

This study provides evidence of cotranslational RNA decay in a plant transcriptome and reveals a number of surprising features and proteins involved in this process.
Abstract
RNA turnover is necessary for controlling proper mRNA levels posttranscriptionally. In general, RNA degradation is via exoribonucleases that degrade RNA either from the 5′ end to the 3′ end, such as XRN4, or in the opposite direction by the multisubunit exosome complex. Here, we use genome-wide mapping of uncapped and cleaved transcripts to reveal the global landscape of cotranslational mRNA decay in the Arabidopsis thaliana transcriptome. We found that this process leaves a clear three nucleotide periodicity in open reading frames. This pattern of cotranslational degradation is especially evident near the ends of open reading frames, where we observe accumulation of cleavage events focused 16 to 17 nucleotides upstream of the stop codon because of ribosomal pausing during translation termination. Following treatment of Arabidopsis plants with the translation inhibitor cycloheximide, cleavage events accumulate 13 to 14 nucleotides upstream of the start codon where initiating ribosomes have been stalled with these sequences in their P site. Further analysis in xrn4 mutant plants indicates that cotranslational RNA decay is XRN4 dependent. Additionally, studies in plants lacking CAP BINDING PROTEIN80/ABA HYPERSENSITIVE1, the largest subunit of the nuclear mRNA cap binding complex, reveal a role for this protein in cotranslational decay. In total, our results demonstrate the global prevalence and features of cotranslational RNA decay in a plant transcriptome.
INTRODUCTION
To maintain dynamic gene regulatory networks in cells, mRNA turnover is an important process in controlling mRNA abundance at the posttranscriptional level. In general, mRNA degradation is mediated by two main mechanisms: exoribonuclease-mediated RNA decay and endonucleolytic cleavage-dependent RNA degradation. In the plant cytoplasm, most mRNAs are degraded by the cytoplasmic 5′ to 3′ exoribonuclease, XRN4, and/or by the 3′ to 5′ exoribonucleolytic exosome complex. In total, the XRN family of 5′ to 3′ exoribonucleases consists of three proteins in plants (Nagarajan et al., 2013). XRN4 is the lone cytoplasm-localized family member, whereas both XRN2 and XRN3 are found in the nucleus (Kastenmayer and Green, 2000; Chiba and Green, 2009). Similarly, the exosome complex has active forms in both the cytoplasm and nucleus (Chekanova et al., 2007; Vanacova and Stefl, 2007).
In general, RNA decay is thought to be dependent on the initial removal of the 3′ poly(A) tail by the process of deadenylation (Garneau et al., 2007). Subsequently, mRNAs can be degraded by either 5′ to 3′ and/or 3′ to 5′ decay pathways. One exception to these general RNA turnover mechanisms in plants is degradation triggered by internal RNA cleavage mediated by microRNA (miRNA)-loaded RNA-induced silencing complexes. In this case, internal cleavage products produced by miRNA-directed ARGONAUTE1 endonucleolytic cleavage results in a 3′ fragment with a free 5′ monophosphate (5′P) and a 5′ fragment with a free 3′ hydroxyl that are degraded by XRN4 and the exosome complex, respectively (Souret et al., 2004). Interestingly, this miRNA-mediated regulatory pathway can also mediate target RNA regulation via translation repression (Chen, 2004; Gandikota et al., 2007; Li et al., 2013).
In the nucleus of eukaryotic cells, the 7-methylguanosine cap is bound by the nuclear cap binding complex (CBC) that consists of two proteins, CAP BINDING PROTEIN20 (CBP20) and CAP BINDING PROTEIN80/ABA HYPERSENSITIVE1. This nuclear CBC is thought to protect the 5′ end of mRNAs from the degradation machinery, but it has also been found to be important in eukaryotic gene transcription, splicing, transcript export, miRNA biogenesis, and translation (Gonatopoulos-Pournatzis and Cowling, 2014). Additionally, the nuclear CBC functions during the initial round of translation that occurs as mRNAs are being exported from the nucleus (Kim et al., 2009). In plants, this complex also plays a role in the biogenesis of miRNAs (Gregory et al., 2008), whereby its absence results in the accumulation of miRNA precursors with a concomitant loss of mature species (Gregory et al., 2008; Raczynska et al., 2010).
Recent studies in Saccharomyces cerevisiae demonstrated that some mRNAs are decapped and undergo 5′ to 3′ exonucleolytic decay while still associated with translating ribosomes (Hu et al., 2009; Pelechano et al., 2015). To do this, one group used an approach they called 5P sequencing (Pelechano et al., 2015) that is similar to a number of approaches, including our genome-wide mapping of uncapped and cleaved transcripts (GMUCT), that were previously developed in plant studies (Gregory et al., 2008; German et al., 2009; Addo-Quaye et al., 2008). These approaches specifically identify cleaved and degrading RNAs by selecting for those molecules that have a free 5′ monophosphate (5′P). However, this initial study in S. cerevisiae did not reveal whether cotranslational RNA degradation also occurs in plants or the features of this process in a multicellular eukaryote (Pelechano et al., 2015). Furthermore, the role of the nuclear CBC in cotranslational decay is also still unclear.
Here, we used GMUCT to reveal that cotranslational mRNA degradation does occur in the Arabidopsis thaliana flower bud transcriptome. We also found some interesting features associated with this process in plants that were not previously found in S. cerevisiae (Pelechano et al., 2015). Finally, we demonstrated that the nuclear CBC was also required for this process in Arabidopsis. In summary, we have provided a comprehensive characterization of cotranslational RNA decay in a plant transcriptome.
RESULTS
GMUCT Is Useful to Study miRNA-Directed Target RNA Cleavage and Cotranslational RNA Decay in Plant Transcriptomes
To globally assess miRNA-mediated cleavage and cotranslational RNA degradation in the Arabidopsis unopened flower bud transcriptome, we performed GMUCT on two biological replicates of Arabidopsis flower bud RNA. This procedure starts with the immediate ligation of a RNA adaptor directly to 5′ monophosphorylated RNA ends, thereby allowing the capture of all uncapped and cleaved transcripts in a transcriptome of interest (Gregory et al., 2008; Willmann et al., 2014). To focus specifically on degrading mRNAs, we aligned the resulting GMUCT reads to full length mature mRNA transcripts (TAIR10 annotation) (Supplemental Figure 1A). We found that 79 to 82% of unique reads were mapped to mRNA transcripts, indicating that cleaved and uncapped mRNAs compose the majority of 5′P intermediates in our poly(A)-selected GMUCT libraries. To determine reproducibility, we used a 100-nucleotide sliding window to determine the correlation of nonredundant sequence read abundance between biological replicates of GMUCT. From this analysis, we observed a high correlation in read counts between the two GMUCT replicate libraries (Pearson correlation = 0.93) (Supplemental Figure 2A), indicating the high quality and reproducibility of our GMUCT experiments. A more careful characterization of the read mapping locations revealed that ∼80% of the 5′P reads were from mRNA coding sequences (CDSs) and ∼10% from the 3′ untranslated region (UTR), whereas <5% mapped to the 5′ UTR (Supplemental Figure 2B). This shows that, in general, 5′P reads tend to accumulate at the 3′ ends of Arabidopsis protein-coding transcripts.
We then focused on two types of cleavage events that are likely present in the 5′P read populations produced by GMUCT: miRNA target sites and sites 16 to 17 nucleotides upstream of mRNA codons. The latter type represents the 5′ end of a ribosome-protected footprint (∼30 nucleotides total) having the codon in its A site. This provides evidence of cotranslational RNA decay in the Arabidopsis transcriptome (Pelechano et al., 2015; Liu et al., 2013b). To study miRNA-mediated cleavage sites, we predicted the target sites of conserved canonical miRNAs using psRNATarget (Dai and Zhao, 2011). We then determined whether there was evidence of cleavage at these predicted miRNA target sites using data from the GMUCT libraries (Supplemental Figure 1B). Additionally, to provide evidence of cotranslational RNA decay in a plant transcriptome, we masked the list of predicted miRNA cleavage sites and determined the accumulation of 5′P reads 16 to 17 nucleotides upstream of stop codons of Arabidopsis mRNAs, which has been shown to be the exact 5′ end of the ribosome footprint during the cotranslational decay process (Supplemental Figure 1C) (Pelechano et al., 2015).
GMUCT Reveals miRNA-Mediated Target RNA Cleavage Efficiency
To identify miRNA cleavage sites genome-wide, we computationally predicted potential target sites of the 45 miRNAs of Arabidopsis that are found throughout the Brassicaceae (Yu et al., 2012). We then used our GMUCT data to validate that we could detect cleavage at these sites. From this analysis, we found that our GMUCT data identified 410 miRNA cleavage sites that were enriched up to 8-fold compared with the 100 nucleotides that flanked these regions (local cleavage efficiency) and 2-fold compared with average coverage of 5′ P reads along the entire length of the transcript (global cleavage efficiency) (Supplemental Figure 1B and Supplemental Data Set 1). More specifically, we identified a significant (P value < 2.2 × 10−100, χ2 test) accumulation of 5′P reads in these validated miRNA cleavage sites compared with their 100-nucleotide flanking regions (Figure 1A).
Figure 1.

GMUCT Provides a Global View of miRNA Target Site Cleavage Efficiency.
(A) Accumulation of 5′P read ends at miRNA target sites. The top of the rectangle indicates the top of the third quartile, and the bottom of the rectangle indicates the bottom of the first quartile. The horizontal bold line near the middle of the rectangle indicates the median value of two biological replicates. The vertical line extending from the top of the rectangle indicates the maximum value, and another vertical line extending from the bottom of the rectangle indicates the minimum value.
(B) Cleavage efficiency of miRNA target sites. The four transcripts (AP2, SPL15, TOE2, and TCP4) with the most efficiently cleaved miRNA target sites are listed. The rest of the identified miRNA target sites (red dots) with efficient miRNA-directed cleavage can be found in Supplemental Data Set 1.
Although most plant miRNAs direct silencing of target mRNAs through nearly perfect complimentary base pairing with their targets, the miRNA-directed cleavage of their target sequences shows very different levels of efficiency. To better understand this characteristic, we defined the set of miRNAs that demonstrated significant local and global miRNA cleavage efficiency as described above (Supplemental Data Set 1). From these analyses, we found that two miR172 target mRNAs, TOE2 and AP2, and two miR156 target mRNAs, SPL15 and SPL3 (Wu et al., 2009; Aukerman and Sakai, 2003; Schwarz et al., 2008), are among the top seven transcripts with the highest identified miRNA cleavage efficiency (Figure 1B; Supplemental Figures 3A to 3C). We also identified three miR396 targets, one of which (GRF4) mediates pistil development (Liang et al., 2014), among the genes with the highest cleavage efficiency (Figure 1B; Supplemental Figure 3D and Supplemental Data Set 1). Interestingly, some genes that demonstrate very high levels of miRNA-mediated target cleavage efficiency, such as AP2 (miR172 target), SPL3 (miR156 target), and CSD1 (miR398 target), are regulated by translational repression (Chen, 2004; Gandikota et al., 2007; Dugas and Bartel, 2008). However, our results suggest that these miRNA target RNAs are regulated by both miRNA-mediated mRNA cleavage and translation repression. Overall, these findings provide a comprehensive analysis of the miRNA-mediated cleavage efficiency for 410 target mRNA transcripts in Arabidopsis unopened flower buds that were identified by this study (Supplemental Data Set 1).
GMUCT Reveals Ribosomal Pausing During Translation Termination and a 3-Nucleotide Periodicity in mRNA Open Reading Frames
Because our main goal in this study was to determine the prevalence and features of cotranslational RNA degradation in the Arabidopsis transcriptome, we did not want miRNA-directed RISC cleavage to dilute the potential signal from this degradation process. Therefore, for all subsequent analyses, we masked all identified miRNA cleavage sites in our GMUCT data sets (Figure 1B), allowing us to ignore signals from these regions. We then searched for other regions of mRNAs that gave very high signal in our GMUCT sequencing results. Using this approach, we observed that 5′P read ends tended to accumulate just upstream of mRNA stop codons (see Supplemental Figure 4 for an example). To determine whether there was a specific pattern in 5′P read ends in this region, we aligned the stop codons of all detectable mRNAs in our samples. This analysis revealed a significant (P value < 2.2 × 10−100, χ2 test) accumulation of 5′P read ends exactly 16 to 17 nucleotides upstream from mRNA stop codons. Significantly, this corresponds precisely to the 5′ boundary of the ribosome with its A site stalled at a stop codon (Figure 2A). Additionally, when focusing this analysis on the 2000 genes with highest levels of 5′P read ends, we again found a significant enrichment (P value < 2.2 × 10−100, χ2 test) of 5′P read ends at positions 16 to 17 nucleotides upstream of these stop codons compared with the flanking 100 nucleotides (Supplemental Figure 5). From this analysis, we also observed a clear 3-nucleotide periodicity of 5′P read ends within mRNA open reading frames (ORFs) (Figure 2A). In total, these results suggested that GMUCT can detect the process of cotranslational RNA decay in the Arabidopsis transcriptome.
Figure 2.

5′P Read Ends Accumulate at The Ribosome Boundary Site of mRNA ORF Stop Codons and Show a 3-Nucleotide Periodicity Pattern throughout ORFs.
(A) The distribution of 5′P read ends relative to stop codons. The first nucleotide of the stop codon is numbered 0 in all detectable mRNA ORFs. The illustration below the graph shows the 5′ ribosome boundary site when the ribosome has an mRNA stop codon in its A site.
(B) A 3-nucleotide periodicity of 5′P read ends is evident in Arabidopsis mRNA ORFs. Asterisks denote a significant difference at a P value < 2.2 × 10−100 as determined by a χ2 test. Error bars represent se of the mean for two biological replicates.
(C) Enrichment of 5′P read ends at the ribosome boundary (16 and 17 nucleotides upstream) of mRNA stop codons. Red dots denote codons with significant enrichment of 5′P read ends at their ribosome boundary sites, while gray dots denote codons that are not significant for this value. Significance was assessed using a χ2 test.
To determine whether this 3-nucleotide periodicity along mRNA ORFs was a global pattern in the GMUCT data, we broke each 3-nucleotide amino acid codon into their three possible coding frames and labeled them 0, 1, and 2, with frame 0 representing the frame used in translation. We then counted the accumulation of 5′P read ends in each frame along all mRNA ORFs. From this analysis, we found that 5′P read ends accumulated at significantly lower levels (P value < 2.2 × 10−100, χ2 test) within frame 0 compared with both other codon frames. Interestingly, frame 0 can be considered the position most likely to be ribosome-protected on each codon, providing further evidence of cotranslational mRNA degradation in Arabidopsis because the ribosome moves in 3-nucleotide intervals along ORFs during translation (Figure 2B). It is noteworthy that this observation is also consistent with our observation that the highest levels of 5′P read ends near stop codons occurs 16 and 17 nucleotides upstream of these sequences (Figure 2A; Supplemental Figure 5).
The observation of the 3-nucleotide periodicity in the GMUCT data led us to examine the levels of 5′P read ends at the ribosome boundary (RB) sites (16 to 17 nucleotides upstream) for each of the 64 codons. This includes both the start and stop codons compared with the flanking median 5′P read ends for each of these sequences. As expected, we found a strong enrichment of 5′P read ends at the ribosome boundary sites (16 to 17 nucleotides upstream) for all three types of stop codons (stop [all three sequences combined], TAA, TAG, and TGA) (Figure 2C). However, we did not observe an enrichment of 5′P read ends at the RB sites of any other codon (see Supplemental Figure 6B for an example showing the GAT codon), including the proline-encoding CCG and CGA codons that were found to promote ribosome stalling in S. cerevisiae (Pelechano et al., 2015). Interestingly, the 3-nucleotide periodicity in our GMUCT data around start codons was relatively weak compared with this pattern around amino acid codons found in the rest of the ORFs (Supplemental Figures 6A and 6B). This is consistent with previous findings from S. cerevisiae (Pelechano et al., 2015), suggesting that the exonuclease required for cotranslational RNA decay does not catch up with the ribosome until the ribosome is beyond the start codon performing translation elongation within the CDS. Additionally, these results suggest that, in Arabidopsis, the ribosome spends a substantial period of time paused only at stop codons, and not at others that code for amino acids.
To investigate ribosome pausing at each type of stop codon (TAA, TGA, and TAG), we looked at the 5′P read end profiles in the regions surrounding each of these three sequences (Supplemental Figure 7). From this analysis, we found that the pattern of 5′P read end accumulation at 16 and 17 nucleotides upstream of each stop codon displayed a distinct pattern, which was not previously observed in S. cerevisiae cotranslational RNA degradation (Pelechano et al., 2015). More specifically, we found that, at TAA stop codons, the majority of 5′P read ends accumulated at 16 nucleotides upstream with less being at 17 nucleotides upstream, whereas TAG showed the exact opposite distribution (Supplemental Figures 7A and 7B). TGA stop codons showed equally high accumulation of 5′P read ends at both 16 and 17 nucleotides upstream (Supplemental Figure 7C). Taken together, these results revealed that there is differential accessibility to 5′ to 3′ exoribonucleases at the 5′ boundary of pausing ribosomes terminating at the three different types of stop codons in Arabidopsis. These results suggest that the termination kinetics are different, and/or a distinct complex of proteins is associated with the terminating ribosomes bound to each class of stop codon in plants.
GMUCT Provides Evidence for Translationally Active uORFs
When further scanning the distribution of 5′P read ends along protein-coding transcripts, we also found a number of transcripts that had peaks at specific positions within their 5′ UTRs. For instance, the transcript of BZIP53 displayed such a peak, and closer inspection revealed that this accumulation of read ends corresponded especially to nucleotides 16 and 17 upstream of the end of an upstream ORF (uORF) known as CPUORF3 found in the 5′ UTR of this mRNA (Figure 3A). We also observed another significant peak of 5′P read ends at 46 as well as 47 nucleotides upstream from the stop codon of CPUORF3 (Figure 3A), indicating the presence of tandem ribosomes pausing near the stop codon of this uORF. These results for CPUORF3 suggested that this dual peak pattern is the one normally observed at high confidence Arabidopsis uORFs. To test this hypothesis, we aligned the stop codons of all high-confidence TAIR10-annotated uORFs and looked at 5′P read end accumulation upstream of these sequences. We identified a very high accumulation of 5′P read ends at 16 as well as 17 nucleotides upstream from the stop codon of this merged set of uORFs. Furthermore, we found a second smaller peak of 5′P read end accumulation exactly 30 nucleotides (46 to 47 nucleotides) upstream of this major cleavage site (Figure 3B). This second major cleavage peak (46 to 47 nucleotides upstream of stop codons) was not observed by this same analysis at major protein-coding ORFs (Figure 2A). Taken together, these results suggest that tandem ribosome occupancy occurs quite frequently at Arabidopsis uORFs but is mostly absent from the main protein-coding ORFs of Arabidopsis mRNAs.
Figure 3.

Accumulation of 5′P Read Ends at the Ribosome Boundary Site Upstream of uORF Stop Codons.
(A) An uORF example (CPUORF3 in the 5′ UTR of BZIP53) that shows accumulation of 5′P read ends at the ribosome boundary of the CPUORF3 stop codon in the two boxes below the transcript model. In the transcript model, the yellow rectangle represents the 5′ UTR upstream of CPUORF3, the light-blue box represents the ORF of CPUORF3, the intervening green box represents the rest of the 5′ UTR before the BZIP53 ORF (blue box), and the last green box represents the 3′ UTR of this transcript.
(B) The distribution of 5′P read ends relative to stop codons (first nucleotide of the stop codon is numbered 0) in all detectable TAIR10 annotated uORFs. The illustration below the graph shows how the two tandem ribosomes pausing at the termination site (stop codon in the A site) gives 5′P read end peaks 16 and 17 nucleotides upstream of the stop codon and secondary peaks 46 and 47 nucleotides upstream of the stop codon.
Recent studies have suggested that near-cognate codons, especially CTG and ACG, could also act as plant uORF translation initiation codons (Laing et al., 2015; Liu et al., 2013b). Given that our results suggest that GMUCT can detect translationally active uORFs that do not overlap with the main ORF in plant transcriptomes (Figures 3A and 3B), we wanted to test this hypothesis. Therefore, we predicted putative open reading frames in the 5′ UTRs of all Arabidopsis mRNAs using three initiation codons (ATG, CTG, and ACG) and then determined which uORFs predicted by GMUCT were translationally active by searching for a peak of 5′P read ends at the stop codon ribosome boundary (16 to 17 nucleotides upstream). From this analysis, we identified a total of 98 uORFs with enrichment of 5′P read ends at their stop codon ribosome boundary compared with their flanking regions (Supplemental Data Set 2). A closer examination of this list demonstrated that, among the top five predicted uORFs, were well-studied examples contained in the 5′ UTR of ATBZIP11 (AT4G34590) and XIPOTL1 (AT3G18000) (von Arnim et al., 2014; Wiese et al., 2004; Tabuchi et al., 2006). Also, found within this list were uORFs previously predicted to be present in the 5′ UTRs of ATCIPK6 (AT4G30960) and BZIP44 (AT1G75390) (Takahashi et al., 2012; Hayden and Jorgensen, 2007; Liu et al., 2013b). This analysis also identified more than 80 putative uORFs that were previously predicted, including those we found in the 5′ UTRs of FCA (AT4G16280) and AGL4/SEP2 (AT3G02310). In total, these results support the idea of utilizing GMUCT as a means of identifying specific, translationally active uORFs in plant transcriptomes.
Following Cycloheximide Treatment, GMUCT Highlights the Accumulation of Cleavage Events at the 5′ Ribosome Boundary of Start Codons
Cycloheximide (CHX) is a translation inhibitor that arrests ribosomes after they complete a single codon addition. Thus, CHX does not inhibit translation initiation but interferes with ribosome translocation and thus blocks translation elongation. To provide additional evidence of cotranslational RNA decay in the Arabidopsis transcriptome, we performed GMUCT on Col-0 leaf tissue following treatment with CHX. From this analysis, we again identified a pattern of 3-nucleotide periodicity within the ORFs of all detectable Arabidopsis protein-coding mRNAs (Figure 4), with a significant enrichment (P value < 2.2 × 10−100, χ2 test) of 5′P read ends in frame 2 compared with frames 0 and 1 (Figure 4A). The observation of a 3-nucleotide periodicity in the GMUCT data across mRNA ORFs led us to examine the levels of 5′P read ends at the ribosome boundary sites for each of the 64 codons. This includes both the start and stop codons compared with the flanking median 5′P read ends for each of these sequences. From this analysis, we found a strong enrichment of 5′P end reads at the ribosome boundary site of start codons, but we did not observe an enrichment of 5′P read ends at the RB sites of any other codon (Figure 4B). This was expected given that CHX treatment allows ribosomes to translocate one codon after inhibition, meaning that all initiating ribosomes should be stalled near the start codon, while terminating ribosomes will be lost from the transcript. After aligning all detectable mRNA transcripts by their start codons, we found a large accumulation of 5′P read ends 13 as well as, to a lesser extent, 14 nucleotides upstream of start codons compared with median 5′P read ends (Figure 4C). Furthermore, a similar analysis focusing on the stop codon revealed no peak of 5′ read end accumulation (Figure 4D). These results are consistent with initiating and terminating ribosomes having moved a single codon downstream of the start and stop codons during inhibition of elongation by CHX treatment, respectively. In total, these results indicate that GMUCT with and without CHX treatment can reveal the genome-wide landscape of cotranslational RNA decay in plant transcriptomes.
Figure 4.

5′P Read Ends Accumulate at the Ribosome Boundary Site of mRNA ORF Start Codons and Show a 3-Nucleotide Periodicity Pattern throughout ORFs in Col-0 Leaf Tissue Treated with the Translation Inhibitor CHX.
(A) A 3-nucleotide periodicity pattern of 5′P read ends is evident in Arabidopsis mRNA ORFs after treatment of Col-0 leaf tissue with CHX. Asterisks denote a significant difference at a P value < 2.2 × 10 to 100 as determined by a χ2 test. Error bars represent se of the mean for two biological replicates.
(B) The average number of 5′P read ends found at the ribosome boundary (16 and 17 nucleotides upstream) of each codon compared with the median coverage in the 50 nucleotide up and downstream. Red dots denote the codons with significant enrichment of 5′P read ends at their ribosome boundary site, while gray dots denote codons that are not significant for this value. Significance was assessed using a χ2 test.
(C) The distribution of 5′P read ends relative to start codons (first nucleotide of the start codon is numbered 0) in all detectable mRNA ORFs. The illustration shows how the ribosome boundary of an initiating ribosome during CHX treatment (start codon in the P site) gives 5′P read end peaks ∼13 nucleotides upstream of the start codon P site. The gray line is the distribution of 5′P read ends relative to start codons in Col-0 without CHX treatment.
(D) The distribution of 5′P read ends relative to stop codons (the first nucleotide of the stop codon is numbered 0) in all detectable mRNA ORFs. The gray line denotes the distribution of 5′P read ends relative to stop codons in Col-0 without CHX treatment.
XRN4 Is the Ribonuclease Required for Cotranslational mRNA Decay in Arabidopsis
Next, we wanted to determine which ribonuclease was required for cotranslational RNA decay in plants. Given that, in S. cerevisiae, Xrn1 is required for this process (Pelechano et al., 2015), the natural candidate is the plant ortholog XRN4. Furthermore, XRN4 is the major cytoplasmic 5′ to 3′ mRNA-degrading exoribonuclease in plants (Chiba and Green, 2009). To address the function of XRN4 in this process, we used previously published GMUCT data from xrn4 mutant plants (Gregory et al., 2008) and found that the pattern of 3-nucleotide periodicity within the ORFs of all detectable Arabidopsis protein-coding mRNAs was completely abrogated in the xrn4 mutant GMUCT data, with no enrichment of 5′P read ends in any of the three frames (Figure 5A). Furthermore, we found that the strong enrichment of 5′P end reads at the ribosome boundary site of all three stop codons compared with the median of flanking read values found for wild-type Col-0 plants (Figure 2C) was also completely lost in the xrn4 mutant GMUCT data (Figure 5B). Significantly, the normal 1.5- to 2.0-fold enrichment of 5′P read ends found at the ribosome boundary at the stop codon sequences was completely lost in the absence of XRN4 function (Figures 5C to 5F). Overall, these results indicate that XRN4 is the 5′ to 3′ exoribonuclease required for the cotranslational RNA decay process in plant transcriptomes.
Figure 5.

XRN4 Is the 5′ to 3′ Exoribonuclease Required for Cotranslational mRNA Decay in Arabidopsis.
(A) The 3-nucleotide periodicity pattern of 5′P read ends is no longer evident in Arabidopsis mRNA ORFs in GMUCT data from xrn4-5 mutant unopened flower buds. NS denotes no significant difference as determined by a χ2 test.
(B) The average number of 5′P read ends found at the ribosome boundary (16 and 17 nucleotides upstream) of each codon compared with the median coverage in the 50 nucleotides up- and downstream. The green dots are codons showing significantly less of an enrichment of 5′P read ends at the ribosome boundary site in GMUCT data from xrn4 mutant compared with wild-type Col-0, and gray dots are other codons. Significance was assessed using a χ2 test.
(C) to (F) The enrichment of 5′P read ends at the RB site of all stop codons (C), TAA (D), TAG (E), and TGA (F) compared with median coverage in the 50 nucleotides up- and downstream in Col-0 compared with xrn4-5 mutant unopened flower buds. Asterisks denote a significant difference at a P value < 2.2 × 10 to 100 as determined by a χ2 test. Error bars represent se of the mean for two biological replicates.
The Nuclear Cap Binding Complex Has a Role in Plant Cotranslational RNA Decay
The nuclear cap binding complex of plants is composed of two proteins: CBP20 and CBP80/ABH1. The presence of both proteins is required for proper functionality, and this complex is known to be involved in multiple biological processes such as mRNA stability and splicing, miRNA processing, and the initial round of protein translation. Given its role in regulating mRNA stability and the initiating round of translation, we wanted to explore the function of this protein complex in plant cotranslational RNA decay. To do this, we performed GMUCT on RNA from unopened flower buds extracted from two independent null alleles of ABH1: abh1-1 and abh1-8. Given that both of these mutant lines have a complete loss of ABH1 function and give highly correlated GMUCT results (Supplemental Figures 8 and 9A), the data from both were averaged together for all analyses. Using this data, we first looked for the pattern of 3-nucleotide periodicity of 5′P read ends. From this analysis, we found a pattern of 3-nucleotide periodicity within the ORFs of all detectable Arabidopsis protein-coding mRNAs (Figure 6), with a significant enrichment (P value < 2.2 × 10−100, χ2 test) of 5′P read ends in frames 1 and 2 compared with frame 0 (Figure 6A). This is similar to the pattern we found in Col-0 (wild type) (Figure 2B). These results demonstrate that the loss of ABH1 does not completely abrogate cotranslational RNA decay in Arabidopsis.
Figure 6.

The Nuclear mRNA Cap Binding Complex Functions in Cotranslational RNA Decay in Arabidopsis.
(A) A 3-nucleotide periodicity pattern of 5′P read ends is evident in Arabidopsis mRNA ORFs in abh1 mutant unopened flower buds. Asterisks denote a significant difference at a P value < 2.2 × 10 to 100 as determined by a χ2 test. Error bars represent se of the mean for two biological replicates.
(B) The average number of 5′P read ends found at the ribosome boundary (16 and 17 nucleotides upstream) of each codon compared with the median coverage in the 50 nucleotides up- and downstream in the abh1 mutant compared with Col-0 unopened flower buds. The red dots are codons showing significantly less of an enrichment of 5′P read ends at the ribosome boundary site in GMUCT data from the abh1 mutant compared with wild-type Col-0, and gray dots are other codons.
(C) The distribution of 5′P read ends relative to stop codons in all detectable mRNA ORFs in Col-0 compared with abh1 mutant unopened flower buds. The first nucleotide of the stop codon is numbered 0.
(D) The enrichment of 5′P read ends at the RB site of all stop codons compared with median coverage in the 50 nucleotides up- and downstream in Col-0 compared with abh1 mutant unopened flower buds. Asterisks denote a significant difference at a P value < 2.2 × 10 to 100 as determined by a χ2 test. Error bars represent se of the mean for two biological replicates.
Next, we compared wild-type and abh1 mutant plants to determine the enrichment of 5′P read ends at ribosome boundary sites (16 to 17 nucleotides upstream) with the median values at the flanking 100 nucleotides of all codons. From this analysis, we found that the enrichment of 5′P read ends at the ribosome boundary of all three stop codons was decreased in abh1 mutant plants compared with the wild type (Figure 6B). As an example, AT2G38670 demonstrated a decrease in 5′P read ends at the ribosome boundary of its stop codon in both abh1 alleles compared with two replicates of the wild type (Supplemental Figure 8). To complement these results, we aligned all detectable Arabidopsis mRNAs by their stop codons and then investigated the pattern of 5′P read ends. We observed that 5′P read ends were increased in abh1 mutant plants compared with the wild type (Figure 6C), likely due to the function of ABH1 in maintaining mRNA stability. However, consistent with the results of a decrease of cleavage at the ribosome boundary at all three stop codons (Figure 6B), we found that the accumulation of 5′P read ends 16 to 17 nucleotides upstream from the stop codon was globally decreased in abh1 mutants compared with the wild type (Figures 6C and 6D; Supplemental Figure 8). In fact, the normal 1.5- to 2.0-fold enrichment of 5′P read ends found at the ribosome boundary of stop codon sequences was significantly (P value < 2.2 × 10−100, χ2 test) decreased (down to ∼1.2-fold) in the absence of ABH1 function (Figure 6D; Supplemental Figures 9B to 9D). Overall, our results reveal a role for the nuclear cap binding complex in plant cotranslational RNA decay.
Global Measurement of Cotranslational RNA Decay in Arabidopsis
The GMUCT approach produces a global view of the uncapped transcript pool, having a mix of all degrading mRNAs that includes targets of cotranslational RNA decay, general ribosome-free degrading mRNAs, and miRNA cleavage products. However, we wanted to get a clear picture of the degree that XRN4 and ABH1 affected the proportion of transcripts specifically undergoing cotranslational RNA decay. To do this, we developed a method to measure the strength of cotranslational RNA decay for each detectable Arabidopsis transcript. A previous study identifying cotranslational RNA decay in S. cerevisiae revealed that Xrn1 was able to cleave only in frame 1, but not in frames 0 or 2 (Pelechano et al., 2015). However, we observed accumulation of 5′P read ends for both frames 1 and 2 compared with 0 (Figure 2B), revealing that Arabidopsis XRN4 is able to cleave in two frames and not just one. Therefore, we defined the cotranslational RNA decay index (CRI) for a given gene as the log2 ratio of average 5′ read ends corresponding to the two unprotected frames ((f1 + f2)/2) with respect to those in the protected frame (f0) (Figure 7A). Therefore, a CRI of 0 reflects a complete absence of cotranslational RNA decay in that plant background. As mentioned previously, ABH1 functions in miRNA biogenesis. Therefore, to avoid interfering signals from this process in the abh1 mutant data, we filtered out all miRNA target genes (Figure 1B; Supplemental Data Set 1) from this analysis. In the wild type, we found the average of the CRI distribution for all detectable transcripts was 0.283 (Figure 7A). Conversely, in xrn4 mutant plants we observed that the peak of the CRI distribution decreased to 0.053, indicating that the loss of XRN4 function almost entirely disrupts cotranslational mRNA decay in Arabidopsis (Figure 7A). As expected, we also found that the peak of CRI distributions decreased to ∼0.184 in abh1 mutant plants. This value is between the CRI peaks of the wild type and xrn4, and that is consistent with a decrease in, but not a complete loss of, the proportion of cotranslational RNA decay in the absence of ABH1 function (Figure 7A). Overall, these results demonstrate that both XRN4 and the nuclear cap binding complex function in cotranslational RNA decay in a plant transcriptome.
Figure 7.

The Levels of Cotranslational RNA Decay Vary between Col-0, xrn4, and abh1.
(A) Histogram of cotranslational RNA decay index values as determined for Col-0 (red line), xrn4 (green line), and abh1 (blue line). The loss of XRN4 almost entirely abolishes cotranslational RNA decay in Arabidopsis, whereas the absence of ABH1 has a more intermediate effect on this process in Arabidopsis.
(B) GO analysis of the group of transcripts with a cotranslational RNA decay index value higher than 1. The length of each bar in the graph is the enrichment ratio of each GO term for genes giving rise to transcripts with high CRI (red bars) or a background control set (blue bars). The value specified for each set of bars is the FDR of enrichment for each denoted GO term in high CRI genes compared with the background control set.
A closer examination of the effect of ABH1 on CRI revealed that ABH1 significantly affects (all P values < 0.05; Student’s t test) the proportion of cotranslational decay for 821 Arabidopsis mRNAs, with 625 and 196 mRNAs showing decreased and increased values, respectively (Figure 7A; Supplemental Data Set 3). These results not only explain the overall negative effect of ABH1 on CRI values in the Arabidopsis transcriptome, but also demonstrate that ABH1 affects only specific cotranslational RNA decay target transcripts. To determine whether ABH1 regulates cotranslational RNA decay target mRNAs that encode proteins with coherent functions, we performed a gene ontology analysis using the list of all transcripts with significantly different CRI values between abh1 mutant and wild-type Col-0 unopened flower buds (Supplemental Data Set 3). From this analysis, we observed a significant enrichment (false discovery rate [FDR] < 0.05) in transcripts encoding proteins involved in response to stress, cold, temperature stimulus, and abiotic stress (Supplemental Figure 10B). This is consistent with the known roles of ABH1 in plant abiotic stress response (Hugouvieux et al., 2001; Daszkowska-Golec et al., 2013). Overall, our results demonstrate that ABH1 is required for cotranslational RNA decay of a specific set of mRNAs that are at least partially degraded by this process and suggest that this regulatory function is required for its roles in various plant abiotic stress responses.
Given the effects that ABH1 had on specific cotranslational RNA decay target transcripts, we wanted to determine if this process in general preferentially targets mRNAs encoding proteins with common cellular functions for turnover. To explore this idea, we performed a gene ontology analysis using the list of all mRNAs with a CRI higher than 1. We focused on these transcripts because they are the ones that display the highest proportion of cotranslational RNA decay in the Arabidopsis transcriptome. From this analysis, we observed a significant enrichment (FDR < 0.05) of transcripts encoding proteins involved in DNA replication, regulation of transcription, response to auxin stimulus, double-stranded RNA binding, protein binding, and organ development (Figure 7B). Overall, these results reveal that transcripts encoding proteins with common cellular functions are highly cotranslationally degraded. Furthermore, these results suggest that this degradation pathway is involved in regulating the proper abundance of transcripts involved in these processes in plants.
Finally, we wanted to explore the possibility that miRNA-directed translation inhibition would affect the proportion of target transcripts regulated by this mechanism within the cotranslational RNA decay process. To test this, we compared CRI values between miRNA target genes known to be translational inhibited (Li et al., 2013) to those that have not been shown to be regulated by this mechanism. From this analysis, we found that those transcripts regulated by translation inhibition showed significantly higher (P value < 0.05; χ2 test) CRI values compared with all other miRNA target mRNAs in Arabidopsis unopened flower buds (Supplemental Figure 10A). In summary, our results revealed an interesting link between miRNA-mediated translation inhibition and cotranslational RNA decay.
DISCUSSION
Several recent studies revealed that heat stress triggers ribosome pausing on the 5′ end of transcripts encoding HSC/HSP70 chaperone targets in both mammals and plants (Shalgi et al., 2013; Liu et al., 2013a; Merret et al., 2013, 2015). For instance, a study in Arabidopsis found that heat stress causes 5′ ribosome pausing that ultimately results in XRN4-mediated decay of translating mRNAs (Merret et al., 2015). These results revealed that cotranslational RNA decay can occur in the plant transcriptome under stress conditions. However, whether cotranslational RNA decay occurs in plants under normal development was largely unknown. In this study, we provide evidence that this degradation process is widespread in the model plant Arabidopsis transcriptome. In support of this and consistent with a recent study on cotranslational RNA decay in S. cerevisiae (Pelechano et al., 2015), our GMUCT approach reveals that 5′P read ends have a pattern of 3-nucleotide periodicity and accumulate 13 and 14 nucleotides upstream of the start codon in GMUCT data from Col-0 plants treated with the translation inhibitor CHX (Figure 4). Furthermore, we observed accumulation of 5′P read ends upstream of all three stop codons, showing that ribosomes pause during the process of translation termination in Arabidopsis (Figure 2; Supplemental Figure 7). However, unlike in S. cerevisiae (Pelechano et al., 2015), we found that, in plants, 5′P read ends were enriched in both frames 1 and 2 compared with frame 0 and that they accumulate at both 16 and 17 nucleotides upstream of stop codons (Figure 2). More interestingly, we found that each stop codon gave a specific 5′P read end accumulation pattern at these two nucleotide positions (Supplemental Figure 7). In fact, the opposite patterns were observed for TAA and TAG at nucleotides 16 and 17. One interpretation of these results is that these two sequences interact with distinct ribosomal termination complexes, each of which confers different accessibility to RNase-mediated cleavage during cotranslational RNA decay. Also, the equal levels of 5′P read ends at both nucleotide positions observed for TGA stop codons suggest that both ribosomal termination complexes are active on transcripts containing this sequence. Alternatively, these findings may reveal that the kinetics of translation termination by the plant ribosome vary depending on the different stop codon sequences.
Consistent with our findings (Supplemental Figure 6), it was previously observed in S. cerevisiae that the 3-nucleotide periodicity of 5′P read ends near the start codon was relatively weak compared with within the rest of the gene and just upstream of stop codons (Pelechano et al., 2015). These findings, in combination with our observation of relatively low 5′P read ends in 5′ UTRs compared with 3′ UTRs (Supplemental Figure 2B), suggest that the exonuclease required for cotranslational RNA decay (XRN4) takes time to catch up with the ribosome during productive translation elongation. This is likely the reason that ribosome pausing at stop but not start codons can be detected by GMUCT without CHX treatment, whereas ribosomal profiling detects a density of higher ribosome footprints within the first 50 codons of the CDS (5′ ramp effect), as well as ribosomes paused at the start codon (Ingolia et al., 2011; Tuller et al., 2010; Lauria et al., 2015). Additionally, we also observed extension of the 3-nucleotide periodicity beyond the 16- to 17-nucleotide upstream of stop codon (stop codon RB) and into the 3′ UTR (Figure 2A), which was also found to occur in S. cerevisiae (Pelechano et al., 2015). These findings are likely the consequence of stop codon read-through by the ribosome at many mRNAs, which has also be found to occur frequently on Drosophila melanogaster and mammalian protein-coding transcripts (Jungreis et al., 2011; Dunn et al., 2013; Loughran et al., 2014).
Unlike in S. cerevisiae, we found that only the stop codon sequences, and none that coded for addition of an amino acid, were able to induce ribosome pausing (Figure 2C). This previous study also demonstrated that the pausing ability of specific amino acid codons changed during S. cerevisiae oxidative stress response (Pelechano et al., 2015). Given these results and the previous results revealing ribosomal pausing during heat stress (Shalgi et al., 2013; Merret et al., 2013, 2015), it will be interesting to use GMUCT to test whether specific amino acid codons can induce ribosomal pausing during various plant stress responses.
Additionally, we found the ability of GMUCT to detect cotranslational mRNA decay in plant transcriptomes allowed us to identify translationally active uORFs that do not overlap with the downstream main ORF in Arabidopsis unopened flower buds (Figure 3). From our analyses, we identified uORFs that had been previously studied and/or predicted, as well as a rather large collection of novel uORFs (Supplemental Data Set 2). For instance, we provide evidence for translationally active uORFs in the 5′ UTRs of FCA (AT4G16280) and AGL4/SEP2 (AT3G02310). It will be interesting in the future to determine whether the uORFs identified by this study play a role in regulating translation from the downstream main ORFs in their parent transcripts.
One other feature that we noticed at translationally active uORFs was a peak of 5′P read ends 46 to 47 nucleotides upstream of their stop codons (Figure 3B), a pattern that was not as prevalent when looking at these same positions of main mRNA ORFs (Figure 2A). This second peak of 5′P read ends indicates the presence of two ribosomes being engaged near the stop codon of uORFs and occurs more frequently in these 5′ UTR localized sequences compared with main ORFs. In combination, these findings reveal that translation termination is the main cause of ribosome pausing during translation elongation in Arabidopsis. Furthermore, the difference between upstream and main ORF 5′P read-end patterns suggests that translation termination occurs more slowly at uORFs compared with main protein-coding ORFs. Determining the molecular details behind the differences in translation termination of upstream compared with main protein-coding ORFs in Arabidopsis will be an important focus for future inquiry.
Interestingly, we did not observe a clear 3-nucleotide periodicity within the coding regions of uORFs (Figure 3B). This is likely a consequence of the short length of their primary nucleotide sequences. For instance, their small size may not provide sufficient time for XRN4 to catch up to the ribosome until it is paused for termination. Alternatively, we observed the GMUCT signature of tandem ribosome pausing in many uORF instances (5′P read ends 46 to 47 nucleotides upstream of their stop codons) (Figure 3B). This may result in better protection of uORF sequences from XRN4 cleavage. Future experiments will be required to determine the lack of 3-nucleotide periodicity observed in uORF coding regions.
Using GMUCT data from xrn4 and abh1 mutant plants (Figures 5 and 6), we demonstrated that both of these proteins functioned in cotranslational RNA decay in plants. In fact, in the absence of XRN4 function we found that all evidence of cotranslational RNA decay in GMUCT data was lost, including the 3-nucleotide periodicity pattern of 5′P read end in ORFs and their accumulation at the ribosome boundary site upstream of stop codons (Figure 5). These findings indicate that plant cotranslational RNA decay is dependent on the cytoplasmic 5′ to 3′ exoribonuclease XRN4, a result that is not surprising because the S. cerevisiae ortholog Xrn1 is required for this process (Pelechano et al., 2015).
The nuclear cap binding complex, which consists of two subunits (CBP20 and CBP80/ABH1), plays multiple roles in RNA metabolism, including functioning in processing of miRNAs, mRNA splicing, and the initial round of translation (Gregory et al., 2008; Kim et al., 2009). These functions in general RNA metabolism and the initiating round of translation spurred us to test whether this mostly nuclear complex of proteins also functions in cotranslational RNA decay. Using GMUCT data from two null alleles of abh1, we discovered an unexpected role of the nuclear mRNA cap binding complex in this process. More specifically, we found that cotranslational RNA decay is decreased ∼50% in the absence of ABH1, unlike in xrn4 mutant plants where this process is almost entirely absent (Figures 5, 6, and 7A). Interestingly, a closer look at this effect on cotranslational RNA decay revealed that 821 transcripts had CRI values that are significantly different in abh1 mutant compared with Col-0 plants. In fact, 76% of these mRNAs had a significant decrease in CRI value, explaining the overall decrease in the proportion of transcripts undergoing cotranslational RNA decay in the absence of ABH1 function. Intriguingly, the transcripts whose CRI is affected by the loss of ABH1 function tend to encode proteins involved in abiotic stress responses, especially those involved with temperature stimulus. Given that ABH1 is known to function in various abiotic stress responses (Hugouvieux et al., 2001; Daszkowska-Golec et al., 2013), these results suggest that its functions in cotranslational RNA decay and abiotic stress response are linked. Future experiments will be required to determine the mechanistic details of this functional link between cotranslational RNA decay and plant abiotic stress response.
As noted, ABH1 functions in multiple general mRNA metabolic processes, including splicing, general RNA stability, and the initiating round of translation. Therefore, there are many mechanisms by which this protein may affect cotranslational RNA decay. We first determined that ABH1 did not affect XRN4 expression. To do this, we looked at the levels of this transcript in expression data from the abh1 mutant background (Gregory et al., 2008). We found no significant difference in XRN4 levels in abh1 mutant compared with Col-0 plants, indicating the change in cotranslational RNA decay in abh1 mutant plants is not merely a consequence of reducing the levels of the exoribonuclease that is required for this process. Other mechanisms that may be involved in the function of ABH1 in cotranslational RNA decay include, but are not limited to (1) determining which transcripts are degraded by a cotranslational or general RNA decay mechanisms; (2) shuttling RNAs into the cotranslational RNA decay pathway during the initiating round of translation; and (3) affecting alternative splicing leading to greater degradation by cotranslational RNA decay of inappropriately spliced transcripts. We note that these models are not mutually exclusive, and we are intrigued by the possibility that the initiating round of translation may have an impact on cotranslational RNA decay in plant transcriptomes. However, significant future work is required to elucidate the mechanistic details of the involvement of ABH1 in this RNA decay pathway.
We also used CRI values to identify a subset of mRNAs that displayed the highest proportion of cotranslational RNA decay products in Arabidopsis unopened flower buds. Interestingly, these transcripts tended to encode proteins with coherent functions in processes such as response to auxin, nucleic acid metabolic processes, DNA replication, and regulation of transcription (Figure 7B). The finding that transcripts involved in auxin response are highly targeted by cotranslational RNA decay is fitting given the recent findings that Arabidopsis ribosomal proteins control developmental programs through translational regulation of auxin response factors (Rosado et al., 2012). Thus, regulating both transcript translation and turnover provides tight control of the developmental effects of auxin in Arabidopsis. The links between these two processes will need to be further investigated by future experiments. Overall, our findings suggest that mRNA transcripts that display extremely high CRI values are likely to be targeted during active translation by the cotranslational RNA decay pathway to regulate their functional output in coherent functional pathways in Arabidopsis. Thus, determining the effect of this pathway on normal plant growth and development should be further examined in the future.
Finally, we compared CRI values of miRNA target genes known to be regulated by translation inhibition with those that are not. This analysis revealed that miRNA-mediated translation inhibition is likely a significant trigger of cotranslational RNA decay in Arabidopsis (Supplemental Figure 10A). The intriguing link between this miRNA-mediated silencing mechanism and cotranslational RNA decay will need to be further investigated in the future to elucidate the mechanistic details.
In summary, our results provided a global view of XRN4-mediated cotranslational mRNA decay in a plant transcriptome. Furthermore, they uncovered a link between the nuclear mRNA cap binding complex and cotranslational mRNA decay and demonstrated that GMUCT provides evidence of translationally active uORFs in plant transcriptomes. Thus, future GMUCT-driven studies of plant translation will undoubtedly discover additional features of this process during both normal development and stress response.
METHODS
Plant Materials and GMUCT Library Construction
GMUCT libraries were constructed using RNA from two biological replicates of leaves 5 to 9 of 4-week-old Arabidopsis thaliana Col-0 (Arabidopsis) that had been treated with the translation inhibitor CHX and unopened flower buds from biological replicates of two abh1 alleles (abh1-1 and abh1-8) using the GMUCT 2.0 protocol (Willmann et al., 2014). In brief, RNA was first subjected to poly(A)+ selection followed by immediate ligation of a 5′ RNA adapter. An additional poly(A)+ selection step was performed to purify the adapter ligated RNAs. These samples were used as the substrates in reverse transcription reactions using a reverse transcription primer that was composed of the 3′ adapter sequence on the 5′ end and a random hexamer on its 3′ end. This allowed for the addition of the 3′ sequencing adapter during reverse transcription. Finally, the GMUCT libraries were amplified and indices were added using a limited PCR amplification reaction.
Mapping GMUCT Reads to mRNA Transcripts
All GMUCT reads (50-nucleotide single-end sequences) were aligned to full-length mature mRNAs extracted from the TAIR10 genome annotation using STAR (version 2.4.0 with parameters “—outFilterMultimapNmax 10 –outFilterMismatchNmax 10–outFilterMismatchNoverLmax 0.10”) (Dobin et al., 2013). Subsequently, the SAM files were converted to BED files containing only the first (most 5′) nucleotide of each read, denoting the cleavage sites resulting in 5′P intermediates. For assessing GMUCT data reproducibility, 5′P reads for each 100-nucleotide bin along all mRNAs were counted. Then a Pearson correlation coefficient was calculated between the two GMUCT replicate libraries, and scatterplots were produced. Additionally, the ratio of total 5′P reads in the annotated 5′ UTR, CDS, and 3′ UTR of mature mRNAs was determined.
Prediction of Canonic miRNA Targets and Precise Cleavage Sites
Sequences for the 45 canonical miRNAs conserved between Arabidopsis and the Brassicaceae were downloaded from miRBase. Their putative targets were predicted using psRNATarget (http://plantgrn.noble.org/psRNATarget/) using default parameters. The cleavage sites of 21-nucleotide miRNAs are between the 10th and 11th nucleotide of the miRNAs as counted from the 5′ end. The 5′P reads within the 100-nucleotide flanking regions of miRNA cleavage sites were also counted. The local cleavage efficiency of miRNAs represents the log2 transformation of the ratio of 5′P read ends at miRNA cleavage sites divided by the median 5′P read end coverage in 100-nucleotide flanking regions. The global cleavage efficiency of miRNAs represents the log2 transformation of the ratio of 5′P read ends at miRNA cleavage sites divided by the average 5′P read-end coverage throughout the entire transcript. These statistics indicate the proportion of cleavage products in a given mRNA that are due to miRNA-mediated cleavage. χ2 tests were performed to assess significant differences between 5′P read ends in miRNA cleavage sites compared with the flanking median 5′P read end coverage or average 5′P read-end coverage throughout the entire transcript.
Defining a 3-Nucleotide Periodicity Pattern in ORFs and Enrichment Analysis of 5′P Read End Accumulation Due to Ribosome Pausing at Each Codon
The three coding frames of each codon are represented as frame 0 (f0), frame 1 (f1), and frame 2 (f2), with frame 0 being the one used in translation. f1 and f2 are cleavage-accessible frames, while f0 is not accessible to cleavage. The abundance of 5′P read ends that accumulated in each frame along all ORFs was quantified. To determine whether the difference in accumulation of 5′P read ends was significantly different between each frame, a χ2 test was performed to assess the significance of each pairwise comparison. To survey ribosome pausing at each type of codon, the enrichment of 5′P read ends at the ribosome boundary (16 to 17 nucleotides upstream from each codon) compared with the median number of 5′P read ends within the flanking 100 nucleotides was calculated for Col-0 with and without CHX treatment, xrn4, and abh1. We also compared the enrichment at these positions upstream of stop codons between Col-0 and xrn4 as well as abh1 mutants. To determine the significance of differences in these comparisons, we used a χ2 test.
Prediction of Putative Translationally Active uORFs Identified by GMUCT
All 5′ UTRs of annotated protein-coding genes were scanned to identify potential ORFs that begin with the start codons ATG, ACG, or CTG and that also contain a defined stop codon following a short ORF. Putative translationally active uORFs that did not overlap with main ORFs were subsequently identified as those where 5′P read ends were enriched at least 2-fold at nucleotide 16 and/or 17 upstream from the stop codon compared with the flanking 100 nucleotides.
Measurement of Cotranslational RNA Decay
We determined the CRI to measure the proportion of cotranslational RNA decay for each gene. The CRI for a given gene represents the log2 ratio of average 5′P read end coverage at cleavage-accessible frames (f1 and f2) divided by the 5′P read coverage of the frame that is not accessible to cleavage (f0). This function is expressed as: 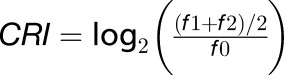 . The CRI distributions for all mRNAs measured in GMUCT samples for Col-0 as well as abh1 and xrn4 mutant plants were plotted. Transcripts with CRI values that were significantly different in abh1 mutant compared with Col-0 plants were identified by Student's t test with P values < 0.05. CRI values were compared between miRNA target genes known to be regulated by translation inhibition with those that are not. χ2 tests were performed to assess significant differences.
. The CRI distributions for all mRNAs measured in GMUCT samples for Col-0 as well as abh1 and xrn4 mutant plants were plotted. Transcripts with CRI values that were significantly different in abh1 mutant compared with Col-0 plants were identified by Student's t test with P values < 0.05. CRI values were compared between miRNA target genes known to be regulated by translation inhibition with those that are not. χ2 tests were performed to assess significant differences.
Gene Ontology Analysis
ORFs of genes with a CRI higher than 1 in Col-0 were extracted, and Gene Ontology (GO) analysis was performed using agriGO (http://bioinfo.cau.edu.cn/agriGO/). GO terms with FDRs less than 0.05 were selected. GO analysis for the group of transcripts that display significantly different CRI values between Col-0 and abh1 mutant plants was also performed as described above.
Accession Numbers
GMUCT sequencing data for the libraries made with RNA extracted from leaves 5 to 9 of 4-week-old Col-0 that had been treated with the translation inhibitor CHX and unopened flower buds of abh1-1 and abh1-8 have been deposited in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE71913. Previously published GMUCT data for Col-0 (GSM1145327 and GSM1145328) and xrn4 (GSM284752) mutant unopened flower buds were obtained from the Gene Expression Omnibus under the accession numbers GSE47121 (Willmann et al., 2014) and GSE11070 (Gregory et al., 2008), respectively. The gene accession numbers for XRN4 and ABH1 are AT1G54490 and AT2G13540, respectively.
Supplemental Data
Supplemental Figure 1. The GMUCT Approach Reveals Both miRNA-Directed Cleavage Sites in Target mRNAs and the Features of Cotranslational RNA Decay in Plant Transcriptomes.
Supplemental Figure 2. GMUCT Is a Highly Reproducible Approach.
Supplemental Figure 3. GMUCT Identifies miRNA-Directed Cleavage Sites in Target mRNAs.
Supplemental Figure 4. GMUCT Reveals Evidence of Cotranslational RNA Decay in the Arabidopsis Transcriptome.
Supplemental Figure 5. Ribosome Pausing at the Stop Codons of mRNA ORFs Provides Evidence for Cotranslational RNA Decay in the Arabidopsis Transcriptome.
Supplemental Figure 6. The Distribution of 5′P Read Ends around the Start and GAT Codons in GMUCT Experiments Using Untreated Arabidopsis Unopened Flower Buds.
Supplemental Figure 7. The Distribution of 5′P Read Ends Is Distinct within Transcripts Containing the Three Different Stop Codon Sequences in Arabidopsis.
Supplemental Figure 8. 5′P Read Ends at RB Sites Near Stop Codons Are Decreased in abh1 Mutant Plants Compared with Wild-Type Col-0.
Supplemental Figure 9. The Nuclear Cap Binding Complex of Arabidopsis Functions in Cotranslational RNA Decay.
Supplemental Figure 10. The Levels of Cotranslational RNA Decay Vary Significantly between miRNA Target Transcripts Regulated by Translation Inhibition and Those That Are Not, and GO Enrichment of Genes with Significantly Different CRI Values between Col-0 and abh1 Mutant Unopened Flower Buds.
Supplemental Data Set 1. The Set of Candidate miRNA Target Sites.
Supplemental Data Set 2. The Set of uORFs With Enrichment of 5′P Read Ends at Their Stop Codon Ribosome Boundary Compared with Their Flanking Regions.
Supplemental Data Set 3. mRNAs Having Significant Differences in Cotranslational RNA Decay Index Values for Col-0 versus abh1 Mutant Plants.
Supplementary Material
Acknowledgments
We thank current and past members of the Gregory lab for their helpful discussions and comments on the manuscript. This work was funded by grants from the National Science Foundation (MCB-1243947 and IOS-1444490) to B.D.G. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
AUTHOR CONTRIBUTIONS
X.Y. and B.D.G. designed the study. M.R.W. and S.J.A. performed the GMUCT experiments with input from B.D.G. X.Y. compiled all sequencing and annotation data and carried out the computational analysis with input from B.D.G. X.Y. and B.D.G. wrote the manuscript. All authors read and approved the manuscript.
Glossary
- miRNA
microRNA
- CBC
cap binding complex
- GMUCT
genome-wide mapping of uncapped and cleaved transcripts
- CDS
coding sequence
- UTR
untranslated region
- ORF
open reading frame
- RB
ribosome boundary
- uORF
upstream ORF
- CHX
cycloheximide
- CRI
cotranslational RNA decay index
- FDR
false discovery rate
- GO
Gene Ontology
Footnotes
Articles can be viewed without a subscription.
References
- Addo-Quaye C., Eshoo T.W., Bartel D.P., Axtell M.J. (2008). Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 18: 758–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aukerman M.J., Sakai H. (2003). Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell 15: 2730–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekanova J.A., et al. (2007). Genome-wide high-resolution mapping of exosome substrates reveals hidden features in the Arabidopsis transcriptome. Cell 131: 1340–1353. [DOI] [PubMed] [Google Scholar]
- Chen X. (2004). A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303: 2022–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba Y., Green P. (2009). mRNA degradation machinery in plants. J. Plant Biol. 52: 114–124. [Google Scholar]
- Dai X., Zhao P.X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39: W155–W159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daszkowska-Golec A., Wojnar W., Rosikiewicz M., Szarejko I., Maluszynski M., Szweykowska-Kulinska Z., Jarmolowski A. (2013). Arabidopsis suppressor mutant of abh1 shows a new face of the already known players: ABH1 (CBP80) and ABI4-in response to ABA and abiotic stresses during seed germination. Plant Mol. Biol. 81: 189–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas D.V., Bartel B. (2008). Sucrose induction of Arabidopsis miR398 represses two Cu/Zn superoxide dismutases. Plant Mol. Biol. 67: 403–417. [DOI] [PubMed] [Google Scholar]
- Dunn J.G., Foo C.K., Belletier N.G., Gavis E.R., Weissman J.S. (2013). Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. eLife 2: e01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandikota M., Birkenbihl R.P., Höhmann S., Cardon G.H., Saedler H., Huijser P. (2007). The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 49: 683–693. [DOI] [PubMed] [Google Scholar]
- Garneau N.L., Wilusz J., Wilusz C.J. (2007). The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8: 113–126. [DOI] [PubMed] [Google Scholar]
- German M.A., Luo S., Schroth G., Meyers B.C., Green P.J. (2009). Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 4: 356–362. [DOI] [PubMed] [Google Scholar]
- Gonatopoulos-Pournatzis T., Cowling V.H. (2014). Cap-binding complex (CBC). Biochem. J. 457: 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory B.D., O’Malley R.C., Lister R., Urich M.A., Tonti-Filippini J., Chen H., Millar A.H., Ecker J.R. (2008). A link between RNA metabolism and silencing affecting Arabidopsis development. Dev. Cell 14: 854–866. [DOI] [PubMed] [Google Scholar]
- Hayden C.A., Jorgensen R.A. (2007). Identification of novel conserved peptide uORF homology groups in Arabidopsis and rice reveals ancient eukaryotic origin of select groups and preferential association with transcription factor-encoding genes. BMC Biol. 5: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., Sweet T.J., Chamnongpol S., Baker K.E., Coller J. (2009). Co-translational mRNA decay in Saccharomyces cerevisiae. Nature 461: 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugouvieux V., Kwak J.M., Schroeder J.I. (2001). An mRNA cap binding protein, ABH1, modulates early abscisic acid signal transduction in Arabidopsis. Cell 106: 477–487. [DOI] [PubMed] [Google Scholar]
- Ingolia N.T., Lareau L.F., Weissman J.S. (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungreis I., Lin M.F., Spokony R., Chan C.S., Negre N., Victorsen A., White K.P., Kellis M. (2011). Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 21: 2096–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenmayer J.P., Green P.J. (2000). Novel features of the XRN-family in Arabidopsis: evidence that AtXRN4, one of several orthologs of nuclear Xrn2p/Rat1p, functions in the cytoplasm. Proc. Natl. Acad. Sci. USA 97: 13985–13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.M., Cho H., Choi K., Kim J., Kim B.-W., Ko Y.-G., Jang S.K., Kim Y.K. (2009). A new MIF4G domain-containing protein, CTIF, directs nuclear cap-binding protein CBP80/20-dependent translation. Genes Dev. 23: 2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing W.A., Martínez-Sánchez M., Wright M.A., Bulley S.M., Brewster D., Dare A.P., Rassam M., Wang D., Storey R., Macknight R.C., Hellens R.P. (2015). An upstream open reading frame is essential for feedback regulation of ascorbate biosynthesis in Arabidopsis. Plant Cell 27: 772–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria F., et al. (2015). RiboAbacus: a model trained on polyribosome images predicts ribosome density and translational efficiency from mammalian transcriptomes. Nucleic Acids Res. 43: e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., et al. (2013). MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis. Cell 153: 562–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G., He H., Li Y., Wang F., Yu D. (2014). Molecular mechanism of microRNA396 mediating pistil development in Arabidopsis. Plant Physiol. 164: 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Han Y., Qian S.B. (2013a). Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol. Cell 49: 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.J., Wu S.H., Wu J.F., Lin W.D., Wu Y.C., Tsai T.Y., Tsai H.L., Wu S.H. (2013b). Translational landscape of photomorphogenic Arabidopsis. Plant Cell 25: 3699–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran G., Chou M.Y., Ivanov I.P., Jungreis I., Kellis M., Kiran A.M., Baranov P.V., Atkins J.F. (2014). Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 42: 8928–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merret R., Descombin J., Juan Y.T., Favory J.J., Carpentier M.C., Chaparro C., Charng Y.Y., Deragon J.M., Bousquet-Antonelli C. (2013). XRN4 and LARP1 are required for a heat-triggered mRNA decay pathway involved in plant acclimation and survival during thermal stress. Cell Reports 5: 1279–1293. [DOI] [PubMed] [Google Scholar]
- Merret R., Nagarajan V.K., Carpentier M.C., Park S., Favory J.J., Descombin J., Picart C., Charng Y.Y., Green P.J., Deragon J.M., Bousquet-Antonelli C. (2015). Heat-induced ribosome pausing triggers mRNA co-translational decay in Arabidopsis thaliana. Nucleic Acids Res. 43: 4121–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan V.K., Jones C.I., Newbury S.F., Green P.J. (2013). XRN 5′→3′ exoribonucleases: structure, mechanisms and functions. Biochim. Biophys. Acta 1829: 590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V., Wei W., Steinmetz L.M. (2015). Widespread co-translational RNA decay reveals ribosome dynamics. Cell 161: 1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raczynska K.D., Simpson C.G., Ciesiolka A., Szewc L., Lewandowska D., McNicol J., Szweykowska-Kulinska Z., Brown J.W., Jarmolowski A. (2010). Involvement of the nuclear cap-binding protein complex in alternative splicing in Arabidopsis thaliana. Nucleic Acids Res. 38: 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado A., Li R., van de Ven W., Hsu E., Raikhel N.V. (2012). Arabidopsis ribosomal proteins control developmental programs through translational regulation of auxin response factors. Proc. Natl. Acad. Sci. USA 109: 19537–19544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz S., Grande A.V., Bujdoso N., Saedler H., Huijser P. (2008). The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant Mol. Biol. 67: 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalgi R., Hurt J.A., Krykbaeva I., Taipale M., Lindquist S., Burge C.B. (2013). Widespread regulation of translation by elongation pausing in heat shock. Mol. Cell 49: 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souret F.F., Kastenmayer J.P., Green P.J. (2004). AtXRN4 degrades mRNA in Arabidopsis and its substrates include selected miRNA targets. Mol. Cell 15: 173–183. [DOI] [PubMed] [Google Scholar]
- Tabuchi T., Okada T., Azuma T., Nanmori T., Yasuda T. (2006). Posttranscriptional regulation by the upstream open reading frame of the phosphoethanolamine N-methyltransferase gene. Biosci. Biotechnol. Biochem. 70: 2330–2334. [DOI] [PubMed] [Google Scholar]
- Takahashi H., Takahashi A., Naito S., Onouchi H. (2012). BAIUCAS: a novel BLAST-based algorithm for the identification of upstream open reading frames with conserved amino acid sequences and its application to the Arabidopsis thaliana genome. Bioinformatics 28: 2231–2241. [DOI] [PubMed] [Google Scholar]
- Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y. (2010). An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141: 344–354. [DOI] [PubMed] [Google Scholar]
- Vanacova S., Stefl R. (2007). The exosome and RNA quality control in the nucleus. EMBO Rep. 8: 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Arnim A.G., Jia Q., Vaughn J.N. (2014). Regulation of plant translation by upstream open reading frames. Plant Sci. 214: 1–12. [DOI] [PubMed] [Google Scholar]
- Wiese A., Elzinga N., Wobbes B., Smeekens S. (2004). A conserved upstream open reading frame mediates sucrose-induced repression of translation. Plant Cell 16: 1717–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmann M.R., Berkowitz N.D., Gregory B.D. (2014). Improved genome-wide mapping of uncapped and cleaved transcripts in eukaryotes--GMUCT 2.0. Methods 67: 64–73. [DOI] [PubMed] [Google Scholar]
- Wu G., Park M.Y., Conway S.R., Wang J.W., Weigel D., Poethig R.S. (2009). The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 138: 750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Wang H., Lu Y., de Ruiter M., Cariaso M., Prins M., van Tunen A., He Y. (2012). Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J. Exp. Bot. 63: 1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.