

Summary
Regulatory T (Treg) cells reside in lymphoid organs and barrier tissues where they control different types of inflammatory responses. Treg cells are also found in human cancers, and studies in animal models suggest that they contribute to cancer progression. However, properties of human intratumoral Treg cells and those present in corresponding normal tissue remain largely unknown. Here, we analyzed features of Treg cells in untreated human breast carcinomas, normal mammary gland, and peripheral blood. Tumor-resident Treg cells were potently suppressive and their gene expression pattern resembled that of normal breast tissue, but not of activated peripheral blood Treg cells. Nevertheless, a number of cytokine and chemokine receptor genes, most notably CCR8, were upregulated in tumor-resident Treg cells in comparison to normal tissue resident ones. Our studies suggest that targeting CCR8 for the depletion of tumor-resident Treg cells may represent a promising immunotherapeutic approach for the treatment of breast cancer.
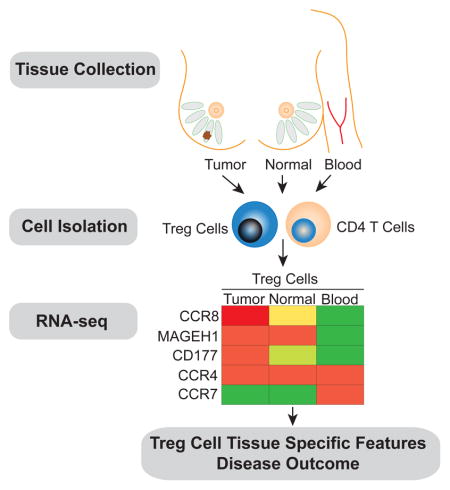
Graphical abstract
Introduction
Regulatory T (Treg) cells expressing the transcription factor Foxp3 play an essential role in controlling autoimmunity and maintain immunological tolerance in mouse and man (Josefowicz et al., 2012). Treg cells are present in secondary lymphoid organs, peripheral blood and in non-lymphoid organs, most prominently at barrier sites including skin, lung, gastrointestinal tract and liver. Under inflammatory conditions, however, Treg cells can be recruited to inflammatory insult sites throughout the body. In addition to secondary lymphoid organs, Treg cells can exert their suppressor function in non-lymphoid tissues as evidenced by specific tissue lesions in mice with selectively impaired Treg cell migration (Sather et al., 2007).
Suppression of distinct types of inflammatory responses by Treg cells is tailored by their sensing of cytokines and other cues resulting in activation of some of the same transcription factors involved in elaboration of pro-inflammatory effector responses (Chaudhry and Rudensky, 2013). Besides sensing distinct types of inflammation, Treg cells residing in non-lymphoid organs can sense unknown tissue cues and exhibit distinct features. Treg cells have also been found in increased numbers in diverse experimental mouse tumors and in human cancers (Nishikawa and Sakaguchi, 2014; Roychoudhuri et al., 2015). While breast carcinomas have not traditionally been considered immunogenic, evidence of tumor infiltrating lymphocytes and their subset composition paralleling disease progression suggest that the underlying interactions of these tumors with immune cells are important (DeNardo and Coussens, 2007). Specifically, the clinical relevance of tumor infiltrating T cells has been intensively studied (Coussens and Pollard, 2011). An increased ratio of CD4+ to CD8+ T cells correlates with lymph node metastases and reduced overall survival (Chin et al., 1992). Increased presence of Treg cells in breast tumor biopsies is associated with an invasive phenotype and diminished relapse-free as well as overall survival (Bates et al., 2006; Bohling and Allison, 2008; Ohara et al., 2009).
It is thought that Treg cells can facilitate tumor growth and metastasis based on the observed regression of established tumors in experimental models of Treg cell depletion ( Joshi et al., 2015; Klages et al., 2010; Pastille et al., 2014; Teng et al., 2010). Transient ablation of Treg cells results in marked reductions in primary and metastatic tumor growth in a poorly immunogenic, oncogene-driven model of mammary carcinoma (Bos et al., 2013). Despite the potential major importance of Treg cells in tumor progression and metastasis and their role as therapeutic targets as established by mouse studies, the properties of Treg cells present in human tumors remain largely unknown. Specifically, it is not clear whether the tumor environment imprints distinct transcriptional and functional features upon Treg cells or whether these cells are similar to activated Treg cells found in corresponding normal tissues or in the peripheral blood.
To address these questions, we explored the functional and transcriptional properties of Treg cells present in breast carcinomas from a large cohort of newly diagnosed patients using flow cytometric and RNA-seq analysis and compared them to peripheral blood or normal breast parenchyma (NBP) resident Treg cells. Our analyses indicated that tumor and normal tissue resident Treg cells exhibit largely shared transcriptional features, distinct from those of activated Treg cells in peripheral blood. Nevertheless, tumor resident Treg cells exhibit increased expression of genes involved in cell activation and cytokine and chemokine signaling including highly augmented expression of chemokine receptor CCR8. CCR8 was also highly expressed by Treg cells present in a number of other cancer types. Analysis of breast cancer gene expression datasets revealed a strong association of CCR8 mRNA amounts normalized to Foxp3, but not Foxp3 mRNA amounts alone, with poor prognosis, implicating highly activated CCR8 expressing Treg cells in breast cancer progression. These findings provide rationale for the therapeutic targeting of Treg cells through a CCR8 depleting antibody in breast cancer, where CTLA-4 and PD-1 focused checkpoint blockade has met limited success so far.
RESULTS
Breast Tumors Have an Increased Presence of Treg Cells
Treg cells variably infiltrate human breast cancers and their frequency in the tumor microenvironment has clinical significance (Demir et al., 2013; Kim et al., 2014; Liu et al., 2011; Liu et al., 2014). To determine the composition of T cell subsets resident in primary human breast tumors we evaluated tissue resident lymphocytes from tumors, NBP, and lymphocytes from peripheral blood by multi-color flow cytometry. The tissue samples were immediately procured from operative specimens of treatment-naïve patients undergoing surgery for primary breast cancer (Table S1). NBP resident cells were isolated from patients undergoing prophylactic mastectomies with no radiographic evidence of disease. Blood samples were from patients undergoing surgery as well as from buffy coats from healthy donors. The frequency of Treg cells among all tumor-infiltrating leukocytes varied widely (Fig. 1D). As compared to NBP and peripheral blood, tumor resident T cell populations exhibited a distinct prevalence and distribution of T cell subsets with an increased abundance of Foxp3+ Treg cells (Fig. 1A). This pattern was observed in multiple patients as evidenced by increased percentage of Treg cells within both CD4+ and total CD3+ T cell populations as well as by a markedly decreased ratio of cytotoxic CD8+ T cells to Treg cells (Fig. 1B). Furthermore, we found that the ratio of CD4+ to CD8+ T cells was markedly different between tumor and NBP with NBP having a greater proportion of CD8+ T cells (Fig. 1C). Our finding was consistent with previously reported analysis of freshly isolated lymphocytes from a small set of normal and malignant breast tissue samples (Ruffell et al., 2012). It is noteworthy that we observed near perfect correlation of high amounts of Foxp3 with high CD25 expression in tumor and NBP resident Treg cells with no detectable Foxp3 found in CD8+ or CD4+ Tconv cells. This enabled highly efficient fractionation of tumor associated Treg and non-Treg cells based on CD25 expression. Our results suggest that the T cell composition in the human tumor microenvironment is distinct from tissue and favors a more immunoregulatory phenotype.
Figure 1. Treg cells densely populate human breast tumors.

Freshly isolated human primary breast tumor, NBP, and peripheral blood mononuclear cells (PBMC) were analyzed by flow cytometry.
(A) Representative flow cytometric analysis of lymphocytes from tumor, NBP, and PBMC isolated from a patient with primary breast cancer. (Data are representative of findings in samples analyzed from 105 patients).
(B) Scatter plots indicating the cumulative frequency of Treg cells among all T cells and the CD8+:Treg cell ratio.
(C) Ratio of CD4+ to CD8+ T cells in tumor, NBP, and PBMC. Data represent the analysis of over 100 individual patients; error bars represent SEM; *p<0.05; **p<0.01, ***p<0.001, ns: non significant (unpaired two-tailed Student’s t test).
(D) Cumulative frequency of Treg cells among all tumor-infiltrating leukocytes. Please also see Table S1.
More Aggressive Breast Cancers are Enriched for Treg Cells
To explore whether any particular clinical correlates were associated with heightened presence of Treg cells, we analyzed the distribution of T cell subsets among the clinically defined surrogates for the molecular subtypes of breast cancer including estrogen receptor positive (ER+), Her2 amplified (Her2+), and triple negative breast cancer (TNBC). In addition, we utilized histologic grade as a variable to gain further insight into the relationship between T cell composition and tumor biology. We found that TNBC had a higher proportion of CD4+ T cells than the other subtypes of breast cancer and that Treg cells were particularly prominent in TNBC (Fig. 2A). Although CD4+ to CD8+ T cell ratio alone could not distinguish between tumors of low to high histologic grade, an increased frequency of Treg cells correlated with higher grade tumors (Fig. 2A). Further analysis of tumor infiltrating Treg cells for expression of a cell division associated marker Ki67 revealed that higher-grade tumors were characterized by higher Treg cell proliferative activity. However, the latter did not correlate with biologic subtype (Fig. 2B). Notably, Treg cells were markedly more proliferative than either conventional CD4+ or CD8+ T cells in breast tumors and NBP and Treg cells isolated from breast tumors were more proliferative than Treg cells isolated from NBP (Fig. 3A). A comparison of expression of CTLA-4 and CD25, two known markers indicative of Treg cell activation, between breast tumor, NBP and peripheral blood Treg and Tconv cells also revealed that tumor resident Treg cells exhibited an activated phenotype consistent with their increased proliferative activity (Fig. 3B). Previous studies of human Treg cells isolated from autoimmune lesions suggested that these tissue resident Treg cells exhibit impaired in vitro suppressor capacity raising questions as to whether tumor resident Treg cells are capable of suppression. Thus, we isolated Treg cells from breast cancers and assessed their ability to suppress proliferation of naive peripheral blood CD4+ T cells isolated from buffy coats. We found that in agreement with their observed activated phenotype, CD25hiCD4+ Treg cells isolated from breast tumors were also strongly suppressive (Fig. 3C). Thus, more advanced grades of different types of breast cancer and the more aggressive TNBC type are associated with a heightened presence of activated Treg cells with potent suppressor function.
Figure 2. Treg cell frequency correlates with grade and type of human breast cancer.

(A) Scatter plots representing the frequency of T cell subsets based on biologic subtype (ER+, Estrogen receptor >1% positive Her2 non-amplified; Her2+, Her2 amplified either by 3+ staining on immunohistochemistry or FISH Her2-to-CEP17 ratio > 2.2; TNBC, ER<1%, PR<1%, Her2 non-amplified) and based on histologic grade.
C) Scatter plots representing the frequency of proliferating Treg cells in tumors based on histologic grade and biologic subtype. Frequencies are the % of Treg cells that are Ki67+. Data represent the analysis of over 100 individual patients; error bars represent SEM; *p<0.05; **p<0.01, ***p<0.001, ns non significant (unpaired two-tailed Student’s T test). The 57 high grade tumors included 33 ER+, 18 TNBC, and 6 Her2+ samples.
Figure 3. Tumor resident Treg cells are activated and potently suppressive.

(A) Representative flow cytometric analysis of lymphocytes from tumor, NBP, and peripheral blood isolated from a patient with primary breast cancer and summary plot indicating the proportion of lymphocytes from tumor, NBP and peripheral blood that are Ki67+. Data represent the analysis of over 70 individual patients; error bars represent SEM; *p<0.05; **p<0.01, ***p<0.001, ns non significant (unpaired two-tailed Student’s T test).
(B) Mean fluorescence intensity (MFI) of staining for activation markers on cells gated on Treg (open histograms) and CD4+ conventional T (Tconv) cells (shaded histograms) in breast tumor, NBP and peripheral blood. Individual dots represent data are from individual tumor specimens.
(C) Tumor resident CD4+CD25hi Treg cells were flow cytometry isolated and their capacity to suppress proliferation of naïve Tconv cells in vitro was assessed by 3H-thymidine incorporation on day 4 (n=3, plot is representative of 3 independent experiments).
Shared and Distinct Features of Breast Tumor and Normal Parenchyma Resident Treg Cells
Next, we sought to explore whether the tumor microenvironment imparts distinct features on Treg cells and on effector CD4+ T (Tconv) cells. Alternatively, it was possible that the transcriptional features of these cell subsets in the tumor resembled those in NBP or in peripheral blood. Thus, we employed RNA-seq analysis to assess genome-wide gene expression features of purified CD25hiCD4+ Treg and effector CD25−CD4+ T cells isolated from breast tumors and from NBP. Activated CD45RO+ Treg cells and Tconv cells were isolated from buffy coats to match the activated phenotype of the corresponding cell subsets found in the tumor and NBP. The gene expression patterns of tumor resident Treg and Tconv cells were very similar to those of NBP resident Treg and Tconv cells, respectively, but distinct from those of the corresponding activated or memory T cell cells isolated from peripheral blood (Fig. 4A–C, Table S2, S3). Thus, residence in a non-lymphoid tissue, regardless of whether it is healthy or has undergone oncogenic transformation, serves as a major determinant of gene expression characteristics of tumor and tissular Treg and Tconv cells.
Figure 4. Tumor Infiltrating Treg and non-Treg CD4 cells are transcriptionally similar to tissue resident cells.

(A) Left: genes differentially expressed in tumor resident Treg cells vs. tumor resident Tconv cells compared to genes differentially expressed in peripheral blood Treg cells vs. Tconv cells (n=4, n=6, n=6, n=4, respectively). Right: genes differentially expressed in tumor resident Treg cells vs. Tconv cells compared to genes differentially expressed in NBP Treg cells vs. NBP Tconv cells (n=8, n=9, n=4, n=5, respectively). The indicated cell subpopulations were sorted based on CD4, CD25 CD45RO and CD127 expression. Gene expression was measured by RNAseq using IonTorrent (left) and Illumina HiSeq (right) platforms; differential gene expression analysis was performed with the DESeq2 package, where p-values represent FDR. A normalized gene count cutoff of 50 was used. Genes down- or up-regulated in tumor resident Treg cells are shown in orange (p<0.01), while genes down- or up-regulated in peripheral blood or NBP Treg cells are shown in blue (p<0.01). Genes down- or up-regulated in both tumor and peripheral blood or NBP Treg cells are shown in red (p<0.01). The numbers of genes differentially expressed are indicated.
(B, C) Left: volcano plot comparing the p-value versus fold-change for genes from tumor resident Treg (B) and Tconv cells (C) relative to peripheral blood Treg (B) and Tconv cells (C) (n=4, n=6 respectively for both comparisons). Right: volcano plot comparing the p-value versus fold-change for genes from tumor resident Treg (B) and Tconv cells (C) relative to NBP Treg (n=8, n=5, respectively) (B) and Tconv cells (n=9, n=6, respectively) (C). Genes labeled in red are significantly differentially expressed between tumor and peripheral blood or NBP Treg and Tconv cells (p<0.01).
(D, E) Gene Set enrichment analysis was performed using the GOrilla bioinformatics tool for significantly up-regulated genes in tumor Treg cells relative to peripheral blood Treg cells on left or NBP Treg cells on right (p-value<0.05, mean normalized read count>50) as the target gene set and all genes expressed in the RNA-seq data as the background set (expressed genes are defined as genes with mean normalized read count>20). Please also see Figure S1, S2.
The observation that breast tumor and tissue Treg cells and breast tumor and tissue Tconv cells were transcriptionally very similar led us to ask whether Treg and Tconv populations in breast tumors are largely shaped by local expansions of pre-existing tissue-resident cells. It was also conceivable that Treg and Tconv cells are recruited into the tumor from draining lymph nodes or peripheral blood and subsequently undergo phenotypic changes in response to the local environment. Thirdly, it was possible that local conversion of Tconv cells into Treg cells within breast tumors largely contribute to the increased presence of Treg cells in breast tumors as compared to NBP. To distinguish between these possibilities, T cell receptor (TCR) beta CDR3 sequences were extracted from the RNA-seq data using MiXCR software (Bolotin et al., 2015). Comparative analysis of TCR repertoires of peripheral blood and tumoral Treg and CD4+ T cells was performed to determine their overlaps within individual patients using VDJtools software (Shugay et al., 2015). These analyses revealed low TCR repertoire overlap between normal tissue and tumor Treg cell subsets, suggesting that local expansion of tissue-resident Treg cells was unlikely the primary origin of intratumoral Treg cells (Fig. 5A). Low TCR repertoire overlap between intratumoral Treg and Tconv cells, which was comparable to that between Treg and Tconv cells in NBP, argued against prominent conversion of Tconv to Treg cells within tumors (Fig. 5A). Furthermore, both normal tissue and tumor Treg subsets contained large expanded clones, similarly to the activated CD45RO+ but not to the resting CD45RA+ peripheral blood Treg cells (Fig. 5B). These results were consistent with the possibility that the majority of Treg cells in breast tumors were recruited from the periphery where their features were affected by the local environment.
Figure 5. TCR beta repertoires of tumor and NBP Treg and Tconv cells are oligoclonal and non-overlapping.

(A) Within-patient similarity of TCR beta CDR3 repertoires between Treg and Tconv cells in breast tumors and NBP was assessed by measuring relative abundance of clones shared between the pairs of samples and displayed as F2 similarity index. P-values were calculated using an unpaired two-tailed Student’s t test.
(B) Fraction of the TCR beta CDR3 repertoire occupied by the 10 most abundant clones. Please also see Figure S3.
Gene ontology (GO) analysis of differentially expressed genes showed that tumor resident Treg cells, when compared to activated Treg cells from peripheral blood, were enriched for cytokine signaling, defense response, inflammatory response and lymphocyte activation categories, among others (Fig. 4D left panel). The gene expression pattern of tumor resident Tconv cells was enriched for a type I interferon signaling gene signature (Fig. 4E left panel). Notably, many chemokine receptors, including CCR5, CCR8, CCR10, CX3CR1, CXCR3 and CXCR6 were robustly and differentially expressed by tumor resident Treg cells at the mRNA and protein levels. Other chemokine receptors, such as CCR4, were highly expressed by both tumor and peripheral blood Treg cells, or were downregulated in tumor resident Treg cells at the mRNA and protein level (CCR7 and CCR9). The wide array of chemokine receptors and cytokine receptors expressed by intratumoral Treg cells suggests that these cells are able to respond to a variety of local factors. As in Treg cells, certain chemokine receptors, such as CCR5, CCR2, CXCR3 and CXCR6 were highly expressed by tumor Tconv cells at the mRNA and protein level. Certain chemokine receptors, such as CCR8, were found to be highly expressed by tumor Treg cells and were much less abundant in Tconv cells, while others, were more abundant in Tconv cells, suggesting that Treg and Tconv cells may rely on both unique and shared pathways to guide their migration to, and retention within the breast tumor environment (Fig. 4B,C left panels, Fig. S1).
Several genes and their products known to be upregulated upon Treg cell activation and associated with increased suppressor function were more highly expressed by Treg cells in breast tumors when compared to peripheral blood Treg cells. These included CD39 (ENTPD1), CD25 (IL2RA), ST2 (IL1RL1), and Epstein-Barr Virus Induced 3 (EBI3). We also found that intratumoral Treg cells exhibited heightened expression of interleukin 1 receptor type II (IL1R2), encoding a decoy IL-1 receptor (Fig. 4A,B left panels). Of these genes, a subset was more highly expressed by Treg cells in tumor as compared to NBP resident Treg cells, including EBI3, OX40 and IL1R2. Likewise, several genes encoding proteins implicated in T cell effector function were differentially expressed by Tconv cells isolated from breast tumors when compared to peripheral blood Tconv cells (Pearce et al., 2003). These include the transcription factor eomesodermin (EOMES), and several genes encoding effector molecules, including granzyme K (GZMK), and interferon-γ (IFNG). Certain genes associated with T cell exhaustion in Tconv cells, PDCD1, TIGIT and TIM3, were differentially expressed by tumor Tconv when compared to those in peripheral blood.
In addition, we found several genes not previously associated with Treg cells or T cells differentially and robustly expressed by tumor Treg cells including melanoma antigen family H1 gene (MAGEH1) and CD177. MAGEH1 belongs to the Type II MAGE protein family, whose are expressed in multiple types of tissues and may act as a E3 ubiquitin ligase, which may regulate Treg cell function or survival within tumors (Doyle et al., 2010). CD177 is a receptor for PECAM1, expressed by a subset of human and mouse neutrophils and implicated in neutrophil extravasation and potentially survival (Xie et al., 2015). CD177 was highly expressed by a subset of intratumoral Treg cells representing 10–50% of total Treg cells in individual tumor samples. In contrast, markedly fewer Treg cells found in NBP expressed CD177 and in lower amounts than those found in tumors (Fig. S1A). Furthermore, our flow cytometric studies showed increased CD177 expression by Treg cells in lung cancer, colorectal cancer and melanoma (Fig. S2). To further examine whether CD177 expression marks a distinct subset of intratumoral Treg cells, we performed RNA-seq analyses of flow cytometry purified CD177+ and CD177− CD25hiCD4+ Treg cells isolated from five breast tumors. CD177+ and CD177− tumor Treg cells were transcriptionally similar to each other (Fig. S3A, Table S4). Furthermore, no correlation was found between the proportion of Treg cells that are CD177+ and histologic grade, nodal status, or biologic subtype (data not shown). However, analysis of TCR beta CDR3 repertoires extracted from RNA-seq data revealed that CD177+ Treg cells contained large clonal expansions that were not prominent in the CD177− populations (Fig. S3B). These observations suggest that CD177 expression is induced in a subset of Treg cells in response to strong TCR stimulation and a yet to be identified accessory signal.
We identified CCR8 as the most robustly and differentially expressed chemokine receptor in breast tumor resident Treg cells as compared to peripheral blood Treg cells or Tconv cells found in either peripheral blood or breast tumors (Fig. 4A,B). Flow cytometric analyses showed that at a protein level, CCR8 was differentially expressed by the entire tumor-resident Treg cell population (Fig. S1A). In contrast, CCR2, which was highly and differentially expressed by intratumoral Treg cells when compared to peripheral blood Treg cells, was equivalently expressed by intratumoral Tconv cells at the protein level (Fig. S1B). Similarly, CCR5, which was differentially expressed by tumor Treg cells, was also highly expressed by a subset of intratumoral Tconv cells and by the majority of intratumoral CD8 T cells (Fig S1B). The robust and differential expression of CCR8 by intratumoral Treg cells provides an opportunity for selective depletion of Treg cells as an immunotherapeutic approach for the treatment of breast cancer.
CCR8 is Expressed by Intratumoral Treg Cells
To assess CCR8 protein expression by diverse types of hematopoietic and non-hematopoietic cells present within the tumor, dissociated tumor cell preparations were analyzed by flow cytometry. We found that CCR8 is expressed exclusively by CD45+ cells, and that within all CD45+ cells CCR8 is largely expressed by Foxp3+ Treg cells (Fig. 6A; data not shown). Further analyses showed that in addition to robust expression by tumor-resident Treg cells and approximately 50% lower expression by Treg cells residing in the NBP, ~ 50% of intratumoral natural killer T (NKT) cells expressed high amounts of CCR8, whereas NK cells, CD8+, γδT cells, myeloid cells and the bulk of CD4+ Tconv cells found in the tumor, NBP and peripheral blood did not (Fig. 6B,C). Although human cord blood NKT cells have been reported to express CCR8, we did not observe any staining in peripheral blood NKT cells (data not shown, Harner et al., 2011). To assess whether CCR8 expression on tumor resident Treg cells is unique to breast cancer or a general feature of Treg cells present in solid tumors of diverse tissue origin, we analyzed tumor-infiltrating T cells by flow cytometry and found that CCR8 was more highly expressed by Treg cells than Tconv cells in several types of cancer, including lung and colorectal cancer, in melanoma and in angiosarcoma (Fig. 6D; data not shown). Analysis of axillary lymph nodes from three patients with nodal metastases from breast cancer revealed a minor subset of Treg cells expressing CCR8 (Fig. S4). These CCR8+ Treg cells may be cells that were poised to migrate into or recently emigrated out of the breast tumor or may represent Treg cells that were residing within the nodal metastases.
Figure 6. CCR8 expression by tumor resident Treg cells is specific and functional.

(A) CCR8 staining of CD45+ cells from a human breast tumor.
(B) MFI of CCR8 expression on multiple cell types in tumor, NBP, and peripheral blood isolated from a patient with primary breast cancer.
(C) Summary plot of MFI ratio of CCR8 expression on Treg cells to Tconv cells in breast tumors, NBP and peripheral blood. Data represent the analysis of over 70 individual patients; error bars represent SEM; *p<0.05; **p<0.01, ***p<0.001, ns non significant (unpaired two-tailed Student’s t test).
(D) MFI of CCR8 expression gated on Treg cells (open histograms) and Tconv cells (shaded histograms) in colorectal adenocarcinoma, melanoma and lung adenocarcinoma.
(E) CD3+ T cells were isolated by flow cytometry from a primary breast tumor and their migratory capacity was assessed in in vitro transwell migration assays. Plot is representative of 5 independent experiments; error bars represent SEM; *p<0.05; **p<0.01, ***p<0.001, ns non significant (unpaired two-tailed Student’s T test).
(F) Effect of breast tumor soluble factors on CCR8 expression by Treg cells was assessed by culturing tumor slices and naïve Treg cells isolated from peripheral blood in transwell chambers separated by a 0.4 um membrane. CCR8 mRNA in Treg cells was measured by qPCR on day 5 with ADAR as a reference gene and calibrated to expression in non-activated Treg cells. Data represent 3 independent experiments; error bars represent SEM; *p<0.05; **p<0.01, ***p<0.001, ns non significant (unpaired two-tailed Student’s T test). Please also see Figure S4, S5, S6.
CCR8 Ligand CCL1 is Expressed by Intratumoral Myeloid Cells
Since the ability of Treg cells to migrate to a specific tissue is essential for their capacity to suppress inflammation, we sought to assess the expression and cellular source of the two known cognate CCR8 ligands, CCL1 and CCL18, in breast tumors (Roos et al., 1997). First, analysis of the Cancer Genome Atlas (TCGA) breast cancer dataset, which includes whole tumor RNA-seq data and clinical correlates for over 1000 patients (Cancer Genome Atlas, 2012) showed that CCL1 mRNA amounts were markedly higher in breast, lung, colorectal and melanoma tumors as compared to adjacent normal tissue (Fig. S5A). To identify cell types producing CCR8 ligands in breast tumors we performed RNA-seq analysis of CD11b+CD14+ myeloid cells isolated from breast tumors and peripheral blood and found that both CCL1 and CCL18 mRNAs were differentially expressed by intratumoral myeloid cells (Fig. S5B, Table S5). To further substantiate these results and to expand our analysis to additional cell subsets we performed CCL1 qPCR on purified fibroblasts, endothelial cells and myeloid cells isolated from breast tumors and found heightened CCL1 and CCL18 mRNA expression by myeloid cells, but not by other cell types analyzed (Fig. S5C).
We next sought to determine the functionality of CCR8 expressed by tumor infiltrating Treg cells by testing their chemotactic response to CCL1. We found that CD25hiCD4+ Treg cells flow cytometry sorted from breast tumors were capable of migrating more robustly than CD25−CD4+ Tconv cells towards CCL1 mediated chemotactic cues (Fig. 6E). To determine whether CCR8 signaling can markedly alter Treg cell suppressor function, the ability of purified intratumoral Treg cells to suppress in vitro proliferative responses of naïve CD4+ T cells upon TCR cross-linking was assessed in the presence of CCL1. We found that CCR8 ligand provision had no appreciable effect on the suppressive capacity of Treg cells (Fig. S6).
To test whether the breast tumor environment has the ability to potentiate CCR8 cexpression in Treg cells, we stimulated CCR8-negative peripheral blood Treg cells with CD3 and CD28 antibodies upon co-culture with tumor or NBP explants separated by a diffusible membrane. We found that stimulation of “naïve” Treg cells isolated from buffy coats in the presence of tumor explants strongly induced CCR8, while Treg cells activated in the presence of NBP or alone induced CCR8 to a much lesser degree (Fig. 6F). Treg cells cultured with tumor or NBP explants without activation failed to induce CCR8. These data suggest that components of the tumor microenvironment may lead to the induction of CCR8 on Treg in tumors or tumor draining lymph nodes and may contribute to the retention of Treg cells within tumors. The strong type I interferon signature in intratumoral T cells led us to test whether type I interferon signaling is able to induce CCR8 on peripheral blood Treg cells. We found that activation of Treg cells in presence of IFN-α inhibited the modest induction of CCR8 upon Treg cell activation (data not shown).
High CCR8 Expression Marks Highly Activated and Proliferative Tumor Treg Cells and Is Associated with Poor Survival
To determine whether CCR8 expression by tumor infiltrating Treg cells has clinical relevance we correlated the expression of CCR8 on Treg cells with available clinicopathologic correlatives of breast cancer outcomes in our patient samples. We found that CCR8 expression on tumor infiltrating Treg cells assessed by flow cytometry did not vary based on the biologic subtype of breast cancer, but that high CCR8 expression on tumor-resident Treg cells was associated with higher-grade tumors (Fig. 7A, data not shown). Several functional Treg cell activation markers, including CTLA-4, CD25 and Foxp3, were more highly expressed by CCR8high tumor Treg cells (Fig. 7B). Consistent with a requirement of Treg cell activation for induction of CCR8 expression in tumor explant co-cultures, we found CCR8 expression correlated strongly with tumor Treg cell proliferative activity assessed by Ki67 staining (Fig. 7C–E).
Figure 7. Treg CCR8 expression correlates with clinic-pathologic features of human breast cancer and is associated with poor disease free and overall survival.

(A) Summary plot of MFI ratio of CCR8 expression on Treg cells to Tconv cells in breast tumors based on histologic grade. Data represent the analysis of 61 individual patients; error bars represent SEM; *p<0.05; **p<0.01, ***p<0.001, ns non significant (unpaired two-tailed Student’s T test).
(B) MFI ratio of various Treg cell activation marker expression by CCR8high vs. CCR8low Treg cells in individual breast tumors. Data represent the analysis of 57 individual patients.
(C) CCR8 and Ki67 staining of CD4+Foxp3+ T cells from a human breast tumor.
(D) Correlation of MFI ratio of CCR8 expression on Treg cells to Tconv cells and % of Ki67+ Treg cells in individual breast tumors. Data represent the analysis of 48 individual patients.
(E) Ratio of percent of CCR8high Treg cells that are Ki67+ to percent of CCR8low Treg cells that are Ki67+ in individual breast tumors. Data represent the analysis of 30 individual patients.
(F) Correlation of CCR8 and FOXP3 normalized mRNA expression in the TCGA breast cancer dataset (Spearman’s r2=0.80, n=1024). mRNA normalization was estimated by the TCGA using the RSEM (RNA-seq by expectation maximization) method.
(G) Boxplots comparing CCR8, FOXP3 or CCR8 to FOXP3 ration normalized mRNA amount in breast tumors versus adjacent NBP. Data represent the analysis of 111 individual patients; *p<0.05; **p<0.01, ***p<0.001, ****p<0.0001, ns non significant (paired two-tailed Student’s T test).
(H) Overall survival of breast cancer patients stratified by median tumor FOXP3 normalized mRNA amount (Log-rank p=0.53, n=1024, Mantel-Cox test)
(I) Overall survival (Log-rank p=0.012, n=1024, Mantel-Cox test) and disease-free survival (Log-rank p=0.002, n=932, Mantel-Cox test) of breast cancer patients stratified by median tumor CCR8/FOXP3 normalized mRNA ratio. Please also see Figure S7.
Since high CCR8 expression on tumor resident Treg cells was associated with higher-grade tumors in the analyzed cohort of patients, we sought to determine whether CCR8 expression by Treg cells in breast cancer correlated with disease prognosis. As CCR8 was expressed almost exclusively by Foxp3+ Treg cells in the analyzed cohort of patients, we turned to the TCGA breast cancer dataset. We first sought to determine whether CCR8 expression correlated with Foxp3 expression in this large cohort of patients. We found a strong positive correlation between normalized CCR8 and Foxp3 mRNA amounts (Fig. 7F). Furthermore, we found that CCR8 and Foxp3 mRNA amounts were markedly higher in breast tumors than in patient-matched adjacent NBP, in full agreement with our observation that breast tumors contain a higher density of Treg cells than NBP (Fig. 7G). When breast cancer patients were stratified into two distinct groups based on median CCR8/Foxp3 mRNA ratio, a high CCR8/Foxp3 mRNA ratio was significantly associated with decreased overall survival (OS) and disease-free survival (DFS, Fig. 7I). To determine if CCR8 to Foxp3 mRNA ratio is an independent prognostic factor for survival after taking into account other clinical variables, we performed multivariate survival analyses. We found that CCR8/FOXP3 mRNA ratio was no longer associated with OS after adjusting for age, stage, and subtype (data not shown). This is not unexpected as we found subtype strongly associated with CCR8 to FOXP3 mRNA ratio and also with OS. Notably, when patients were stratified into two groups based on median Foxp3 mRNA amount alone, a high Foxp3 mRNA amount was not associated with worse survival outcome (Fig. 7H). Since CCR2, CCR4, CCR5 mRNAs are also highly expressed by tumor Treg cells, we assessed whether expression of these chemokine receptors in breast tumors was associated with survival. We found a strong positive correlation between normalized CCR2, CCR4 or CCR5 and Foxp3 mRNA amounts, but when patients were stratified into two groups based on median chemokine receptor/Foxp3 mRNA ratio, a high chemokine receptor/Foxp3 mRNA was not associated with worse survival outcome in any case (Fig. S7). Similarly, when patients were stratified into two groups based on median mRNA amount of CCR2, CCR4 or CCR5 alone, high chemokine receptor expression was not associated with worse survival outcome (data not shown). Interpretation of this result is complicated due to expression of these chemokine receptors by other cell types. In summary, high CCR8 expression by Treg cells may mark a subset of highly activated and suppressive cells, which, when present in breast tumors, is associated with decreased survival. These results implicate activated CCR8 expressing Treg cells in the pathogenesis of human breast cancer.
DISCUSSION
The presence of Treg cells in the tumor microenvironment has recently emerged as a characteristic feature of human tumors including breast carcinomas (Gobert et al., 2009; Hossain et al., 2013; Senovilla et al., 2012; Shang et al., 2015). Here, through the analysis of primary tumors surgically removed from over 100 treatment-naïve patients, we found that Treg cells were abundant in human breast cancers as compared to normal breast parenchyma and peripheral blood. In addition their frequency is increased in the more aggressive triple-negative subset of breast cancers and also correlates with higher-grade lesions across all subsets. These findings support the notion that Treg cells can directly contribute to the natural progression of cancer rather than accumulating merely as an indirect consequence of other mechanisms underlying cancer progression.
Studies in mice have revealed that tissue-resident Treg cells differ from their counterparts residing in the secondary lymphoid organs (Burzyn et al., 2013). Some of these distinguishing characteristics mirror the gene expression features of the particular niche within which the Treg cells reside. In contrast to mice, it remains unknown whether in humans Treg cells residing in non-lymphoid organs or tissues are markedly different in their gene expression patterns from those in peripheral blood. The unique metabolic nature of the tumor microenvironment including relative hypoxia, scarcity of nutrients, and heightened lactate production, raises the possibility that this environment imparts specific properties on Treg cells distinct from those in normal tissue or from activated circulating Treg cells, enabling Treg cells to optimally support tumor growth. An alternative possibility is that active recruitment of Treg cells from circulation might lead to intratumoral Treg cells that largely resemble activated peripheral blood Treg cells. Finally, it is possible that tumor resident Treg cells are similar to those found in normal tissue with few, if any distinguishing features. Our RNA-seq analysis of highly purified Treg cells from breast carcinomas showed major differences in gene expression when compared to activated peripheral blood Treg cells. In contrast, Treg cells from breast carcinomas shared the bulk of features characteristic of normal breast tissue resident Treg cells. The remaining differences between the two subsets pointed to heightened activation and proliferation of Treg cells in tumors. The gene expression profile of tumor resident Treg cells (and Tconv cells) closely mirrored that of their normal breast parenchyma resident counterparts. These findings suggest that, despite many fundamental differences, the normal breast tissue and breast tumor environment attract and impact tissue-resident Treg cells through overlapping sets of cues and mechanisms. This observation raises the possibility that local expansion of pre-existing tissue resident Treg cells may account for the high Treg cell density in breast tumors. However, comparison of TCR repertoires revealed low clonal overlap between normal tissue and tumor Treg cell subsets arguing against this hypothesis. Furthermore, a small number of genes were markedly upregulated on tumor Treg cells relative to normal breast Treg cells. GO pathway enrichment analysis revealed that these genes were enriched for cytokine-mediated signaling and type I interferon response pathways. We also found MAGEH1 gene, previously identified in several cancer types including melanoma, to be upregulated by intratumoral Treg cells. Although its function remains to be explored, MAGEH1 expression has also been observed in Treg cells isolated from colorectal cancer and lung adenocarcinoma resident Treg cells (M. Pagani, personal communication).
The enhanced state of activation of intratumoral Treg cells may reflect more potent TCR stimulation, which is required for Treg cell activation, proliferation and suppressor function (Levine et al., 2014). We also found chemokine signaling and cell migration as a major single category of genes enriched in intratumoral Treg cells. Treg cell expression of homing receptors defines distinct populations that can direct migration to a range of tissues or sites of inflammation (Huehn et al., 2004). Trafficking receptors and molecules that have been reported to be expressed by Treg populations found in non-lymphoid and inflamed tissues include CCR2, CCR4, CCR5, CCR6, CCR8, CCR9, CCR10, CXCR3, several integrins, selectin ligands and GPCRs (Campbell, 2015). Coordinated expression of these receptors and ligands enables the homing of Treg cells to non-lymphoid organs and may contribute to other aspects of Treg cell function (Lee et al., 2007). Characterization of homing receptors expressed by Treg and effector T cells within tumors may help shift the effector to suppressor cell balance. This has been demonstrated experimentally for CCR4, whose ligands CCL17 and/or CCL22 can be expressed by tumor cells or tumor associated myeloid cells (Curiel et al., 2004; Faget et al., 2011; Sugiyama et al., 2013).
It is unlikely that targeting a single chemokine receptor will mitigate the migration or retention of Treg cells in the tumor microenvironment as there is considerable overlapping functionality and promiscuity of ligand binding between the different chemokine receptors. The differential expression of chemokine receptors between Treg cells and T effector cells, however, raises the opportunity to develop selective depletion strategies (Ueda, 2015). Although we found that CCR4 was highly expressed by activated Treg cells in peripheral blood, normal breast tissue and breast tumors, it was also present on activated effector CD4+ T cells, albeit in somewhat lower amounts. Accordingly, diminution in both Treg and effector CD4+ T cells was observed in patients treated with a CCR4 depleting antibody (Kurose et al., 2015)(G.P. and A.R., unpublished observations). High expression of CCR8, facilitated by yet to be identified factors present in the tumor environment, was a unique feature of intratumoral Treg cells with markedly lower amounts observed in normal breast tissue resident Treg cells and negligible expression by Treg cells in peripheral blood. Importantly, CCR8 was not found on effector αβT, NK and γδT cells, and myeloid cells in the tumor with the exception of CCR8 expression by ~50% of NKT cells. The biologic importance of high CCR8 expression by intratumoral Treg cells in breast cancer was strongly suggested by the observed correlation of high CCR8/Foxp3 mRNA expression ratio in 1000 tumor samples with markedly decreased disease free and overall survival whereas high Foxp3 expression alone failed to reach statistical significance. While CCR8 represents a promising means, by which to selectively deplete Treg cells in the tumor microenvironment with likely less efficient depletion in normal tissue secondary to the different expression amounts, the role of CCR8 on Treg cell function remains unclear.
Another noteworthy finding was high expression of CD177, another cell adhesion and migration related protein, by a subset of intratumoral Treg cells in breast cancer patients as well as in lung cancer and melanoma (Fig. S1). CD177 is a glycosyl-phosphatidylinositol (GPI)-linked cell surface glycoprotein expressed by neutrophils that has not been previously linked to T cells. Although the biological role of CD177 expression by Treg cells restricted to human tumors remains to be explored, it is reasonable to assume that CD177 likely enables interactions of the positive Treg cell subset with its ligand PECAM-1 and may endow these cells with distinct functionality.
In conclusion, our studies show that the tissue of residence, i.e. normal breast parenchyma, rather than tumor environment itself largely defines distinct properties of intratumoral Treg cells. Nevertheless, breast tumor-resident Treg cells exhibited few distinct features including high amounts of chemokine receptor CCR8. The latter was also expressed by Treg cells in diverse cancers of mesenchymal and epithelial origin, including melanoma, lung and colorectal cancer, and angiosarcoma. High CCR8/Foxp3 mRNA ratio, but not high Foxp3 amount alone correlated with poor prognosis suggesting that CCR8 expression by Treg cells likely plays a role in breast cancer progression either by itself or by marking highly activated pathogenic intra-tumoral Treg cells. Our results suggest that CCR8 may serve as a promising target for cancer immunotherapy.
EXPERIMENTAL PROCEDURES
Human Samples
Samples were collected from 105 adult treatment-naïve women undergoing surgery for primary breast cancer after informed consent and approval from the Institutional Review Board (IRB) at MSKCC. The clinical characteristics of the patients are shown in Table S1. NBP was obtained from patients undergoing contralateral prophylactic mastectomies. Peripheral blood mononuclear cells (PBMCs) were obtained from patients prior to their surgical procedures and from buffy coats obtained from the New York Blood Center (NYBC).
Flow Cytometry and Cell Isolation
Lymphocytes form tumor and normal tissues were isolated by mincing the freshly obtained surgical specimens and subsequent enzymatic digestion using Liberase TL (Sigma) for 20 minutes at 37°C. After passing through a 100 μm filter cells were washed twice with PBS prior to staining. PBMC were first enriched for CD4 T cells through negative selection with RosetteSep antibody cocktails (Stem Cell Technologies). Lymphocytes were stained at 1 x 106 cells per ml for 20 minutes. Definitions used for cell sorting were for conventional CD4+ T cells: CD45+CD3+CD4+CD8−CD25−; for Treg cells: CD45+CD3+CD4+CD8−CD127−CD25high. Tissue Treg cells were largely of an activated phenotype and for a fair comparison to their peripheral blood counterparts activated Treg cells were defined as CD45+CD3+CD4+CD8−CD45RO+CD127−CD25high, while resting Treg cells were defined as CD45+CD3+CD4+CD8−CD45RA+CD127−CD25high. Post sort purity was routinely > 95% pure for the sorted populations. For RNA sequencing, cells were sorted directly into Trizol LS (ThermoFisher) and stored at −80°C prior to RNA extraction. For downstream in vitro assays, cells were sorted into fetal bovine serum (FBS). All antibodies were purchased from eBioscience or BioLegend. Stained cells were either analyzed on a LSRII flow cytometer (BD) or sorted using a FACSAria II (BD). Flow cytometry data were analyzed with FlowJo software (TreeStar).
Transcriptional Profiling
RNA sequencing and data processing is described in detail in the Supplemental Experimental Procedures.
In vitro Assays
For in vitro suppression assays, naive CD4+ T cells were isolated from PBMC using the human naïve CD4+ T cell isolation kit II (Miltenyi) and cultured with graded numbers of CD4+CD25hi Treg cells flow cytometry-sorted from breast tumors on 96-well round-bottom plates pre-coated with CD3 and CD28 antibodies in RPMI1640 supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate, 10 mM HEPES, 2 × 10−5 M 2-mercaptoethanol, 100 U ml−1 penicillin, and 100 mg ml−1 streptomycin for 80 h. Proliferation of T cells was assessed by[3H]-thymidine incorporation during the final 8 h of culture. For in vitro migration assays, CD4+ T cells were flow cytometry-sorted from breast tumors and resuspended in serum-free RPMI1640 supplemented with 0.1% bovine serum albumin (BSA) and plated on 8 μM Transwell inserts (Corning) placed in 24-well plates containing RPMI1640 containing 0.1% BSA alone or containing RPMI1640 with 0.1% BSA and with either CCL19 as a positive control (100ng/mL) or CCL1 (1, 10, 100 or 1000 ng/mL) added. Migration of T cells was assessed by flow cytometry-based staining and cell counting of CD4+Foxp3+ Treg cells and CD4+Foxp3− Tconv cells that had passed through the Transwell after 3 h of culture at 37°C for 3 h. Chemotactic index was calculated as number of migrated cells in the given condition normalized to number of migrated cells in the RPMI1640 with 0.1% BSA alone condition. Additional in vitro assays are described in detail in the Supplemental Experimental Procedures.
Statistical Analysis
GraphPad Prism was used for statistical analysis to compare outcomes using a two-tailed unpaired Student’s t test; significance was set to p < 0.05 and represented as *< 0.05, **< 0.01, ***< 0.001, and **** < 0.0001. Error bars show SEM. For TCGA whole tumor and patient-matched NBP gene expression comparisons, a two-tailed paired Student’s t test was used.
Accession Numbers
The accession number for the sequencing data reported in this paper is GSE89225.
Supplementary Material
Acknowledgments
We thank Rachel Fromme and Emily Zabor for technical assistance and the specimen donors at MSKCC and the TCGA. The results published here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. This study was supported by MSKCC Flow Cytometry Core, Tissue Procurement Service and Integrated Genomics Operation at MSKCC, NIH grants R37 AI034206 (A.Y.R), Cancer Center Support Grant P30 CA008748, the Hilton-Ludwig Cancer Prevention Initiative funded by the Conrad N. Hilton Foundation and Ludwig Cancer Research, and Russian Science Foundation project 14-14-00533 (D.M.C). G.P. is the recipient of a Breast Cancer Alliance Young Investigator Grant. C.K. was a recipient of a Hutham S. Olayan Graduate Fellowship. A.Y.R. is an investigator with the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
G.P, C.K., M.M, and A.Y.R. conceived the experiments. G.P, C.K, P.B, and K.W. performed experiments. C.K. performed computational analyses. D.M.C, and E.V.P. performed TCR-b CDR3 sequences extraction and analysis. A.Y.R. oversaw the experiments. G.P., C.K. and A.Y.R. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:5373–5380. doi: 10.1200/JCO.2006.05.9584. [DOI] [PubMed] [Google Scholar]
- Bohling SD, Allison KH. Immunosuppressive regulatory T cells are associated with aggressive breast cancer phenotypes: a potential therapeutic target. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2008;21:1527–1532. doi: 10.1038/modpathol.2008.160. [DOI] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, Chudakov DM. MiXCR: software for comprehensive adaptive immunity profiling. Nature methods. 2015;12:380–381. doi: 10.1038/nmeth.3364. [DOI] [PubMed] [Google Scholar]
- Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. The Journal of experimental medicine. 2013;210:2435–2466. doi: 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nature immunology. 2013;14:1007–1013. doi: 10.1038/ni.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DJ. Control of Regulatory T Cell Migration, Function, and Homeostasis. Journal of immunology. 2015;195:2507–2513. doi: 10.4049/jimmunol.1500801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Rudensky AY. Control of inflammation by integration of environmental cues by regulatory T cells. The Journal of clinical investigation. 2013;123:939–944. doi: 10.1172/JCI57175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin Y, Janseens J, Vandepitte J, Vandenbrande J, Opdebeek L, Raus J. Phenotypic analysis of tumor-infiltrating lymphocytes from human breast cancer. Anticancer research. 1992;12:1463–1466. [PubMed] [Google Scholar]
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Pollard JW. Leukocytes in mammary development and cancer. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature medicine. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Demir L, Yigit S, Ellidokuz H, Erten C, Somali I, Kucukzeybek Y, Alacacioglu A, Cokmert S, Can A, Akyol M, et al. Predictive and prognostic factors in locally advanced breast cancer: effect of intratumoral FOXP3+ Tregs. Clinical & experimental metastasis. 2013;30:1047–1062. doi: 10.1007/s10585-013-9602-9. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast cancer research : BCR. 2007;9:212. doi: 10.1186/bcr1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JM, Gao J, Wang J, Yang M, Potts PR. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Molecular cell. 2010;39:963–974. doi: 10.1016/j.molcel.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faget J, Biota C, Bachelot T, Gobert M, Treilleux I, Goutagny N, Durand I, Leon-Goddard S, Blay JY, Caux C, Menetrier-Caux C. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer research. 2011;71:6143–6152. doi: 10.1158/0008-5472.CAN-11-0573. [DOI] [PubMed] [Google Scholar]
- Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, Biota C, Doffin AC, Durand I, Olive D, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer research. 2009;69:2000–2009. doi: 10.1158/0008-5472.CAN-08-2360. [DOI] [PubMed] [Google Scholar]
- Harner S, Noessner E, Nadas K, Leumann-Runge A, Schiemann M, Faber FL, Heinrich J, Krauss-Etschmann S. Cord blood Valpha24-Vbeta11 natural killer T cells display a Th2-chemokine receptor profile and cytokine responses. PloS one. 2011;6:e15714. doi: 10.1371/journal.pone.0015714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain DM, Panda AK, Manna A, Mohanty S, Bhattacharjee P, Bhattacharyya S, Saha T, Chakraborty S, Kar RK, Das T, et al. FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity. 2013;39:1057–1069. doi: 10.1016/j.immuni.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF, Lauber J, Frey O, Przybylski GK, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. The Journal of experimental medicine. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annual review of immunology. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, et al. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity. 2015;43:579–590. doi: 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee A, Lim W, Park S, Cho MS, Koo H, Moon BI, Sung SH. Zonal difference and prognostic significance of foxp3 regulatory T cell infiltration in breast cancer. Journal of breast cancer. 2014;17:8–17. doi: 10.4048/jbc.2014.17.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MW, Ngiow SF, Smyth MJ, Hamann A, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Research. 2010;70:7788–7799. doi: 10.1158/0008-5472.CAN-10-1736. [DOI] [PubMed] [Google Scholar]
- Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, Doi T, Suzuki S, Isobe M, Funakoshi T, et al. Phase Ia Study of FoxP3+ CD4 Treg Depletion by Infusion of a Humanized Anti-CCR4 Antibody, KW-0761, in Cancer Patients. Clinical Cancer Research. 2015;21:4327–4336. doi: 10.1158/1078-0432.CCR-15-0357. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kang SG, Kim CH. FoxP3+ T cells undergo conventional first switch to lymphoid tissue homing receptors in thymus but accelerated second switch to nonlymphoid tissue homing receptors in secondary lymphoid tissues. Journal of Immunology. 2007;178:301–311. doi: 10.4049/jimmunol.178.1.301. [DOI] [PubMed] [Google Scholar]
- Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nature Immunology. 2014;15:1070–1078. doi: 10.1038/ni.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lang R, Zhao J, Zhang X, Pringle GA, Fan Y, Yin D, Gu F, Yao Z, Fu L. CD8(+) cytotoxic T cell and FOXP3(+) regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Research and Treatment. 2011;130:645–655. doi: 10.1007/s10549-011-1647-3. [DOI] [PubMed] [Google Scholar]
- Liu S, Foulkes WD, Leung S, Gao D, Lau S, Kos Z, Nielsen TO. Prognostic significance of FOXP3+ tumor-infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast cancer research : BCR. 2014;16:432. doi: 10.1186/s13058-014-0432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Current opinion in immunology. 2014;27:1–7. doi: 10.1016/j.coi.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Ohara M, Yamaguchi Y, Matsuura K, Murakami S, Arihiro K, Okada M. Possible involvement of regulatory T cells in tumor onset and progression in primary breast cancer. Cancer immunology, immunotherapy : CII. 2009;58:441–447. doi: 10.1007/s00262-008-0570-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastille E, Bardini K, Fleissner D, Adamczyk A, Frede A, Wadwa M, von Smolinski D, Kasper S, Sparwasser T, Gruber AD, et al. Transient ablation of regulatory T cells improves antitumor immunity in colitis-associated colon cancer. Cancer research. 2014;74:4258–4269. doi: 10.1158/0008-5472.CAN-13-3065. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- Roychoudhuri R, Eil RL, Restifo NP. The interplay of effector and regulatory T cells in cancer. Current opinion in immunology. 2015;33:101–111. doi: 10.1016/j.coi.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2796–2801. doi: 10.1073/pnas.1104303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, Campbell DJ. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. The Journal of experimental medicine. 2007;204:1335–1347. doi: 10.1084/jem.20070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science. 2012;337:1678–1684. doi: 10.1126/science.1224922. [DOI] [PubMed] [Google Scholar]
- Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3(+) regulatory T cells in cancers: a systematic review and meta-analysis. Scientific reports. 2015;5:15179. doi: 10.1038/srep15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI, et al. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLoS computational biology. 2015;11:e1004503. doi: 10.1371/journal.pcbi.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, Ezoe S, Kanakura Y, Sato E, Fukumori Y, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17945–17950. doi: 10.1073/pnas.1316796110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MW, Ngiow SF, von Scheidt B, McLaughlin N, Sparwasser T, Smyth MJ. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer research. 2010;70:7800–7809. doi: 10.1158/0008-5472.CAN-10-1681. [DOI] [PubMed] [Google Scholar]
- Ueda R. Clinical Application of Anti-CCR4 Monoclonal Antibody. Oncology. 2015;89(Suppl 1):16–21. doi: 10.1159/000431059. [DOI] [PubMed] [Google Scholar]
- Xie Q, Klesney-Tait J, Keck K, Parlet C, Borcherding N, Kolb R, Li W, Tygrett L, Waldschmidt T, Olivier A, et al. Characterization of a novel mouse model with genetic deletion of CD177. Protein & cell. 2015;6:117–126. doi: 10.1007/s13238-014-0109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.