Scholar]
- Jansen LET, et al. Propagation of centromeric chromatin requires exit from mitosis. The Journal of Cell Biology. 2007;176(6):795–805. doi: 10.1083/jcb.200701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, Bai Y. A Conserved Mechanism for Centromeric Nucleosome Recognition by Centromere Protein CENP-C. Science. 2013a;340(6136):1110–1113. doi: 10.1126/science.1235532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klare K, et al. CENP-C is a blueprint for constitutive centromere-associated network assembly within human kinetochores. The Journal of Cell Biology. 2015;210(1):11–22. doi: 10.1083/jcb.201412028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ST, et al. Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. The Journal of Cell Biology. 2006;175(1):41–53. doi: 10.1083/jcb.200606020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon GA, et al. Both tails and the centromere targeting domain of CENP-A are required for centromere establishment. The Journal of Cell Biology. 2015;208(5):521–531. doi: 10.1083/jcb.201412011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Cheeseman IM. The molecular basis for centromere identity and function. Nature Reviews Molecular Cell Biology. 2015:1–14. doi: 10.1038/nrm.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, et al. The CENP-L-N Complex Forms a Critical Node in an Integrated Meshwork of Interactions at the Centromere-Kinetochore Interface. MOLCEL. 2015;60(6):886–898. doi: 10.1016/j.molcel.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga KH, et al. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Research. 2014;24(4):697–707. doi: 10.1101/gr.159624.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, et al. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nature Methods. 2009;6(12):917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Okada M, et al. The CENP-H–I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nature Cell Biology. 2006;8(5):446–457. doi: 10.1038/ncb1396. [DOI] [PubMed] [Google Scholar]
- Prendergast L, et al. Premitotic Assembly of Human CENPs -T and -W Switches Centromeric Chromatin to a Mitotic State D. Cleveland, ed. PLoS Biology. 2011;9(6):e1001082. doi: 10.1371/journal.pbio.1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Régnier V, et al. CENP-A is required for accurate chromosome segregation and sustained kinetochore association of BubR1. Molecular and Cellular Biology. 2005;25(10):3967–3981. doi: 10.1128/MCB.25.10.3967-3981.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Shelby RD, Vafa O, Sullivan KF. Assembly of CENP-A into Centromeric Chromatin Requires a Cooperative Array of Nucleosomal DNA Contact Sites. The Journal of Cell Biology. 1997;136(3):501–513. doi: 10.1083/jcb.136.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoak EM, et al. Long-Term Retention of CENP-A Nucleosomes in Mammalian Oocytes Underpins Transgenerational Inheritance of Centromere Identity. Current Biology. 2016:1–8. doi: 10.1016/j.cub.2016.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachiwana H, et al. HJURP Involvement in De Novo CenH3CENP-A and CENPC Recruitment. Cell reports. 2015;11(1):22–32. doi: 10.1016/j.celrep.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Weir JR, et al. Insights from biochemical reconstitution into the architecture of human kinetochores. Nature. 2016;537(7619):249–253. doi: 10.1038/nature19333. [DOI] [PubMed] [Google Scholar]
- Wood L, et al. Auxin/AID versus conventional knockouts: distinguishing the roles of CENP-T/W in mitotic kinetochore assembly and stability. Open Biology. 2016;6(1):150230. doi: 10.1098/rsob.150230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda K, et al. A human centromere protein, CENP-B, has a DNA binding domain containing four potential alpha helices at the NH2 terminus, which is separable from dimerizing activity. The Journal of Cell Biology. 1992;119(6):1413–1427. doi: 10.1083/jcb.119.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.