

Abstract
Inhibition or genetic deletion of poly(ADP-ribose) (PAR) polymerase-1 (PARP-1) is protective against toxic insults in many organ systems. The molecular mechanisms underlying PARP-1–dependent cell death involve release of mitochondrial apoptosis-inducing factor (AIF) and its translocation to the nucleus, which results in chromatinolysis. We identified macrophage migration inhibitory factor (MIF) as a PARP-1–dependent AIF-associated nuclease (PAAN). AIF was required for recruitment of MIF to the nucleus, where MIF cleaves genomic DNA into large fragments. Depletion of MIF, disruption of the AIF-MIF interaction, or mutation of glutamic acid at position 22 in the catalytic nuclease domain blocked MIF nuclease activity and inhibited chromatinolysis, cell death induced by glutamate excitotoxicity, and focal stroke. Inhibition of MIF's nuclease activity is a potential therapeutic target for diseases caused by excessive PARP-1 activation.
Poly(ADP-ribose) (PAR) polymerase-1 (PARP-1) is a nuclear enzyme that is activated by DNA damage and facilitates DNA repair (1). Excessive activation of PARP-1 causes an intrinsic caspase-independent cell death program designated parthanatos (2, 3), which occurs after toxic insults in many organ systems (4, 5), including ischemia-reperfusion injury after stroke and myocardial infarction; inflammatory injury; reactive oxygen species–induced injury; glutamate excitotoxicity; and neurodegenerative diseases, such as Parkinson's disease and Alzheimer's disease (2, 4, 6). Consistent with the idea that PARP-1 is a key cell-death mediator, PARP inhibitors or genetic deletion of PARP-1 protect against such cellular injury in models of human disease (2, 4, 5, 7).
During parthanatos, PAR causes release of apoptosis-inducing factor (AIF) from the mitochondria and its translocation to the nucleus, resulting in fragmentation of DNA into 20- to 50-kb fragments (2, 8–11). AIF itself has no obvious nuclease activity (2). Although it has been suggested that CED-3 protease suppressor (CPS)–6, an endonuclease G (EndoG) homolog in Caenorhabditis elegans, cooperates with the worm AIF homolog (WAH-1) to promote DNA degradation (12), mammalian EndoG does not seem to have an essential role in PARP-dependent chromatinolysis and cell death (13, 14) and after transient focal cerebral ischemia in mammals (15). Thus, the nuclease responsible for the chromatinolysis during parthanatos is not known.
PARP-1–dependent cell death requires MIF
To confirm that the EndoG is dispensable for parthanatos, the CRISPR-Cas9 system was used to deplete (knockout) EndoG from human neuroblastoma cell line (SH-SY5Y) cells (fig. S1A). We found that knockout of EndoG failed to block N-methyl-N′-nitro-N-nitrosoguanidine (MNNG)–induced parthanatos (fig. S1B) and large DNA fragmentation (fig. S1C); thus, EndoG is unlikely to be the main contributor to PARP-1–dependent large DNA fragmentation and MNNG-induced cell death (fig. S1) (13, 15).
To identify a PARP-1–dependent AIF-associated nuclease (PAAN), we probed protein chips containing more than 16,000 and 5000 human recombinant proteins in duplicate along with several control proteins (16) with recombinant mouse AIF. The 160 strongest interacting proteins were depleted with small interfering RNA (siRNA) in cultured human HeLa cells to screen for modifiers of parthanatos induced by MNNG (2, 9, 11) (Fig. 1, A and B). We further tested whether depletion of these potential AIF-interacting proteins provided protection equivalent to that of depletion of PARP-1 and whether the proteins exhibited sequence and structure similarity consistent with possible nuclease activity. Depletion of AIF interactor 18 was as protective as depletion of PARP-1 (Fig. 1B). AIF interactor 18 is previously known under various synonyms (glycosylation-inhibiting factor, phenylpyruvate tautomerase, l-dopachrome tautomerase, l-dopachrome isomerase), and it is collectively known as macrophage migration inhibitory factor (MIF or MMIF) (17, 18). Effects of three different short hairpin RNA (shRNA) constructs against human and mouse MIF confirmed that depletion of MIF protected against parthanatos induced by MNNG toxicity in HeLa cells or N-methyl-d-aspartate (NMDA) excitotoxicity in mouse primary cortical neurons (fig. S2, A to F). To rule out off-target effects from the shRNA, we prepared MIF constructs that were resistant to shRNA 1 (RshRNA1) and 3 (RshRNA3). Cells expressing these constructs were impervious to effects of the shRNAs (fig. S2, G and H).
Fig. 1. Identification of MIF as a key cell-death effector mediating PARP-1–dependent cell death.

(A) Strategy for identifying AIF-associated proteins involved in PARP-1–dependent cell death. (B) siRNA-based PARP-1–dependent cell viability high-throughput screening in HeLa cells 24 hours after MNNG treatment (50 μM, 15 min); n = 8. The experiments were repeated in four independent tests ***P < 0.001, one-way ANOVA. (C) Schematic representation of MIF's PD-D/E(X)K domains. Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; Y, Tyr; and X, any amino acid. (D) Alignment of the nuclease domain of human MIF and other nucleases. Arrows above the sequence indicate β strands and rectangles represent α helices. Amino acid residues that were mutated are indicated with an arrow and number (see Results). Nuclease and CxxCxxHx(n)C domains are highlighted in green and pink, respectively. (E) Crystal structure of MIF trimer (pdb:1GD0) (left) and MIF PD-D/E(x)K motif in trimer (right).
MIF contains three PD-D/E(X)K superfamily motifs that are found in many nucleases (19–21) (Fig. 1, C and D) and are highly conserved across mammalian species (fig. S3A). It also contains a CxxCxxHx(n)C zinc finger domain (Fig. 1C and fig. S3B), which is commonly found in DNA damage-response proteins (20). MIF exists as a trimer (22–24). The core PD-D/E(X)K topology structure in the MIF trimer consists of four β strands next to two α strands (Fig. 1E and fig. S3, C to G, and supplementary text), which is similar to those of well-characterized nucleases, including Eco RI, Eco RV, Exo III, and Pvu II (fig. S3, H to O, and supplementary text). These sequence analyses and three-dimensional (3D) modeling results indicated that MIF belongs to the PD-D/E(X)K nuclease-like superfamily (25, 26).
MIF is a nuclease
To determine whether MIF has nuclease activity, we incubated a plasmid c–promoter DNA (pcDNA) vector with recombinant human MIF. Supercoiled pcDNA was cleaved by MIF—but not by its nuclease-deficient mutant MIF E22Q [in which glutamine (Q) replaces glutamic acid (E) at position 22] identified in the nuclease assays below—into an open circular form and, further, to a linear form (Fig. 2A). Moreover, MIF cleaved human genomic DNA in a concentration- and time-dependent manner (fig. S4, A and B). Addition of 10 mM Mg2+, 2 mM Ca2+, or 1 mM Mn2+ was required for MIF nuclease activity (fig. S4C), consistent with the divalent cation concentrations required for in vitro activity of other similar nucleases (27). EDTA blocked MIF's nuclease activity against human genomic DNA (Fig. 2B). In the absence of the divalent cation or with the cation at 2 to 10 μM, MIF had no nuclease activity (fig. S4C). Addition of 200 μM Zn2+ precipitated genomic DNA in the presence of MIF, whereas 2 μM Zn2+ had no effect. Na+ had no effect on MIF's nuclease activity (fig. S4C). Pulsed-field gel electrophoresis indicated that MIF cleaves human genomic DNA into large fragments comparable to those of DNA purified from HeLa cells treated with MNNG (Fig. 2B, lane 8). Depletion of MIF with shRNA prevented MNNG-induced DNA cleavage, which was similar to the effect of PARP inhibition by 3,4-dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline (DPQ) (Fig. 2C). Because MIF has been reported to have tautomerase activity, we tested the effects of the MIF tautomerase inhibitor ISO-1 (28). ISO-1 failed to prevent MNNG-induced DNA damage (Fig. 2C). Moreover, the MIF P2G (also known as the P1G) tautomerase mutant, which lacks tautomerase activity (29), had no effect on MIF's nuclease activity (fig. S4D). These data indicate that MIF is a nuclease that functions in PARP-1–dependent DNA fragmentation.
Fig. 2. MIF is a nuclease that cleaves genomic DNA.

(A) In vitro MIF (2 μM) nuclease assay with pcDNA as substrate. (B) In vitro pulsed-field gel electrophoresis MIF (4 μM) nuclease assay with human genomic DNA (hgDNA) as a substrate in buffer containing Mg2+ (10 mM) with or without EDTA (50 mM) or Ca2+ (2 mM) with or without EDTA (25 mM). (C) Pulsed-field gel electrophoresis assay of MNNG-induced DNA damage in MIF-deficient HeLa cells and wild-type (WT) HeLa cells treated with or without DPQ (30 μM) or ISO-1 (100 μM). NT/MNNG (nontargeting shRNA/MNNG). (D) Nuclease assay of MIF WT and mutants (4 μM) using human genomic DNA as the substrate.
To identify amino acid residues critical for MIF's nuclease activity, we mutated key aspartate, glutamate, and proline residues within the PD-D/E(X)K domains of MIF. E22Q, but not Glu replaced by Asp (E22D), inhibited MIF's nuclease activity, whereas replacement with Ala (E22A) partially reduced MIF's nuclease activity (Fig. 2D; fig. S4, E to H; and supplementary text). Thus, this glutamic acid residue (E22) in the first α helix of MIF is critical for its nuclease activity, which is consistent with reports that this glutamic acid in the first α helix of many exonuclease-endonuclease-phosphatase (EEP) domain superfamily nucleases is highly conserved and that it is the active site for nuclease activity (25, 26). MIF has both oxidoreductase and tautomerase activities (28, 30, 31). MIF active site mutants E22Q and E22A had no effect on MIF's oxidoreductase or tautomerase activities (fig. S5, A and B, and supplementary text). The lack of effect indicated that MIF nuclease activity is independent of its oxidoreductase and tautomerase activities. Moreover, MIF's protein conformation was unaffected by the E22Q and E22A mutations as determined by far-ultraviolet (UV) circular dichroism (CD) and near UV CD spectroscopy (fig. S5, C to M, and supplementary text). The purity of MIF proteins was confirmed by Coomassie blue staining, fast protein liquid chromatography (FPLC), and mass spectrometry (MS) assays (fig. S4G and fig. S5, C and D; Materials and methods; and supplementary text). No adventitious nuclease contamination was observed.
MIF preferentially binds to stem-loop single-stranded DNA
To determine the characteristics of DNA sequences bound by MIF in an unbiased manner, HeLa cells were treated with dimethyl sulfoxide (DMSO) or MNNG (50 μM, 15 min), followed by anti-MIF chromatin immunoprecipitation (ChIP) assays and deep sequencing (fig. S6 and supplementary text). We used the multiple Em for motif elicitation (MEME) program, which performs comprehensive motif analysis on large sets of nucleotide sequences (32), and we identified two classes of MIF-binding motifs (Fig. 3A). The first class (sequences 1 through 3) represents a highly related family of overlapping sequences (Fig. 3A and fig. S7A). The sequence features of this family are best captured in sequence 1 with 30 nucleotides and designated PS30, the most statistically significant motif identified, as determined by the MEME program (E-value = 1.4e–051) (Fig. 3A and fig. S7A). The second class identified was a poly(A) sequence.
Fig. 3. MIF binds and cleaves ssDNA.
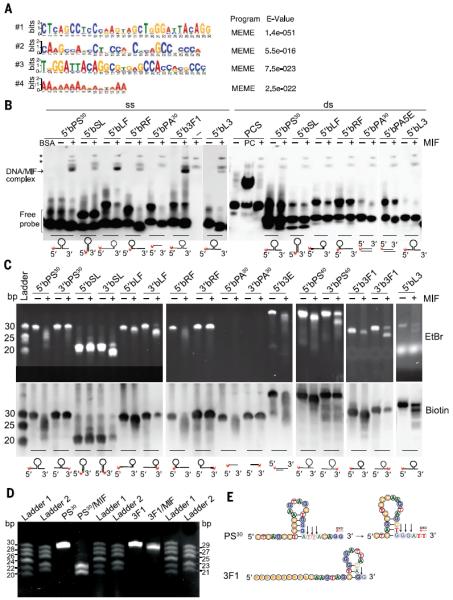
(A) MIF DNA binding motif determined by ChIP-seq. (B) Binding of MIF to biotin-labeled small DNA substrates with different structures or different sequences in an EMSA assay (see fig. S8 for illustrations of substrates, and tables S1 and S2 for sequences). Arrow indicates the DNA-MIF complex. Asterisk indicates nonspecific bands. PCS, positive control substrate from the LightShift Chemiluminescent EMSA Kit (Thermo Scientific) containing 60 base pairs (bp) of 5′ biotin-labeled duplex. With or without BSA, bovine serum albumin; PC, Epstein-Barr nuclear antigen extract from the LightShift Chemiluminescent EMSA Kit or MIF. (C) MIF cleavage of unpaired bases at the 3′ end of the stem loop of 5′ or 3′ biotin-labeled small DNA substrates with various structures or sequences in a nuclease assay (see fig. S8 for illustrations of substrates, and tables S1 and S2 for sequences). Experiments were replicated four times with three independent preparations of MIF protein. (D) MIF cleavage of 3′ unpaired bases from nonlabeled PS30 and 3F1 substrates. DNA ladders 1 and 2 were customized with PS30 and its cleavage products by removing its 3′ nucleotides one by one. DNA ladder 1 was prepared using PS30, PS28, PS26, PS24, PS22, and PS20. DNA ladder 2 was prepared using PS29, PS27, PS25, PS23, and PS21. (E) MIF cleavage sites on nonlabeled PS30 and 3F1 substrates.
We performed 3D modeling to determine likely points of DNA interaction with MIF's PD-D/E(X)K motif. Within the PD-D/E(X)K motif, P16 and D17 on MIF are predicted to be positioned close to double-stranded DNA (dsDNA), whereas E22 is close to ssDNA, indicating MIF might bind single-stranded DNA (ssDNA), dsDNA, or both (fig. S7B). We examined both single-stranded and double-stranded forms of MIF DNA substrates for MIF binding and cleavage specificity. We synthesized the ssPS30 sequence with a 5′ biotin label and subjected it to an electrophoretic mobility shift assay (EMSA) (fig. S7C). MIF bound to the biotin-labeled ssPS30, forming one major complex in the presence of 10 mM Mg2+ (fig. S7C), which was completely disrupted by the addition of excess unlabeled DNA substrate (PS30) or a polyclonal antibody to MIF (fig. S7C). MIF E22Q, E22A, P16A, P17A, and P17Q mutants still formed MIF/ssPS30 complexes (fig. S7C).
Because ssPS30 has the potential to form a stem-loop structure with unpaired bases at the 5′ and 3′ ends, we tested whether MIF binds to ssDNA with sequence or structure specificity. We used 5′ biotin-labeled ssPS30 and sequence-related substrates with different structures created by removing unpaired bases at the 5′ end, 3′ end, or both 5′ and 3′ ends, or by eliminating the stem loop in the EMSA (Fig. 3B and fig. S8). Completely removing the 3′ unpaired bases (5′bLF) had no effect on the DNA-MIF complex formation (Fig. 3B). In contrast, removing the 5′ unpaired bases (5′bRF) reduced, but did not abolish DNA-MIF binding. Similar results are observed when both 5′ and 3′ unpaired bases were removed (5′bSL). Thus MIF appears to mainly bind to 5′ unpaired bases in ssDNA with stem-loop structures. We also used a poly(A) sequence that has no stem loop (5′bPA30) and a short poly(A) sequence at the 5′ end of a stem-loop structure (5′b3F1) as the substrates. MIF failed to bind to 5′bPA30 but did bind to 5′b3F1. These results indicated that a stem loop is required for MIF-ssDNA binding (Fig. 3B and fig. S8). We also tested a substrate unrelated in sequence but that had a stem loop–like structure (5′bL3). MIF bound weakly to 5′bL3. But its binding efficiency was much lower than that of 5′bPS30. These data indicate that MIF preferentially binds to ssDNA with a stem loop and that its specificity is not entirely determined by the sequence. We also tested whether MIF bound to dsDNA with PS30; poly(A); substrates with sequence similarity to PS30 (5′bPS30, 5′bSL, 5′bLF, 5′bRF, 5′bPA30, and 5′bPA5E); and others with nonrelated sequences (PCS and 5′bL3) (Fig. 3B and fig. S8). MIF failed to bind to any of these double-stranded substrates (Fig. 3B).
MIF cleaves 3′ unpaired bases of stem-loop ssDNA
To determine whether MIF cleaves ssDNA or dsDNA, we added 35 random nucleotides to both the 5′ and 3′ ends of the PS30 DNA binding motif (designated PS100) and, under identical conditions, measured cleavage of ssDNA (ssPS100) or dsDNA (dsPS100). MIF cleaved ssPS100 and its complementary strand ssPS100R, but not dsPS100 (fig. S9, A and B). The MIF DNA binding motif identified by ChIP sequencing (PS30) appeared to be sufficient for MIF cleavage because MIF cleaved ssPS30 in a concentration-dependent manner (fig. S9C). MIF cleavage of ssPS30 required Mg2+, and MIF E22Q and E22A mutations blocked the cleavage of ssPS30 (fig. S9D). MIF cleaved ssPS30 in a time-dependent manner with a t1/2 of 12 min, and it cleaved ssPS30 in a concentration-dependent manner with an affinity for substrate (Km) of 2 μM and a maximum initial velocity (Vmax) of 41.7 nM/min (fig. S9, E to G). These kinetic properties are similar to those of other PD-D/E(X)K nucleases, such as Eco RI (27, 33). MIF also cleaved dsPS30 (fig. S9H), but required at least 4 times as high MIF concentrations and a four-fifths reduction in substrate concentration (compare lane 2 of fig. S9C to lane 2 of fig. S9H). MIF failed to cleave its related sequence dsRF, or the nonrelated sequence dsL3 (fig. S9H). MIF's preference for ssDNA is consistent with the 3D model of ssDNA binding to MIF's active site (fig. S7B) and our MIF-DNA binding assays (Fig. 3B). In the presence of AIF, MIF more efficiently cleaved genomic DNA and dsPS30 (fig. S10, A to C), which might be because of the observation that AIF enhanced MIF binding to dsDNA (fig. S10D).
To determine whether MIF has sequence- or structure-specific endonuclease or exonuclease activity, we synthesized a series of variants labeled at the 5′ and 3′ ends with biotin, on the basis of the secondary structure of the DNA substrate ssPS30, and measured their cleavage by MIF (Fig. 3C and fig. S8). MIF had 3′ exonuclease activity and preferentially recognized and degraded unpaired bases at the 3′ end of ssPS30. This was blocked by biotin modification at the 3′ end (lanes 2 to 5 in Fig. 3C, fig. S8, and tables S1 and S2). MIF's 3′ exonuclease activity was also supported by cleavage assays in which the 5′bRF or 5′b3E substrates were used (Fig. 3C, fig. S8, and tables S1 and S2). Moreover, we used poly(A) (PA30), which lacks secondary structure and cannot be stained by ethidium bromide (EtBr) (Fig. 3C, top). We found that MIF's 3′ exonuclease activity allowed it to cleave 5′ biotin–poly(A) (5′bPA30), but not 3′ biotin–poly(A) (3′bPA30), so that MIF's 3′ exonuclease activity can occur independently of secondary structure (Fig. 3C, bottom, and fig. S8). MIF endonuclease activity was also influenced by secondary structure, because it cleaved short unpaired bases of ssDNA at the 3′ end adjacent to the stem loop (5′bPS40, 3′bPS40, 5′b3F1, 3′b3F1, and 5′bL3), as well as 3′-OH or 3′-biotin adjacent to the stem loop (3′bSL and 3′bLF) (Fig. 3C and fig. S8). In contrast to its exonuclease activity, MIF's endonuclease activity was not blocked by biotin modification at the substrate's 3′ end (3′bSL, 3′bLF, 3′bPS40, and 3′b3F1). However, 5′bL3, a sequence not related to PS30 but with a similar stem-loop structure, was cleaved by MIF, but with less efficiency (Fig. 3C and fig. S8). These results indicate that MIF has both 3′ exonuclease and endonuclease activities and cleaves unpaired bases of stem-loop ssDNA at the 3′ end. In the presence of AIF, AIF also increased the binding of MIF to ssDNAs, including 5′bPS30, as well as 5′bSL, which has no 5′ unpaired bases (fig. S10D). Nevertheless, we found that AIF increased both exonuclease and endonuclease activities of MIF (0.5 μM) on 5′bPS30, 3′bPS30, and 3′bSL (fig. S10E). However, AIF has a rather weak effect, if any, on the nuclease activity of MIF at 4 μM (fig. S10F). At this higher concentration, MIF itself can efficiently bind and cleave ssDNAs. These data suggest that AIF may enhance MIF nuclease activity by increasing its binding to ssDNAs.
To further study where MIF cleaves DNA and to avoid the potential interference of biotin labeling, we used non-labeled PS30 and 3F1, which has only one unpaired base at the 3′ end of the stem-loop structure as substrates and customized two different DNA ladders based on PS30. After incubation of these substrates with MIF (2 μM) for 2 hours, two major products of 20 and 22 nucleotides were detected (Fig. 3D). Faint bands of higher molecular mass were also observed. These bands were more obvious in the experiment in which PS30 was biotin labeled and the incubation time was 1 hour (Fig. 3D). MIF cleavage of the 3F1 substrate yielded only a 29-nucleotide (nt) band consistent with cleavage of one unpaired base at the 3′ end of the stem-loop structure (Fig. 3, D and E). These data indicate that PS30 is initially cleaved by MIF after “A23↓T24↓T25↓” (arrow indicates cleavage) by both 3′ exonuclease and endonuclease activity (Fig. 3E, left). Then the resulting product appears to form a new stem-loop structure, as predicted by the online RNA/DNA structure prediction software (http://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Predict1/Predict1.html) (Fig. 3E, right). MIF then cleaves at the new unpaired bases at the 3′ end of this stem-loop structure after “G20↓G21↓G22↓”. We conclude that MIF cleaves unpaired bases at the 3′ end adjacent to the stem loop at the +1 to ~+3 positions through both 3′ exonuclease and endonuclease activities.
AIF interacts with MIF and recruits MIF to the nucleus
Wild-type (WT) glutathione S-transferase–tagged AIF (GST-AIF) associated with MIF, and wild-type GST-MIF associated with AIF in GST pulldown analyses from cell lysates (Fig. 4A; fig. S11, A to D; and supplementary text). We mapped the MIF-AIF binding domain. MIF bound to AIF at amino acids 567 to 592 (fig. S11, A to C, and supplementary text). Conversely, the MIF E22A mutant showed reduced binding to GST-AIF, whereas the E22D and E22Q mutants still bound to GST-AIF (Fig. 4, A and B, and fig. S11D). The other PD-D/E(X)K and C57A;C60A mutations still bound GST-AIF (fig. S11D). Thus, MIF E22 appears to be critical for AIF binding. Endogenous AIF also coimmunoprecipitated with MIF from cortical neurons treated with NMDA (500 μM) but was barely detectable in untreated cultures (Fig. 4, C and D).
Fig. 4. Requirement of AIF for the recruitment of MIF to the nucleus in NMDA excitotoxicity.

(A) Binding of immobilized GST-MIF WT and GST-MIF variants to AIF. (B) Nuclease activity and AIF-binding activity of MIF WT and MIF variants. (C and D) Coimmunoprecipitation (IP) of MIF and AIF in control (CSS) and NMDA-treated cortical neurons. Asterisk indicates IgG. Ab, antibody. (D) Intensity of treated versus untreated cultures. *P < 0.05, Student's t test. (E) Images of nuclear translocation of AIF and MIF after NMDA treatment in WT, AIF knockdown, and MIF knockdown cortical neurons. AIF shRNA (AIF sh) and MIF shRNA (MIF sh) caused a 71.3 ± 5.2% and 73.3 ± 6.1% protein reduction, respectively. White color indicates the overlay of AIF, MIF, and 4′,6′-diamidino-2-phenylindole (DAPI), showing the nuclear translocation of AIF and MIF. Purple color indicates the overlay of AIF and DAPI, showing the nuclear translocation of AIF. Z stacks illustrating the x,z and y,z axis are provided to demarcate the nucleus. Arrowheads indicate cells with the high magnification. (F) Quantification of the percentage of cells with nuclear translocation of MIF and AIF after NMDA treatment in WT, AIF knockdown, and MIF knockdown cortical neurons. CSS, control salt solution. ****P < 0.0001, versus its CSS control; ####P < 0.0001, versus its WT treated with NMDA, one-way ANOVA. (G) Immunoblots of nuclear translocation of AIF and MIF after NMDA treatment in WT, AIF knockdown, and MIF knockdown cortical neurons. Compare total protein (T), post-nuclear fraction (PN), nuclear fraction (N), and Mito, mitochondrial antibody. (H and I) Relative levels of AIF and MIF in T, PN, and N. Means ± SEM. Experiments were replicated at least three times; ****P < 0.0001, versus its CSS control; ##P < 0.01, ###P < 0.0001, versus its total protein, one-way ANOVA.
MIF was localized predominantly to the cytosol of both cortical neurons (Fig. 4E) and HeLa cells (fig. S12A). Both MIF and AIF translocated to the nucleus in cortical neurons treated with NMDA (Fig. 4, E and F) and HeLa cells stimulated with MNNG (fig. S12A). Depletion of AIF with shRNA led to a loss of MIF translocation to the nucleus, but depletion of MIF did not prevent translocation of AIF to the nucleus in cells exposed to NMDA (Fig. 4, E and F). Subcellular fractionation into nuclear and postnuclear fractions confirmed the translocation of MIF and AIF to the nucleus in cultured cortical neurons exposed to NMDA (Fig. 4, G to I). AIF was required for MIF translocation (Fig. 4, E to I). DPQ prevented accumulation of both MIF and AIF in the nucleus in HeLa cells treated with MNNG (fig. S12, A to C) and cortical neurons treated with NMDA (fig. S13, A to C). Consistent with the notion that NMDA excitotoxicity involves nitric oxide production, the nitric oxide synthase inhibitor nitro-arginine (N-Arg) prevented accumulation of both MIF and AIF in the nucleus (fig. S13, A to C).
We transduced primary cortical cultures from WT MIF knockout mice with lentivirus carrying Flag-tagged MIF (MIF-WT-Flag) or MIF mutants (MIF-E22Q-Flag and MIF-E22A-Flag) to confirm that AIF and MIF binding is required for MIF nuclear accumulation after NMDA administration (Fig. 5, A and B). Wild-type MIF and E22Q interacted with AIF, but MIF E22A did not bind to AIF (Fig. 5B). In nontransduced MIF knockout cultures and in MIF knockout cultures transduced with MIF-WT-Flag, MIF-E22Q-Flag, and MIF-E22A-Flag, AIF translocated to the nucleus when cells were exposed to NMDA (Fig. 5, C and D). Both MIF wild-type and MIF E22Q also translocated to the nucleus; however, the MIF E22A mutant, which is deficient in AIF binding, failed to do so (Fig. 5, C and D). Separation of nuclear and post-nuclear fractions confirmed the observations made by immunofluorescence (Fig. 5, E to G). These results indicate that MIF's interaction with AIF is required for the nuclear translocation of MIF.
Fig. 5. MIF E22A mutant prevents AIF's recruitment of MIF to the nucleus in NMDA-excitotoxicity.

(A) Expression of MIF in WT and knockout (KO) neurons. (B) Coimmunoprecipitation of Flag-tagged MIF variants and AIF in cortical neurons after NMDA treatment. (C) Images of nuclear translocation of AIF and exogenous MIF WT and MIF variants after NMDA treatment in MIF KO cortical neurons. Scale bar, 20 μm. White color indicates the overlay of AIF, MIF, and DAPI, showing the nuclear translocation of AIF and MIF. Purple color indicates the overlay of AIF and DAPI, showing the nuclear translocation of AIF. Z stacks illustrating the x,z and y,z axis are provided to demarcate the nucleus. (D) Quantification of the percentage of cells with nuclear translocation of AIF and exogenous MIF WT and MIF variants after NMDA treatment in MIF KO cortical neurons. ****P < 0.0001, versus KO group; ###P < 0.001, versus KO-WT group, one-way ANOVA. (E) Immunoblots of nuclear translocation of AIF and exogenous MIF WT and MIF variants after NMDA treatment in MIF KO cortical neurons. H4, histone H4; mito, mitochondrial antibody. (F and G) Relative levels of AIF and MIF in total protein (T), postnuclear fraction (PN) and nuclear fraction (N). Means ± SEM. Experiments were replicated at least three times. **P <0.01, ****P < 0.001, versus KO control group; ##P < 0.01, ####P < 0.0001, versus its total protein, one-way ANOVA.
MIF nuclease activity is required for chromatinolysis and parthanatos
To determine whether MIF's nuclease activity and AIF-mediated recruitment are required for parthanatos, we transduced MIF knockout cultures with the nuclease-deficient MIF E22Q mutant and the AIF binding–deficient MIF E22A mutant. Consistent with the shRNA experiments, cortical cultures lacking MIF were resistant to NMDA excitotoxicity (Fig. 6, A and B). Transduction of cells with wild-type MIF or the tautomerase-deficient mutant MIF P2G fully restored NMDA excitotoxicity; conversely, neither MIF E22Q nor MIF E22A restored NMDA excitotoxicity (Fig. 6, A and B). By the comet assay, a method to measure DNA damage, we found that NMDA administration in wild-type cortical neurons resulted in substantial numbers of neurons with DNA damage, whereas no such damage was detected in MIF knockout neurons (Fig. 6, C to F). Transduction of knockout neurons with wild-type MIF, but not with MIF E22Q or MIF E22A, restored DNA damage in cells treated with NMDA (Fig. 6, C to F). Depletion of MIF with shRNA in HeLa cells with two different shRNAs resulted in a reduced number of cells showing damaged DNA after treatment with MNNG compared with DNA in cells treated with nontargeted shRNA (fig. S14, A to D). A pulsed-field gel electrophoresis assay of genomic DNA confirmed that NMDA administration caused large DNA fragments in wild-type cortical neurons but not in MIF knockout cortical neurons (Fig. 6G). No obvious large DNA fragments were observed in MIF knockout neurons transduced with MIF E22Q or MIF E22A (Fig. 6G). Transduction of knockout neurons with wild-type MIF or MIF P2G restored NMDA-induced formation of large DNA fragments (Fig. 6G). HeLa cells lacking MIF after we used CRISPR-Cas9 were resistant to MNNG toxicity (fig. S15, A to C). Transduction of knockout HeLa cells with wild-type MIF or MIF P2G restored MNNG-induced formation of large DNA fragments and toxicity (fig. S15). These results indicate that MIF is the major nuclease involved in large-scale DNA fragmentation during MNNG- or NMDA-induced parthanatos, which is independent from MIF's tautomerase activity.
Fig. 6. MIF nuclease activity is critical for DNA damage and PARP-1–dependent cell death in cortical neurons.

(A) Representative images and (B) quantification of NMDA-induced (500 μM for 5 min) excitotoxicity in MIF WT, KO, and KO cortical neurons expressing MIF WT, E22Q, E22A, or P2G. Scale bar, 200 μm. (C) Representative images and (D to F) quantification of NMDA-induced DNA damage 6 hours after treatment determined by the comet assay in MIF WT, KO, and KO neurons expressing MIF WT, E22Q, E22A, or P2G. Dashed lines indicate the center of the head and tail. Scale bar, 20 μm. (G) Pulsed-field gel electrophoresis assay of NMDA-induced DNA damage 6 hours after treatment in MIF WT and KO neurons and KO neurons expressing MIF WT, E22Q, E22A, or P2G. Means ± SEM are shown in (B), (D), (E), and (F). *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA; ns, nonsignificant.
To evaluate the requirement of MIF nuclease activity and MIF binding to AIF in cell death due to parthanatos in vivo, we transduced MIF knockout mice with adeno-associated virus serotype 2 virus (AAV2) containing wild-type MIF, or the nuclease-deficient MIF E22Q mutant or the AIF-binding-deficient MIF E22A mutant by injecting the different AAV2 MIFs into the intracerebroventricular zone of newborn mice. The effectiveness of transduction was confirmed by immunostaining for MIF-Flag in the cortex, striatum, and hippocampus in adult mice (fig. S16, A and B). Two-month old male mice were then subjected to 45-min transient occlusion of the middle cerebral artery (MCAO). Despite the similar intensity of the ischemic insult (fig. S16C), infarct volume as previously reported (34) was reduced in MIF knockout mice in the cortex, striatum, and hemisphere by about 75% compared to that in their wild-type counterparts (Fig. 7, A to D). Moreover, the neuroprotection in MIF knockout mice remained for at least 7 days (Fig. 7, C and D). Expression of wild-type MIF, but not MIF E22Q or MIF E22A, in the MIF knockout mice restored infarct volume to that observed in wild-type animals (Fig. 7, A to D). We assessed behavior by spontaneous activity in the open field task on days 1, 3, and 7 after MCAO. Consistent with the infarct data, MIF knockout mice had improved behavioral scores compared to those of wild-type mice. MIF knockout mice expressing wild-type MIF had behavioral scores equivalent to those of wild-type mice whereas expression of MIF E22Q or MIF E22A had no effect (Fig. 7, E and F). Over 3 and 7 days, the behavioral scores of MIF knockout mice remained higher than those of wild-type treated mice (Fig. 7, F and G). A corner test measuring sensorimotor function showed that all mice do not show a side preference before MCAO surgery. However, wild-type mice and MIF knockout mice expressing wild-type MIF had significantly (P < 0.05 to P < 0.001, one-way analysis of variance (ANOVA)] increased turning toward the non-impaired side at days 1, 3, and 7 after MCAO (Fig. 7G), indicating these mice have more severe sensory and motor deficits. No preference was observed in MIF knockout mice and MIF knockout mice with expression of MIF E22Q or MIF E22A (Fig. 7G).
Fig. 7. MIF nuclease activity is critical for DNA damage and ischemic neuronal cell death in vivo.

(A) Representative images of triphenyl tetrazolium chloride staining of MIF WT, KO, and KO mice that were injected with AAV2-MIF WT, E22Q, or E22A 24 hours after 45 min of middle cerebral artery occlusion (MCAO). (B to D) Quantification of infarction volume in cortex, striatum, and hemisphere 1 day or 7 days after 45-min MCAO. WT MCAO (n = 29); KO MCAO (n = 20); KO-WT MCAO (n = 23). KO-E22Q (n = 22) and KO-E22A MCAO (n = 19). *P < 0.05, ***P < 0.001, versus KO group at the same time point; ##P < 0.01, ###P < 0.001, the same group at 7 days versus at 1 day after 45-min MCAO, one-way ANOVA. (E to G) Neurological deficit was evaluated by [(E) and (F)] open field on a scale of 0 to 5 and (G) corner test evaluated by percentage of right turns at 1 day, 3 days, or 7 days after MCAO surgery. WT MCAO (n = 16); KO MCAO (n = 12); and KO-WT MCAO (n = 16). KO-E22Q MCAO (n = 16) and KO-E22A MCAO (n = 16). Means ± SEM. *P < 0.05, ***P < 0.001, one-way ANOVA in (E) and (G). **P < 0.01, two-way ANOVA in (F), WT and KO-WT versus KO, KO-E22Q, and KO-E22A at different time points. (H) DNA fragmentation determined by pulsed-field gel electrophoresis in the penumbra 1 day, 3 days, or 7 days after 45-min MCAO surgery in MIF WT, KO, and KO mutant mice, which were injected with AAV2-MIF WT, E22Q, or E22A. WT MCAO (n = 15); KO MCAO (n = 15); and KO-WT MCAO (n = 15). KO-E22Q (n = 15) and KO-E22A MCAO (n = 15). (I) Quantification of noncleaved genomic DNA. Means ± SEM. ****P < 0.0001, versus its sham treatment group, one-way ANOVA.
Significant (P < 0.0001, one-way ANOVA) DNA damage as assessed by pulse field gel electrophoresis was observed at days 1, 3, and 7 after MCAO in wild-type mice or MIF knockout mice expressing wild-type MIF (Fig. 7, H and I). DNA damage was reduced in the MIF KO mice and MIF knockout mice expressing E22Q or E22A MIF (Fig. 7, H and I). We examined the localization of AIF and MIF by confocal microscopy in the penumbra region of the stroke (fig. S17, A and B). Consistent with the observation in cultured cortical neurons, AIF significantly (P < 0.001, one-way ANOVA) translocated to the nucleus at 1, 3, and 7 days after MCAO in wild-type animals. In MIF knockout animals as well as MIF knockout mice injected with MIF wild-type, E22Q, and E22A AIF significantly (P < 0.001, one-way ANOVA) translocated to the nucleus at 1 and 3 days after MCAO and there was reduced translocation of AIF at 7 days (fig. S17, A and B). Both MIF wild-type and MIF E22Q also significantly (P < 0.001, one-way ANOVA) translocated to the nucleus at 1 and 3 days after MCAO and there was reduced translocation at 7 days; however, the AIF binding–deficient mutant MIF E22A failed to do so (fig. S17, A and B). These data indicate that MIF is required for AIF-mediated neurotoxicity and DNA cleavage and that AIF is required for MIF translocation in vivo.
Conclusion
We identified MIF as a PAAN. Prior crystallization studies of MIF allowed us to show via 3-D modeling that MIF is structurally similar to PD-D/E(x)K nucleases (25, 26). The MIF monomer, which has pseudo 2-fold symmetry does not contain the core PD-D/E(X)K structure since the MIF monomer has four β strands next to the two α helices, and the orientations of the β-strands within an isolated monomer do not fit the requirement of the PD-D/E(x)K topology (23). However, our structure-activity analyses based on the MIF trimer, which has 3-fold symmetry, indicated that the interactions of the β strands of each monomer with the other monomers results in a MIF PD-D/E(x)K structure that consists of four β strands next to two α strands (23). Two of the β strands are parallel (β-4 and β-5), whereas the other two strands (β-6 and β-7) (from the adjacent monomer) are antiparallel. This topology exquisitely supports the idea that MIF's nuclease activity requires the trimer as the monomers do not support the required topology and is consistent with MIF existing as a trimer. The PD-D/E(X)K domains in MIF are highly conserved in vertebrates. The glutamic acid residue (E22) in the first α helix of MIF is critical for its nuclease activity, which is consistent with prior reports that this glutamic acid in the first α helix of many exonuclease-endonuclease-phosphatase (EEP) domain superfamily nucleases is highly conserved, and it is the active site for nuclease activity (25, 26).
MIF has both 3′ exonuclease and endonuclease activity. It preferentially binds to 5′ unpaired bases of ssDNA with the stem-loop structure and cleaves its 3′ unpaired bases. AIF interacts with MIF and recruits MIF to the nucleus where MIF binds and cleaves genomic DNA into large fragments similar to the size induced by stressors that activate parthanatos. MIF binding to AIF facilitates its cleavage of double-stranded genomic DNA, and, based on the chromatin immunoprecipitation sequencing ChiP-seq data, the average distance of MIF binding is about 15 to 60 kb, which is comparable to the size of large DNA fragments caused by MIF. MIF's cleavage of genomic DNA into 20- to 50-kb fragments is likely due to its rare binding on genomic DNA. Knockout of MIF reduces DNA fragmentation induced by stimuli that activate PARP-1–dependent cell death. Mutating a key amino acid residue, glutamic acid residue (E22), in the PD-D/E(X)K motif eliminates MIF's nuclease activity and protects cells from parthanatos both in vitro and in vivo. Disruption of the AIF and MIF protein-protein interaction prevents the translocation of MIF from the cytosol to the nucleus, which also protects against PARP-1–dependent cell death both in vitro and in vivo. Neither MIF's thiol-protein oxidoreductase activity nor tautomerase activity are involved in its actions as a nuclease. Knockout of MIF, a MIF nuclease–deficient mutant and a MIF AIF binding–deficient mutant all reduce infarct volume and have long-lasting behavioral rescue in the focal ischemia model of stroke in mice. Thus, MIF is a PAAN that is important in cell death because of activation of PARP-1 and the release of AIF (2). Future studies are required to further determine whether the stem-loop–ssDNA binding activity or the 3′ exonuclease and endonuclease activities of MIF, is important for its in vivo PAAN activity. In addition, our stroke data from MIF knockout mice indicate that other nucleases other than MIF might be involved in ischemic neuronal cell death. However, how these nucleases interact with MIF and contribute to PARP-1–induced cell death requires future studies.
MIF has a variety of pleiotropic actions. It is widely distributed throughout the brain (35, 36). It functions as a nonclassically secreted cytokine and may play important roles in cancer biology, immune responses, and inflammation (18, 37). MIF also has important roles in cellular stress and apoptosis (34, 38, 39). How MIF's nuclease activity relates to its role in the immune system and its other actions requires future studies.
Like PARP, inhibition of MIF nuclease activity is an attractive target for acute neurologic disorders. However, it may have advantages over PARP inhibition in chronic neurodegenerative diseases where long-term inhibition of PARP could impair detection and repair of DNA damage. Inhibition of MIF's nuclease activity could bypass this potential concern and offer a therapeutic opportunity for various disorders.
Materials and methods
Human protein chip high-throughput screening
Human protein chips (16K and 5K), which were prepared by spotting more than 16,000 or 5000 highly purified proteins onto special nitrocellulose-coated slides (16), were incubated in renaturation buffer containing 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM DTT, 0.3% Tween 20 for 1 hour at 4°C. After Blocking with 5% non-fat dry milk for 1 hour at room temperature, protein chips were incubated with purified mouse AIF protein (50 nM, NP_036149) in 1% milk for 1 hour. Protein interaction was then determined either by sequentially incubating with rabbit anti-AIF antibody (JH532, JHU) (9, 11) and Alexa Fluor 647 donkey anti-rabbit IgG, or Alexa Fluor 647 donkey anti-rabbit IgG only as negative control. Protein microarrays were scanned with GenePix 4000B Microscanner (Tecan) using the Cy5 image and the median fluorescence of each spot was calculated. We used the same procedure described previously to identify interacting proteins (16).
Reverse transfection format siRNA-based screen for PARP-1–dependent cell viability
On-Target plus SMARTpool siRNAs targeting AIF-interacting proteins resulting from human protein chip high throughput screening were customized in 96-well plates from Dharmacon. The plates were rehydrated using DharmaFECT 1 transfection reagent at room temperature for 30 min. HeLa cells were then seeded in the plates with the cell density at 1 × 104/well. 48 hours after transfection, cells were treated with MNNG (50 μM) or DMSO for 15 min and then incubated in normal complete medium for 24 hours. After adding Alamar Blue for 1–4 hours, cell viability was determined by fluorescence at excitation wavelength 570 nm and Emission wavelength 585 nm. PARP-1 siRNAs were used as the positive control and non-target siRNAs as the negative control.
Nuclease assays
Human genomic DNA (200 ng/reaction, Promega), pcDNA (200 ng/reaction) or PS30 and its related and non-related substrates (1 μM) was incubated with wild-type MIF or its variants at a final concentration of 0.25–8 μM as indicated in 10 mM Tris-HCl buffer (pH 7.0) containing 10 mM MgCl2 or specific buffer as indicated, for 1 hour (with pcDNA and small DNA substrates) or 4 hours (with human genomic DNA) at 37°C. The reaction was terminated with loading buffer containing 10 mM EDTA and incubation on ice. The human genomic DNA samples were immediately separated on a 1.2% pulse field certified agarose in 0.5 × TBE buffer with initial switch time of 1.5 s and a final switch time of 3.5 s for 12 hours at 6 V/cm. pcDNA samples were determined by 1% agarose gel. Small DNA substrates were separated on 15% or 25% TBE-urea polyacrylamide (PAGE) gel or 20% TBE PAGE gel. The gel was then stained with 0.5 μg/ml Ethidium Bromide (EtBr) followed by electrophoretic transfer to a nylon membrane. Biotin-labeled DNA was further detected by chemiluminescence using the Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific).
Electrophoretic mobility shift assay (EMSA)
EMSA assays were performed using the Light-Shift Chemiluminescent EMSA kit (Thermo Scientific) following the manufactures instruction. Briefly, purified MIF protein (2 μM) was incubated with biotin-labeled DNA substrates (10 nM) in the binding buffer containing 10 mM MgCl2 for 30 min on ice. Then samples were separated on 6% retardation polyacrylamide followed by electrophoretic transfer to a nylon membrane. Biotin-labeled DNA was further detected by chemiluminescence using the Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific).
Comet assay
Comet assays were conducted following protocols provided by Trevigen (Gaithersburg, MD). Briefly, HeLa cells with or without MNNG treatment and cortical neurons with or without NMDA treatment were washed with ice-cold PBS 6 hours after the treatment, harvested by centrifugation at 720 g for 10 min and re-suspended in ice-cold PBS (Ca2+ and Mg2+ free) at 1 × 105 cells/ml. Cells were then combined with 1% low melting point agarose in PBS (42°C) in a ratio of 1:10 (v/v), and 50 μl of the cell-agarose mixture was immediately pipetted onto the Comet Slide and placed flatly at 4°C in the dark for 30 min to enhance the attachment. After being lysed in lysis buffer, slides were immersed with alkaline unwinding solution (200 mM NaOH, pH >13, 1 mM EDTA) for 1 hour at RT. The comet slides were transferred and electrophoresed with 1 L of alkaline unwinding solution at 21 Volts for 30 min in a horizontal electrophoresis apparatus. After draining the excess electrophoresis buffer, slides were rinsed twice with dH2O and then fixed with 70% ethanol for 5 min and stained with SYBR Green for 5 min at 4°C. Cell images were captured using a Zeiss epifluorescent microscope (Axiovert 200M) and image analysis was performed with CASP software (version 1.2.2). The length of the “comet tail,” which is termed as the length from the edge of the nucleus to the end of the comet tail, for each sample, was measured.
Protein expression and purification
Human EndoG (NM_004435), cyclophilin A (NM_021130), mouse AIF (NM_012019), human MIF (NM_002415) cDNA and their variants were subcloned into glutathione S-transferase (GST)-tagged pGex-6P-1 vector (GE Healthcare) by EcoRI and XhoI restriction sites and verified by sequencing. The protein was expressed and purified from Escherichia coli by glutathione sepharose. The GST tag was subsequently proteolytically removed for the nuclease assay. MIF point mutants were constructed by polymerase chain reaction (PCR) and verified by sequencing. The purity of MIF proteins that were used in the nuclease assays was further confirmed by mass spectrometry. MIF proteins purified by FPLC were also used in the nuclease assays and no obvious difference was observed between FPLC MIF and non-FPLC MIF proteins. GST protein was used as a negative control in the nuclease assay.
Middle cerebral artery occlusion (MCAO)
Cerebral ischemia was induced by 45 min of reversible MCAO as previously described (40). Adult male MIF knockout (KO) mice (2 to 4 months old, 20 to 28 g) were anesthetized with isoflurane and body temperature was maintained at 36.5 ± 0.5°C by a feedback-controlled heating system. A midline ventral neck incision was made, and unilateral MCAO was performed by inserting a 7.0 nylon monofilament into the right internal carotid artery 6–8 mm from the internal carotid/pterygopalatine artery bifurcation via an external carotid artery stump. Sham-operated animals were subjected to the same surgical procedure, but the suture was not advanced into the internal carotid artery. After 1 day, 3 days or 7 days of reperfusion, mice were perfused with PBS and stained with triphenyl tetrazolium chloride (TTC). The brains were further fixed with 4% PFA and sliced for immunohistochemistry (9, 11, 41).
ChIP-seq
We preformed ChIP-seq as previously described (42, 43). Briefly, HeLa Cells were first treated with DMSO or MNNG (50 μM, 15 min). 5 hours after MNNG treatment, cells were cross-linked with 1% formaldehyde for 20 min at 37°C, and quenched in 0.125 M glycine. Chromatin extraction was performed before sonication. The anti-MIF antibody (ab36146, Abcam) was used and DNA was immunoprecipitated from the sonicated cell lysates. The libraries were prepared according to Illumina's instructions accompanying the DNA Sample kit and sequenced using an Illumina HiSEq. 2000 with generation of 50 bp single-end reads. Detailed procedures are as follows. HeLa cells were treated with DMSO or MNNG (50 μM) for 15 min and cultured in the fresh medium for an additional 5 hours. Cells were then cross-linked with 1% formaldehyde for 10 min at 37°C, and the reaction was quenched in 0.125 M glycine for 20 min at room temperature. Chromatin was extracted using SimpleChIP Enzymatic Chromatin IP kit from Cell Signaling Technology (Cat# 9003), and sonicated 30 s on and 30 s off for 15 cycles using a Bioruptor Twin (Diagenode). The quality and size of sheared chromatin DNA were examined on an agarose gel by DNA electrophoresis. 10% of chromatin was kept as input and the rest of the chromatin was diluted and pre-cleared using 10 μl Magnetic protein G agarose slurry for 30 min at 4°C to exclude nonspecific binding to protein G agarose beads directly. The pre-cleared chromatin was incubated overnight with an anti-MIF antibody (3 μg/ml, ab36146, Abcam) or control IgG (3 μg/ml) in the presence of Magnetic protein G agarose slurry (30 μl) at 4°C. After washing the protein G agarose beads for 3 times, half of the protein G agarose/antibody complex was subjected to immunoblot assays to check the quality of the immunoprecipitation. Another half of the protein G agarose/antibody complex was eluted in 170 μl of elution buffer containing 1% SDS, 0.1 M NaHCO3 at 65°C. The eluates as well as the chromatin input were treated with 1 mg/ml RNase A at 37°C for 30 min, and reverse-crosslinked by incubating at 65°C for 4 hours after adding 3 μl of 5 M NaCl and 1 μl of 10 mg/ml proteinase K. Finally the chromatin DNA was purified using phenol/chloroform/isoamyl alcohol and precipitated by ethanol. The ChIP and input DNA libraries were prepared using Illumina's Truseq DNA LT Sample Prep Kit according to the instructions. The final product was amplified for 15 cycles. The quality and the size of the insert was analyzed using a bioanalyzer. Sequencing was performed in the Next Generation Sequencing Center at Johns Hopkins using an Illumina HiSEq. 2000 with generation of 50 bp single-end reads. The ChIP-seq raw data have been deposited in the GEO database accession #: GSE65110.
ChIP-seq data analysis
Raw data from the HiSEq. 2000 was converted to FASTQ using CASAVA v1.8 and demultiplexed. Reads were mapped to the human genome (hg19) using Bowtie2 (v2.0.5) using the default parameters. Converted SAM files were passed to MACS (v1.4.1) for peak calling using the default parameters. Peaks from DMSO- and MNNG-treated libraries were reported in .bed format and are provided in GEO. Peaks differentially identified in the DMSO- and MNNG-treated groups were parsed by a custom R script. Sequence corresponding to peaks identified in only MNNG-treated, but not DMSO-treated libraries were fed into SeSiMCMC_4_36, Chipmunk v4.3+, and MEMEchip v4.9.0 for motif discovery using default parameters.
Data transfer: The CASAVAv1.8 software was used to convert the raw files into FASTQ files as well demultiplex the lanes.
MIF-DNA docking methods
A DNA duplex structure (44) (PDB accession 1BNA) and a single-stranded DNA structure [PDB accession 2RPD (45)] were docked onto the surface of MIF [PDB accession 1FIM (24)] using Hex-8.0. protein-DNA docking program (46, 47). The HEX program uses a surface complementarity algorithm to identify contact between protein and DNA. MIF surfaces were generated using Pymol. All images were viewed and labeled with pdb viewer, Pymol. The MIF-DNA docked models are shown as obtained from the HEX program.
Lentivirus, adeno-associated virus (AAV) construction and virus production
Mouse MIF-WT-Flag (NM_010798), MIF-E22Q-Flag and MIF-E22A-Flag were subcloned into a lentiviral cFugw vector by AgeI and EcoRI restriction sites, and its expression was driven by the human ubiquitin C (hUBC) promoter. Human MIF and mouse MIF shRNAs were designed using the website <http://katahdin.cshl.org/siRNA/RNAi.cgi?type=shRNA>. The program gave 97 nt oligo sequences for generating shRNAmirs. Using PacI SME2 forward primer 5' CAGAAGGTTAATTAAAAGGTATATTGCTGTTGACAGTGAGCG 3' and NheI SME2 reverse primer 5' CTAAAGTAGCCCCTTGCTAGCCGAGGCAGTAGGCA 3', we then PCR amplified them to generate the second strand and added PacI and NheI restriction sites to clone the products into pSME2, a construct that inserts an empty shRNAmir expression cassette in the pSM2 vector with modified restriction sites into the cFUGw backbone. This vector expresses GFP. The lentivirus was produced by transient transfection of the recombinant cFugw vector into 293FT cells together with three packaging vectors: pLP1, pLP2, and pVSV-G (1.3:1.5:1:1.5). The viral supernatants were collected at 48 and 72 hours after transfection and concentrated by ultracentrifugation for 2 hours at 50,000 g. MIF-WT-Flag, MIF-E22Q-Flag and MIF-E22A-Flag were subcloned into a AAV-WPRE-bGH (044 a.m./CBA-pI-WPRE-bGH) vector by BamHI and EcoRI restriction sites, and its expression was driven by chicken β-actin (CBA) promoter. All AAV2 viruses were produced by the Vector BioLabs.
Sequences of MIF substrates, templates, and primers
Sequences of MIF substrates, templates and primers used for shRNA constructs and point mutation constructs are provided in Table S1.
Cell culture, transfection, lentiviral transduction, and cytotoxicity
HeLa cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (HyClone). V5-tagged MIF was transfected with Lipofectamine Plus (Invitrogen). Primary neuronal cultures from cortex were prepared as previously described (9). Briefly, the cortex was dissected and the cells were dissociated by trituration in modified Eagle's medium (MEM), 20% horse serum, 30 mM glucose, and 2 mM l-glutamine after a 10-min digestion in 0.027% trypsin/saline solution (Gibco-BRL). The neurons were plated on 15-mm multiwell plates coated with polyornithine or on coverslips coated with polyornithine. Neurons were maintained in MEM, 10% horse serum, 30 mM glucose, and 2 mM l-glutamine in a 7% CO2 humidified 37°C incubator. The growth medium was replaced twice per week. In mature cultures, neurons represent 70 to 90% of the total number of cells. Days in vitro (DIV) 7 to 9, neurons were infected by lentivirus carrying MIF-WT-Flag, MIF-E22Q-Flag, or MIF-E22A-Flag [1 × 109 units (TU)/ml] for 72 hours. Parthanatos was induced by either MNNG (Sigma) in HeLa cells or NMDA (Sigma) in neurons. HeLa cells were exposed to MNNG (50 μM) for 15 min, and neurons (DIV 10 to 14) were washed with control salt solution [CSS, containing 120 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 25 mM tris-Cl, and 20 mM glucose (pH 7.4)], exposed to 500 μmM NMDA plus 10 μmM glycine in CSS for 5 min, and then exposed to MEM containing 10% horse serum, 30 mM glucose, and 2 mM l-glutamine for various times before fixation, immunocytochemical staining, and confocal laser scanning microscopy. Cell viability was determined the following day by unbiased objective computer-assisted cell counting after staining of all nuclei with 7 μM Hoechst 33342 (Invitrogen) and dead cell nuclei with 2 μM propidium iodide (Invitrogen). The numbers of total and dead cells were counted with the Axiovision 4.6 software (Carl Zeiss). At least three separate experiments using at least six separate wells were performed with a minimum of 15,000 to 20,000 neurons or cells counted per data point. For neuronal toxicity assessments, glial nuclei fluoresced at a different intensity than neuronal nuclei and were gated out. The percentage of cell death was determined as the ratio of live to dead cells compared with the percentage of cell death in control wells to account for cell death attributed to mechanical stimulation of the cultures.
Pull-down, coimmunoprecipitation, and immunoblotting
For the pull-down assay, GST-tagged MIF or AIF proteins immobilized glutathione Sepharose beads were incubated with 500 μg of HeLa cell lysates, washed in the lysis buffer, and eluted in the protein loading buffer. For coimmunoprecipitation, 1 mg whole-cell lysates were incubated overnight with AIF antibody (1 μg/ml) in the presence of protein A/G Sepharose (Santa Cruz Biotechnology), followed by immunoblot analysis with mouse anti-Flag antibody (Clone M1, Sigma), mouse anti-V5 (V8012, Sigma) or Goat anti-MIF (ab36146, Abcam). The proteins were separated on denaturing SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked and incubated overnight with primary antibody (50 ng/ml; mouse anti-Flag; rabbit anti-AIF; or goat anti-MIF) at 4°C, followed by horseradish peroxidase (HRP)–conjugated donkey anti-mouse, anti-rabbit or anti-goat for 1 hour at RT. After washing, the immune complexes were detected by the SuperSignalWest Pico Chemiluminescent Substrate (Pierce).
Subcellular fractionation
The nuclear extracts (N) and postnuclear cell extracts (PN), which is the fraction prepared from whole-cell lysates after removing nuclear proteins, were isolated in hypotonic buffer (9, 11). The integrity of the nuclear and postnuclear subcellular fractions was determined by monitoring histone H3 or H4 and MnSOD or mitochondria antibody (MTC02) (Mito) immunoreactivity (9, 11).
Immunocytochemistry, immunohistochemistry, and confocal microscopy
For immunocytochemistry, cells were fixed 4 hours after MNNG or NMDA treatment with 4% paraformaldehyde, permeabilized with 0.05% Triton X-100, and blocked with 3% BSA in PBS. AIF was visualized by Donkey anti-Rabbit Cy3 or donkey anti-rabbit 647. MIF was visualized by donkey anti-mouse cy2 (2 μg/ml), donkey anti-goat Cy2 or donkey anti-goat 647. Immunohistochemistry was performed with an antibody against Flag. Immunofluorescence analysis was carried out with an LSM710 confocal laser scanning microscope (Carl Zeiss) as described (9).
Quantification of relative percentage levels of AIF and MIF in subcellular fractions
The relative levels of AIF and MIF in different fractions were quantified and calculated as the percentage of their total proteins based on the intensity of protein signals relative to the protein amount prepared from the same number of cells (6 million). The detailed calculation is as follows: 1) The signal intensity of each interested band was measured and normalized to their mitochondrial and nuclear markers, with the total proteins of CSS in Fig. 4, G to I and knockout neurons treated with NMDA in Fig. 5 E to G. A volume factor was used to calculate the relative amount of total protein (T), post-nuclear protein (PN) and nuclear protein (N) prepared from the same number (6 million) of cells. As such, the relative ratio of different samples in the same fractions and the same sample in different fractions will be calculated as the relative intensity of total protein (Ti), post-nuclear fraction (PNi) and nuclear fraction (Ni). 2) A Z factor for the adjusted total proteins for each sample was determined via Z = (PNi + Ni)/Ti. 3) Relative protein levels in PN and N fractions were calculated as follows: PN% = (PNi × Z)/ Ti × 100%; N% = (Ni × Z)/Ti × 100%; T% = (Ti × Z)/ Ti × 100%.
FPLC
The native state and purity of the purified recombinant MIF were determined using standard calibration curve between elution volume and molecular mass (kD) of known molecular weight native marker proteins in Akta Basic FPLC (Amersham-Pharmacia Limited) using Superdex 200 10/300GL column (GE Healthcare, Life Sciences). The gel filtration column was run in standard PBS buffer at a flow rate of 0.5 ml/min. The following molecular weight standards were used: Ferritin (440 kD), aldolase (158 kD), conalbumin (75 kD), ovalbumin (43 kD), carbonic anhydrase (29 kD), and ribonuclease (13.7 kD) respectively (GE Healthcare, Life Sciences). Eluted fractions containing MIF were resolved on 12% SDS-PAGE and stained with commassie blue to check the purity of the protein.
Mass spectrometry analysis for MIF protein purity
MIF proteins used for nuclease assays were also examined by mass spectrometry in order to exclude any possible contamination from other known nucleases. We performed analyses using different criteria at a 95% and lower confidence levels in order to capture any remote possibility of a nuclease. Analysis and search of the NCBI database using all species reveal that no known nuclease that can digest single or double-stranded DNA was detected in the MIF protein that was used in the nuclease assays.
Circular dichroism (CD) spectroscopy
CD spectroscopy was performed on a AVIV 420 CD spectrometer (Biomedical Inc., Lakewood, NJ, USA). Near-UV CD spectra were recorded between 240–320 nm using a quartz cuvette of 0.5 cm path length with protein samples at a concentration of 2 mg/ml at room temperature. Far UV CD spectra were also recorded at room temperature between 190–260 nm using quartz sandwich cuvettes of 0.1 cm path length with protein samples at a concentration of 0.2 mg/ml (48). The proteins were suspended in PBS buffer with or without magnesium chloride (5.0 mM) and/or zinc chloride (0.2 mM). The CD spectra were obtained from 0.5 nm data pitch, 1 nm/3 s scan speed and 0.5 s response time were selected for the recordings.
Oxido-reductase activity assay
The thiol-protein oxidoreductase activity of MIF was measured using insulin as the substrate as described previously (30). Briefly, the insulin assay is based on the reduction of insulin and subsequent insolubilization of the insulin β-chain. The time-dependent increase in turbidity is then measured spectrophotometrically at 650 nm. The reaction was started by adding 5 μM MIF to WT, E22A, E22Q, C57A;C60A or and P2G mutants dissolved in 20 mM sodium phosphate buffer (pH 7.2), and 200 mM reduced glutathione (GSH) to ice-cold reaction mixture containing 1 mg/ml insulin, 100 mM sodium phosphate buffer (pH 7.2) and 2 mM EDTA. MIF insulin reduction was measured against the control solution (containing GSH) in the same experiment.
Tautomerase activity assay
Tautomerase activity was measured using d-dopachrome tautomerase as the substrate as described previously (49). Briefly, a fresh solution of d-dopachrome methyl ester was prepared by mixing 2 mM l-3,4 dihydroxyphenylalanine methyl ester with 4 mM sodium peroxidate for 5 min at room temperature and then placed directly on ice before use. The enzymatic reaction was initiated at 25°C by adding 20 μl of the dopachrome methyl ester substrate to 200 μl of MIF WT, E22A, E22Q, C57A;C60A (final concentration 5 μM) or and P2G mutants prepared in tautomerase assay buffer (50 mM potassium phosphate, 1 mM EDTA, pH 6.0). The activity was determined by the semi-continuous reduction of OD 475 nm using a spectrophotometer.
Quantification of noncleaved genomic DNA
Noncleaved genomic DNA was quantified as percentage (%) of the total genomic DNA that included both noncleaved genomic DNA and cleaved genomic DNA in each individual group.
Quantification of cells with AIF and MIF nuclear translocation
Nuclear translocation of AIF and MIF was calculated as the percentage of total cells in each individual immunostained image. At least 5 to 12 images were quantified for each group. 500 or more neurons were counted for each condition. White indicates the overlay of AIF (red), MIF (green) and 4′,6′-diamidino-2-phenylindole (DAPI) (blue) suggesting the nuclear translocation of both AIF and MIF. Pink indicates the overlay of AIF (red) and DAPI (blue) suggesting the nuclear translocation of AIF only. Cyan indicates the overlay of MIF (green) and DAPI (blue) suggesting the nuclear translocation of MIF. Representative immunostaining images of MIF and AIF nuclear translocation were shown in Figs. 4E and 5C and fig. S17A.
Intracerebroventricular (ICV) injection
Three microliters of AAV2-MIF WT, E22Q and E22A (1 × 1013 GC/ml, Vector BioLabs) were injected into both sides of intracerebroventricular of the newborn MIF KO mice (41). The expression of MIF and its variants were checked by immunohistochemistry after MCAO surgery at 8–16 weeks of age.
Neurobehavioral activity
Spontaneous motor activity was evaluated 1 day, 3 days and 7 days after MCAO by placing the animals in a mouse cage for 5 min. A video camera was fitted on top of the cage to record the activity of a mouse in the cage. Neurological deficits were evaluated by an observer blinded to the treatment and genotype of the animals with a scale of 0–5 (0, no neurological deficit; 5, severe neurological deficit). The following criteria were used to score deficits: 0 = mice appeared normal, explored the cage environment and moved around in the cage freely; 1 = mice hesitantly moved in cage but could occasionally touch the walls of the cage, 2 = mice showed postural and movement abnormalities, and did not approach all sides of the cage, 3 = mice showed postural and movement abnormalities and made medium size circles in the center of cage, 4 = mice with postural abnormalities and made very small circles in place, 5 = mice were unable to move in the cage and stayed at the center. Recordings were evaluated by observers blinded to the treatment and genotype of the animals.
The corner test was performed 1 day, 3 days and 7 days after MCAO to assess sensory and motor deficits following both cortical and striatal injury. A video camera was fitted on top of the cage to record the activity of a mouse in the cage for 5 min. The mice were placed between two cardboards each with a dimension of 30 cm × 20 cm × 0.5 mm attached to each other from the edges with an angle of 30°. Once in the corner, the mice usually rear and then turn either left or right. Before stroke mice do not show a side preference. Mice with sensory and motor deficits following stroke will turn toward the non-impaired side (right). Percent of right turn = right turns/total turns × 100 was calculated and compared. Recordings were evaluated by observers blinded to the treatment and genotype of the animals.
Animals
The Johns Hopkins Medical Institutions are fully accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). All research procedures performed in this study were approved the Johns Hopkins Medical Institutions Institutional Animal Care and Use Committee (IACUC) in compliance with the Animal Welfare Act regulations and Public Health Service (PHS) Policy. All animal studies were performed in a blinded fashion. Mouse genotype was determined by K.N. Stroke surgery was performed by R.A. Mouse genotypes were decoded after the stroke surgery, mouse behavior tests and data analysis. Based on their genotype, mice were grouped as WT, KO, KO-WT, KO-E22Q and KO-E22A. Within each group, mice were randomly assigned to subgroups including sham, 1 day-post stroke, 3 days- or 7 days-post stroke.
Statistical analysis
Unless otherwise indicated, statistical evaluation was carried out by Student's t test between two groups and by one-way analysis of variance (ANOVA) followed by post hoc comparisons with the Bonferroni test using GraphPad Prism software within multiple groups. Data are shown as means ± SEM. P <0.05 is considered significant. Experiments for quantification were performed in a blinded fashion. In order to ensure adequate power to detect the effect, at least 3 independent tests were performed for all molecular biochemistry studies and at least 5 mice from 3 different litters were used for animal studies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant K99/R00 NS078049, American Heart Association (AHA) National Scientist Development Grant 12SDG11900071, and the University of Texas Southwestern Medical Center Department of Pathology Startup funds and UT Rising Stars to Y.W.; and grants from National Institute on Drug Abuse, NIH, DA000266; and National Institute of Neurological Disorders and Stroke, NIH, R01 NS067525, R37 NS067525, and NS38377 to T.M.D. and V.L.D. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with the Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the foundation's Parkinson's Disease Program M-2016. ChIP-seq data are archived in National Center for Biotechnology Information, NIH, GEO, GSE65110. Y.W. contributed to all aspects of the project. R.A., G.K.U., K.N., H.P., B.K., L.B., M.M.H., C.C., R.C., S.M.E., M.M.H., T.-I.K., S.N., G.M., and H.S. helped with some experiments. J.S.J., S.B., and H.Z. provided protein chips. Z.X. and J.Q. helped with the bioinformatics analysis. Y.W., V.L.D., and T.M.D. designed experiments and wrote the paper. The study was conceived and scientifically directed by V.L.D. and T.M.D. Y.W., H.P., V.L.D., and T.M.D. are inventors on a patent application submitted by Johns Hopkins University that covers the use of inhibitors of MIF nuclease activity to treat stroke and other disorders. The supplementary materials contain additional data.
Footnotes
SUPPLEMENTARY MATERIALS www.sciencemag.org/content/354/6308/aad6872/suppl/DC1
REFERENCES AND NOTES
- 1.Bai P. Biology of poly(ADP-ribose) polymerases: The factotums of cell maintenance. Mol. Cell. 2015;58:947–958. doi: 10.1016/j.molcel.2015.01.034. doi: 10.1016/j.molcel.2015.01.034; pmid: 26091343. [DOI] [PubMed] [Google Scholar]
- 2.Fatokun AA, Dawson VL, Dawson TM. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014;171:2000–2016. doi: 10.1111/bph.12416. doi: 10.1111/bph.12416; pmid: 24684389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Dawson VL, Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp. Neurol. 2009;218:193–202. doi: 10.1016/j.expneurol.2009.03.020. doi: 10.1016/j.expneurol.2009.03.020; pmid: 19332058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am. J. Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. doi: 10.2353/ajpath.2008.080019; pmid: 18535182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szabó C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol. Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. doi: 10.1016/S0165-6147(98)01193-6; pmid: 9703762. [DOI] [PubMed] [Google Scholar]
- 6.Martire S, Mosca L, d'Erme M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer's and Parkinson's diseases. Mech. Ageing Dev. 2015;146–148:53–64. doi: 10.1016/j.mad.2015.04.001. doi: 10.1016/j.mad.2015.04.001; pmid: 25881554. [DOI] [PubMed] [Google Scholar]
- 7.Virág L, Robaszkiewicz A, Rodriguez-Vargas JM, Oliver FJ. Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med. 2013;34:1153–1167. doi: 10.1016/j.mam.2013.01.007. doi: 10.1016/j.mam.2013.01.007; pmid: 23416893. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, et al. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci. 2004;24:10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. doi: 10.1523/JNEUROSCI.3461-04.2004; pmid: 15574746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1–dependent cell death (parthanatos) Sci. Signal. 2011;4:ra20. doi: 10.1126/scisignal.2000902. doi: 10.1126/scisignal.2000902; pmid: 21467298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu SW, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. U.S.A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. doi: 10.1073/pnas.0606528103; pmid: 17116881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu SW, et al. Mediation of poly(ADP-ribose) polymerase-1–dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. doi: 10.1126/science.1072221; pmid: 12114629. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Yang C, Chai J, Shi Y, Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298:1587–1592. doi: 10.1126/science.1076194. doi: 10.1126/science.1076194; pmid: 12446902. [DOI] [PubMed] [Google Scholar]
- 13.David KK, Sasaki M, Yu SW, Dawson TM, Dawson VL. EndoG is dispensable in embryogenesis and apoptosis. Cell Death Differ. 2006;13:1147–1155. doi: 10.1038/sj.cdd.4401787. doi: 10.1038/sj.cdd.4401787; pmid: 16239930. [DOI] [PubMed] [Google Scholar]
- 14.Irvine RA, et al. Generation and characterization of endonuclease G null mice. Mol. Cell. Biol. 2005;25:294–302. doi: 10.1128/MCB.25.1.294-302.2005. doi: 10.1128/MCB.25.1.294-302.2005; pmid: 15601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Z, et al. Endonuclease G does not play an obligatory role in poly(ADP-ribose) polymerase-dependent cell death after transient focal cerebral ischemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;299:R215–R221. doi: 10.1152/ajpregu.00747.2009. doi: 10.1152/ajpregu.00747.2009; pmid: 20427721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu S, et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell. 2009;139:610–622. doi: 10.1016/j.cell.2009.08.037. doi: 10.1016/j.cell.2009.08.037; pmid: 19879846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calandra T, et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat. Med. 2000;6:164–170. doi: 10.1038/72262. doi: 10.1038/72262; pmid: 10655104. [DOI] [PubMed] [Google Scholar]
- 18.Merk M, Mitchell RA, Endres S, Bucala R. d-Dopachrome tautomerase (d-DT or MIF-2): Doubling the MIF cytokine family. Cytokine. 2012;59:10–17. doi: 10.1016/j.cyto.2012.03.014. doi: 10.1016/j.cyto.2012.03.014; pmid: 22507380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kosinski J, Feder M, Bujnicki JM. The PD-(D/E)XK superfamily revisited: Identification of new members among proteins involved in DNA metabolism and functional predictions for domains of (hitherto) unknown function. BMC Bioinformatics. 2005;6:172. doi: 10.1186/1471-2105-6-172. doi: 10.1186/1471-2105-6-172; pmid: 16011798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kratz K, et al. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell. 2010;142:77–88. doi: 10.1016/j.cell.2010.06.022. doi: 10.1016/j.cell.2010.06.022; pmid: 20603016. [DOI] [PubMed] [Google Scholar]
- 21.MacKay C, et al. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell. 2010;142:65–76. doi: 10.1016/j.cell.2010.06.021. doi: 10.1016/j.cell.2010.06.021; pmid: 20603015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugimoto H, Suzuki M, Nakagawa A, Tanaka I, Nishihira J. Crystal structure of macrophage migration inhibitory factor from human lymphocyte at 2.1 A resolution. FEBS Lett. 1996;389:145–148. doi: 10.1016/0014-5793(96)00553-4. doi: 10.1016/0014-5793(96)00553-4; pmid: 8766818. [DOI] [PubMed] [Google Scholar]
- 23.Sun HW, Bernhagen J, Bucala R, Lolis E. Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc. Natl. Acad. Sci. U.S.A. 1996;93:5191–5196. doi: 10.1073/pnas.93.11.5191. doi: 10.1073/pnas.93.11.5191; pmid: 8643551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki M, et al. Crystal structure of the macrophage migration inhibitory factor from rat liver. Nat. Struct. Biol. 1996;3:259–266. doi: 10.1038/nsb0396-259. doi: 10.1038/nsb0396-259; pmid: 8605628. [DOI] [PubMed] [Google Scholar]
- 25.Laganeckas M, Margelevicius M, Venclovas C. Identification of new homologs of PD-(D/E)XK nucleases by support vector machines trained on data derived from profile-profile alignments. Nucleic Acids Res. 2011;39:1187–1196. doi: 10.1093/nar/gkq958. doi: 10.1093/nar/gkq958; pmid: 20961958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steczkiewicz K, Muszewska A, Knizewski L, Rychlewski L, Ginalski K. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 2012;40:7016–7045. doi: 10.1093/nar/gks382. doi: 10.1093/nar/gks382; pmid: 22638584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pingoud V, et al. On the divalent metal ion dependence of DNA cleavage by restriction endonucleases of the EcoRI family. J. Mol. Biol. 2009;393:140–160. doi: 10.1016/j.jmb.2009.08.011. doi: 10.1016/j.jmb.2009.08.011; pmid: 19682999. [DOI] [PubMed] [Google Scholar]
- 28.Lubetsky JB, et al. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J. Biol. Chem. 2002;277:24976–24982. doi: 10.1074/jbc.M203220200. doi: 10.1074/jbc.M203220200; pmid: 11997397. [DOI] [PubMed] [Google Scholar]
- 29.Fingerle-Rowson G, et al. A tautomerase-null macrophage migration-inhibitory factor (MIF) gene knock-in mouse model reveals that protein interactions and not enzymatic activity mediate MIF-dependent growth regulation. Mol. Cell. Biol. 2009;29:1922–1932. doi: 10.1128/MCB.01907-08. doi: 10.1128/MCB.01907-08; pmid: 19188446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kudrin A, et al. Human macrophage migration inhibitory factor: A proven immunomodulatory cytokine? J. Biol. Chem. 2006;281:29641–29651. doi: 10.1074/jbc.M601103200. doi: 10.1074/jbc.M601103200; pmid: 16893895. [DOI] [PubMed] [Google Scholar]
- 31.Rosengren E, et al. The macrophage migration inhibitory factor MIF is a phenylpyruvate tautomerase. FEBS Lett. 1997;417:85–88. doi: 10.1016/s0014-5793(97)01261-1. doi: 10.1016/S0014-5793(97)01261-1; pmid: 9395080. [DOI] [PubMed] [Google Scholar]
- 32.Machanick P, Bailey TL. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. doi: 10.1093/bioinformatics/btr189; pmid: 21486936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao G, Zhao B, Tong Z, Mu R, Guan Y. Effects of 2′-O-methyl nucleotide substitution on EcoRI endonuclease cleavage activities. PLOS ONE. 2013;8:e77111. doi: 10.1371/journal.pone.0077111. doi: 10.1371/journal.pone.0077111; pmid: 24194862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inácio AR, Ruscher K, Leng L, Bucala R, Deierborg T. Macrophage migration inhibitory factor promotes cell death and aggravates neurologic deficits after experimental stroke. J. Cereb. Blood Flow Metab. 2011;31:1093–1106. doi: 10.1038/jcbfm.2010.194. doi: 10.1038/jcbfm.2010.194; pmid: 21063426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bacher M, et al. MIF expression in the rat brain: Implications for neuronal function. Mol. Med. 1998;4:217–230. pmid: 9606175. [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, et al. Expression of macrophage migration inhibitory factor in the mouse neocortex and posterior piriform cortices during postnatal development. Cell. Mol. Neurobiol. 2014;34:1183–1197. doi: 10.1007/s10571-014-0094-1. doi: 10.1007/s10571-014-0094-1; pmid: 25118614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calandra T, Roger T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat. Rev. Immunol. 2003;3:791–800. doi: 10.1038/nri1200. doi: 10.1038/nri1200; pmid: 14502271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hudson JD, et al. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J. Exp. Med. 1999;190:1375–1382. doi: 10.1084/jem.190.10.1375. doi: 10.1084/jem.190.10.1375; pmid: 10562313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleemann R, et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. 2000;408:211–216. doi: 10.1038/35041591. doi: 10.1038/35041591; pmid: 11089976. [DOI] [PubMed] [Google Scholar]
- 40.Andrabi SA, et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat. Med. 2011;17:692–699. doi: 10.1038/nm.2387. doi: 10.1038/nm.2387; pmid: 21602803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glascock JJ, et al. Delivery of therapeutic agents through intracerebroventricular (ICV) and intravenous (IV) injection in mice. J. Vis. Exp. 2011;2001:2968. doi: 10.3791/2968. pmid: 21988897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, et al. Systematic evaluation of factors influencing ChIP-seq fidelity. Nat. Methods. 2012;9:609–614. doi: 10.1038/nmeth.1985. doi: 10.1038/nmeth.1985; pmid: 22522655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. doi: 10.1038/nprot.2012.101; pmid: 22936215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drew HR, et al. Structure of a B-DNA dodecamer: Conformation and dynamics. Proc. Natl. Acad. Sci. U.S.A. 1981;78:2179–2183. doi: 10.1073/pnas.78.4.2179. doi: 10.1073/pnas.78.4.2179; pmid: 6941276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masuda T, Ito Y, Terada T, Shibata T, Mikawa T. A non-canonical DNA structure enables homologous recombination in various genetic systems. J. Biol. Chem. 2009;284:30230–30239. doi: 10.1074/jbc.M109.043810. doi: 10.1074/jbc.M109.043810; pmid: 19729448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghoorah AW, Devignes MD, Smaïl-Tabbone M, Ritchie DW. Protein docking using case-based reasoning. Proteins. 2013;81:2150–2158. doi: 10.1002/prot.24433. doi: 10.1002/prot.24433; pmid: 24123156. [DOI] [PubMed] [Google Scholar]
- 47.Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW. HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010;38:W445–W449. doi: 10.1093/nar/gkq311. Web Server. doi: 10.1093/nar/gkq311; pmid: 20444869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reddy PP, et al. Molecular dynamics of the neuronal EF-hand Ca2+-sensor Caldendrin. PLOS ONE. 2014;9:e103186. doi: 10.1371/journal.pone.0103186. doi: 10.1371/journal.pone.0103186; pmid: 25058677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bendrat K, et al. Biochemical and mutational investigations of the enzymatic activity of macrophage migration inhibitory factor. Biochemistry. 1997;36:15356–15362. doi: 10.1021/bi971153a. doi: 10.1021/bi971153a; pmid: 9398265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.