

SUMMARY
Reactive α-dicarbonyls (α-DCs), like methylglyoxal (MGO) accumulate with age, and have been implicated in aging and various age-associated pathologies such as diabetic complications and neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease. Evolutionarily conserved glyoxalases are responsible for α-DC detoxification; however their core biochemical regulation have remained unclear. We have established a Caenorhabditis elegans model, based on an impaired glyoxalase (glod-4/GLO1), to broadly study α-DC-related stress. We show that in comparison to wild-type (N2, Bristol), glod-4 animals rapidly exhibit several pathogenic phenotypes including hyperesthesia, neuronal damage, reduced motility, and early mortality. We further demonstrate TRPA-1/TRPA1 as a sensor for α-DCs, conserved between worms and mammals. Moreover, TRPA-1 activates SKN-1/Nrf via calcium-modulated kinase signaling, ultimately regulating the glutathione-dependent (GLO1) and co-factor-independent (DJ1) glyoxalases to detoxify α-DCs. Interestingly, this pathway is in stark contrast to the TRPA-1 activation and the ensuing calcium flux implicated in cold sensation in C. elegans, whereby DAF-16/FOXO gets activated via complementary kinase signaling. Finally, a phenotypic drug-screen using C. elegans identified podocarpic acid as a novel activator of TRPA1 that rescues α-DC-induced pathologies in C. elegans and mammalian cells. Our work thus identifies TRPA1 as a bonafide drug target for amelioration of α-DC stress, which represents a viable option to address aging-related pathologies in diabetes and neurodegenerative diseases.
Graphical abstract
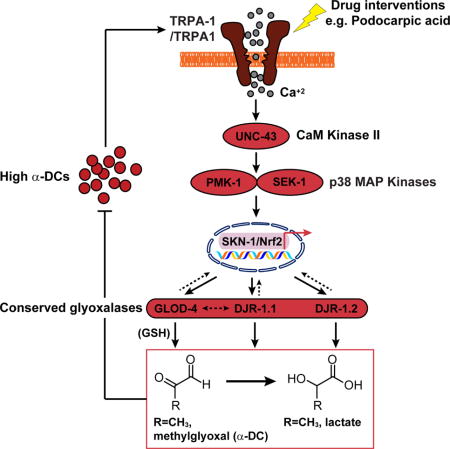
INTRODUCTION
Aging-associated pathologies such as diabetes mellitus and neurodegenerative disorders, such as Alzheimer’s and Parkinson’s disease results in several metabolic and biochemical aberrations, most importantly elevation of a series of highly reactive α-dicarbonyl compounds (α-DCs, e.g. glyoxal/GO, methylglyoxal/MGO, and 3-deoxyglucosone/3DG) [1]. These α-DCs are unavoidable byproducts of anaerobic glycolysis and lipid peroxidation and react non-enzymatically with proteins, lipids, and DNA to yield a heterogeneous group of molecules, collectively called advanced glycation endproducts (AGEs) [2]. AGE formation renders irreversible damage to these biological macromolecules, altering their structural and functional integrity [3]. In particular, a large body of evidence has linked accelerated AGE formation via the in vivo accumulation of reactive α-DCs, specifically MGO, to the pathogenesis of many forms of diabetic complications. These include peripheral neuropathy, neurodegenerative conditions, cardiomyopathy, nephropathy, retinopathy, microvascular damage, and early mortality [2–5]. Given these deleterious physiological effects of α-DC stress, cellular detoxification of these metabolites is crucial. The evolutionarily conserved glutathione-dependent glyoxalase system, comprised of glyoxalase I and II (human GLO1 and 2), is believed to be primarily responsible for α-DC detoxification and has garnered significant scientific interest in the context of diabetic complications (Fig. 1A) [6].
Figure 1. Establishing C. elegans glod-4 as a viable model for studying α-DC-related pathologies.
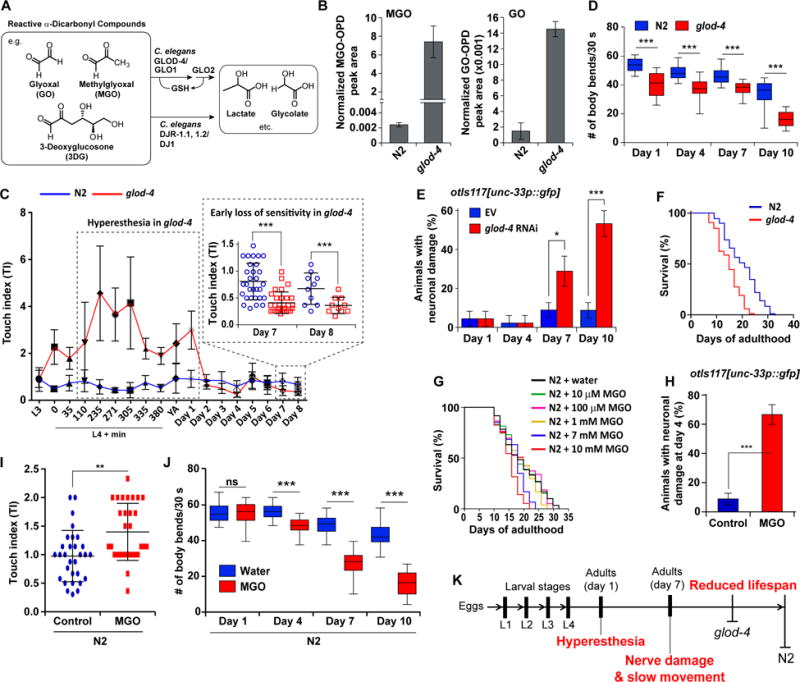
(A) Structures of endogenous reactive α-DCs. In vivo α-DC detoxification is primarily mediated via glutathione (GSH)-dependent glyoxalase I/II (human GLO1/2) and the co-factor-independent glyoxalase, DJ1 (human). GLOD-4 and DJR-1.1, DJR-1.2 are the C. elegans orthologs, of the mammalian GLO1 and DJ1, respectively.
(B) Levels of MGO (left) and GO (right) in wild-type (N2) and glod-4 at day 4 of adulthood.
(C) Age-dependent change in sensitivity to touch, quantified by the touch index (TI) in N2 and glod-4.
(D) Age-dependent change in number of body bends (swim bends) in liquid media for N2 and glod-4. n = 30.
(E) Neuronal damage in unc-33p∷gfp (pan-neuronal GFP) at days 1, 4, 7, and 10 of adulthood reared on empty vector (EV, L4440) or glod-4 RNAi. n = 45.
(F) Survival curves for N2 and glod-4 animals reared on OP50-1.
(G) Survival curves for N2 animals supplemented with water (control) or MGO at 10 μM, 100 μM, 1 mM, 7 mM, and 10 mM.
(H) Quantification of the extent of neuronal damage in pan-neuronal GFP animals (unc-33p∷gfp) at day 4 of adulthood reared supplemented with water (control) or 7 mM MGO. n = 45.
(I) Touch index (TI) during young adult stage (~L4 + 8–10 h) for N2 animals, supplemented with water (control) or 7 mM MGO.
(J) Number of body bends in liquid media for N2 animals supplemented with water (control) or 7 mM MGO.
(K) Progression of age-dependent phenotypes observed in glod-4 mutants, which form the basis of using this mutant as a model to study diabetes-related pathologies.
Data are represented as mean ± SD. Significance: *P<0.05, **P<0.005 and ***P<0.0005.
See also Figure S1 and Tables S1 and S2.
Recent projections [7] have created an enormous urgency for the discovery of novel therapeutics and thus an immediate necessity for developing model systems that allow rapid assessment of the consequences of in vivo α-DC accumulation. In vertebrate models such as mice, it is generally difficult to perform causation studies due to their comparatively long lifespan. Hence, developing a genetically tractable invertebrate model for studying α-DC stress, enabling rapid discovery is urgently required. To that end, we have established a Caenorhabditis elegans model for studying complications associated with α-DC buildup that is amenable to high throughput genetic and drug screens. Recent studies have shown that Glo1 knockdown in non-diabetic mice result in elevated MGO levels and oxidative stress, ultimately causing pathologies reminiscent of diabetic neuropathy and nephropathy [8–10]. Similarly, our model, based on the mutant glod-4/GLO1 [11], exhibits several pathogenic phenotypes reminiscent of α-DC stress-induced age-associated disorders.
C. elegans is an ideal model for understanding complex molecular networks because of the readily available and powerful genetic tools, ease of culture, and relatively short lifespan [12]. We show that TRPA1 acts as a conserved sensor for α-DCs and identify several components of an ensuing signaling pathway that triggers α-DC detoxification via Nrf activation. A phenotypic drug screen using this C. elegans model, identified potential candidates for amelioration of neuropathic and age-related complications associated with α-DC stress. Ultimately, this work exemplifies the utility of invertebrate systems, such as C. elegans in modeling mammalian diseases, facilitating rapid drug discovery.
RESULTS
A C. elegans model to study pathologies associated with α-DC stress
We set out to establish an invertebrate model that accumulates α-DCs and recapitulates phenotypes reminiscent of α-DC stress-associated pathologies. A preliminary LC-MS/MS assay (Figures S1A and S1B) based screen, developed based on previous reports [13, 14], revealed that the C. elegans mutant glod-4(gk189) significantly accumulates several α-DCs (GO, MGO, and 3DG) (Figure 1B; Figure S1C). Further, glod-4 animals are hypersensitive to touch (hyperesthetic) in an age-dependent fashion (Figure 1C), which is among the first symptoms experienced by diabetics, who are likely to develop neuropathy later in life [15]. During early adulthood, glod-4 animals exhibited significantly elevated touch indices (TIs, see Experimental Procedures) as compared to wild-type (N2, Bristol), which is replaced by a loss of sensitivity to touch later in life (Figure 1C). Further, as the glod-4 animals aged, we observed reduced motility (Figure 1D) and neuronal damage (Figure 1E; Figure S1D), as compared to N2. Finally, glod-4 animals exhibited a significantly shorter lifespan compared to N2 as previously demonstrated [11, 16] (Figure 1F).
Among the α-DCs assayed, the accumulation of MGO (several thousand fold) was exceedingly more prominent than GO (5–10 fold) or 3DG (1.2–1.5 fold) in glod-4 mutants (Figure 1B; Figure S1C). A modest increase of 3DG may simply imply that 3DG is not a direct substrate for glod-4/GLO1 [17]. Hence, we examined if exogenous MGO supplementation could recapitulate glod-4-like phenotypes. As previously demonstrated for exogenous drug assays [18], a dose-response experiment suggested that a relatively high exogenous MGO concentration (at least 7 mM) was required to show significant lifespan shortening in N2 animals (Figure 1G). 7 mM MGO treatment on N2 animals further leads to hyperesthesia, lower motility, and neuronal damage (Figures 1H–1J; Figures S1E and S1F), similar to glod-4 mutants. Further, the hyperesthesia phenotype, observed with MGO was not observed with another potent oxidative stressor, paraquat (methyl viologen chloride) (Figure S1F). In addition, the pathogenic phenotypes in glod-4, i.e. accumulation of MGO, neuronal damage, and early mortality, were exacerbated when the animals were reared on a high glucose (2% glucose) diet (Figures S1G–S1J); our results are in agreement with the observed lifespan reduction in C. elegans upon glucose supplementation [16, 19]. These findings (Figure 1K) lay the foundation to utilize glod-4 as a viable model for studying the relation of α-DC stress to age-related diseases including diabetes.
TRPA1-Nrf protects against α-DC stress
Next, we set out to identify key biochemical pathways that respond to α-DC stress. Among the various conserved components, a protective role of Nrf in diabetes-induced oxidative stress have been implicated [20]. We first checked if Nrf also responds to α-DC stress and found a significant SKN-1/Nrf activation upon glod-4 mutation or exogenous MGO treatment. This was evident from the upregulation of SKN-1 canonical target genes, gst-4 (Glutathione S-Transferase) and gcs-1 (γ-glutamyl-cysteine synthase heavy chain), using GFP reporter, as well as RT-PCR-based expression analyses (Figures 2A–2E). Further, as reported for various stressors [21, 22], we found SKN-1 to undergo nuclear localization due to α-DC stress (Figures S2A and S2B). However, exogenous MGO failed to trigger DAF-16/FOXO nuclear localization and there was no observable expression difference between glod-4 RNAi and control for DAF-16/FOXO target gene sod-3 (Superoxide Dismutase 3) (Figures S2A and S2C). These findings rule out the direct involvement of insulin/IGF-1 signaling in this process and thus a generic stress response. Phenotypically, we observed that the presence of SKN-1 is beneficial for α-DC stress: glod-4 knockdown in skn-1(zu135) mutants resulted in elevated touch sensitivity and lifespan shortening as compared to N2, respectively (Figures 2F and 2G). This protection was achieved tissue-specifically, as only the intestinal SKN-1C isoform expression can rescue the exacerbated touch sensitivity and short lifespan phenotypes of skn-1 animals under glod-4 RNAi (Figures 2G and 2H). Intestinal SKN-1 activation has been previously reported to be dependent on the p38/Mitogen Activated Protein Kinase (MAPK) pathway [23]. We found that in glod-4;gst-4p∷gfp animals, the expression of gst-4 was significantly suppressed by sek-1 or pmk-1 knockdown (C. elegans MAP kinases), but not by knockdown of sgk-1 (Figure 2I; Figure S2D), a kinase involved in mediating DAF-2/InR responses in C. elegans [24]. Accordingly, glod-4 knockdown significantly reduces the lifespans of pmk-1 and sek-1, but not sgk-1 (Figure 2J; Figure S2E). These results indicate a specific role for p38 MAPK in activating SKN-1 in response to α-DC stress.
Figure 2. SKN-1 renders physiological protection against α-DC-induced toxicity in C. elegans.
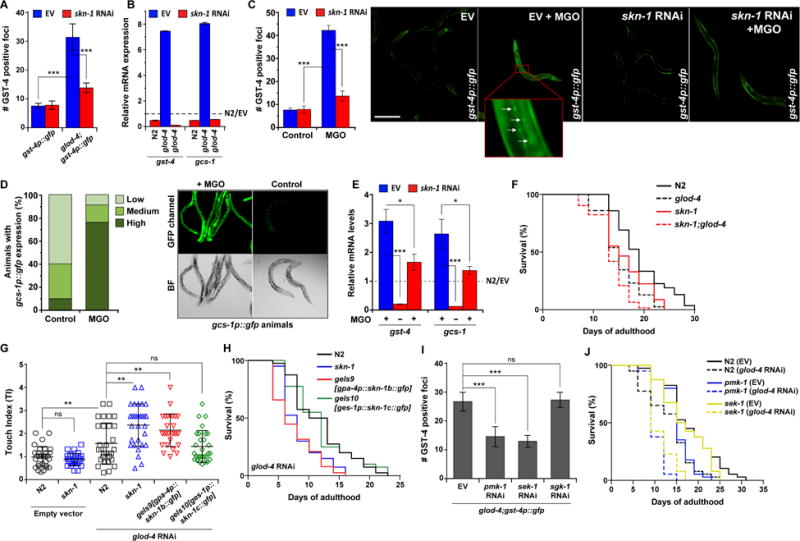
(A) Quantification of GFP foci in gst-4p∷gfp and glod-4;gst-4p∷gfp animals reared on empty vector (EV, L4440) or skn-1 RNAi. n = 15.
(B) qPCR analysis of SKN-1 target genes, gcs-1 and gst-4 in N2 and glod-4 reared on EV or skn-1 RNAi. The data is normalized to the corresponding expression levels in N2 reared on EV (represented by the dotted line).
(C) Quantification (left) and fluorescence microscopy images (right) of GFP foci in gst-4p∷gfp animals reared on empty vector (EV, L4440) or skn-1 RNAi supplemented with water (control) or 7 mM MGO. Foci are marked by white arrows. n = 91. Scale bar 0.15 mm.
(D) Relative quantification of animals exhibiting different fluorescence levels (left) and microscopy images (right) of GFP intensities in gcs-1p∷gfp animals supplemented with water (control) or 7 mM MGO. Animals were categorized as follows: ‘high’ for strong GFP signal throughout the intestine, ‘medium’ for GFP signal in the anterior or posterior section of the intestine and ‘low’ for weak or no signal. n = 100.
(E) qPCR analysis of SKN-1 target genes, gst-4 and gcs-1 in N2 animals reared on EV or skn-1 RNAi supplemented with water (control, −) or 7 mM MGO (+). The data is normalized to the corresponding expression levels in N2 (EV, water), represented by the dotted line.
(F) Survival curves for N2, glod-4, skn-1, and skn-1;glod-4 mutant animals reared on OP50-1.
(G) Touch indices during young adult stage (~L4 stage + 8–10 h) for N2, skn-1, and animals with transgenic expression of skn-1 only in ASI neurons (skn-1b) or only in the intestine (skn-1c). gpa-4 and ges-1 promoters were used to drive skn-1 expression in the ASI neuron and intestine, respectively.
(H) Survival curves for N2, skn-1, and animals with transgenic expression of skn-1 only in ASI neurons (skn-1b) or only in the intestine (skn-1c) under glod-4 RNAi.
(I) Quantification of GFP foci in glod-4;gst-4p∷gfp animals reared on EV, pmk-1, sek-1, or sgk-1 RNAi. n = 15.
(J) Survival curves for N2, pmk-1, and sek-1 mutant animals reared on EV or glod-4 RNAi.
Data are represented as mean ± SD. Significance: *P<0.05, **P<0.005 and ***P<0.0005.
Next, we examined additional upstream components, required for SKN-1 activation under α-DC stress. Several prior studies have implicated ion channels, such as Nav1.8 [10] and TRPA1 [25] in MGO-induced nociception. In general, TRP (transient receptor potential) family of ion channels has been implicated in mechanical, thermal, and pain sensation in both vertebrates and invertebrates [26]. Among the various TRP channel mutants, only trpa-1/TRPA1 showed a significant lifespan shortening, compared to N2 under glod-4 RNAi (Figure 3A; Figure S3A), suggesting a similar beneficial role of TRPA-1 under α-DC stress. In apparent contradiction to this protective role of TRPA-1, knockdown of trpa-1 reduced the hypersensitivity to touch response in glod-4 mutants (Figure 3B). This is not surprising as TRPA-1 is also implicated in mediating C. elegans mechanosensation [27]. However, trpa-1 knockdown resulted in an accelerated neuronal damage specifically in glod-4, similar to skn-1 knockdown (Figure 3C; Figure S3B and S3C), suggesting that the presence of a functional TRPA-1 is ultimately neuroprotective, corroborating the lifespan effects. Interestingly, expression of the SKN-1 reporters, gst-4 and gcs-1, were significantly reduced on trpa-1 knockdown both in glod-4 and MGO-treated wild-type animals (Figures 3D–3G). This suggests that TRPA-1 may be an upstream sensor for α-DC accumulation, required to trigger a SKN-1-dependent stress response.
Figure 3. TRPA-1 is a sensor for α-DCs and result in SKN-1 activation.
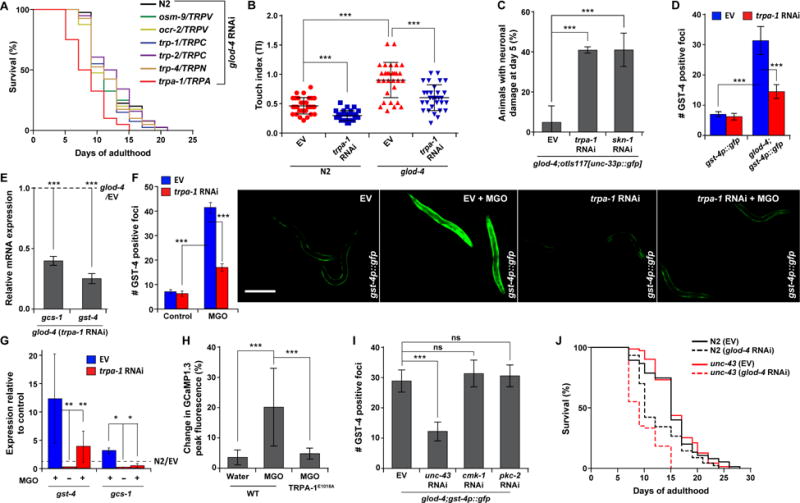
(A) Survival curves for RNAi knockdown of N2 and various C. elegans TRP channel mutants under glod-4 RNAi.
(B) Touch indices during young adult stage for glod-4 animals reared on empty vector (EV, L4440) or trpa-1 RNAi.
(C) Neuronal damage in glod-4;unc-33p∷gfp (pan-neuronal GFP) on day 5 of adulthood, reared on EV, trpa-1, or skn-1 RNAi.
(D) Quantification of GFP foci in gst-4p∷gfp and glod-4;gst-4p∷gfp animals reared on EV or trpa-1 RNAi. n = 15.
(E) qPCR analysis of SKN-1 target genes, gcs-1 and gst-4 in glod-4 mutants reared on EV or trpa-1 RNAi. The data is normalized to the corresponding expression levels in glod-4 mutants reared on EV (represented by the dotted line).
(F) Quantification (left) and fluorescence microscopy images (right) of GFP foci in gst-4p∷gfp animals reared on EV or trpa-1 RNAi supplemented with water (control) or 7 mM MGO. n = 91. Scale bar 0.15 mm.
(G) qPCR analysis of SKN-1 target genes, gst-4 and gcs-1 in N2 animals reared on EV or trpa-1 RNAi supplemented with water (control, −) or 7 mM MGO (+). The data is normalized to the corresponding expression levels in N2 (EV, water) represented by the dotted line.
(H) Percentage change in peak fluorescence intensity observed in transgenic animals expressing intestinal GCaMP1.3 (Ca2+ sensor) and containing a wild-type TRPA-1 or TRPA-1E1018A (Ca2+ impermeable) mutant channel in response to MGO or water (control). n = 15.
(I) Quantification of GFP foci in glod-4;gst-4p∷gfp animals reared on EV, or several Ca+2-sensitive kinase RNAis: unc-43, cmk-1, and pkc-2. n = 15.
(J) Survival curves for N2 and unc-43 animals reared on EV or glod-4 RNAi condition.
Data are represented as mean ± SD. Significance: *P<0.05, **P<0.005 and ***P<0.0005.
Then, we set out to identify the intermediary signaling components that transduce the signal from TRPA-1 to SKN-1. Previous studies have shown that permeability of Ca+2 through TRPA-1 is critical for mediating nociception [25, 28]. Similarly, using a G-CaMP1.3 sensor, we observed a robust Ca+2 response only with a functional TRPA-1 channel with MGO; while control (water) treatment or animals harboring the Ca+2-impermeable TRPA-1E1018A channel resulted in basal responses (Figure 3H; Figure S3D). This suggests the specificity of TRPA-1 in modulating a MGO-induced Ca+2 flux, which cannot be compensated for by any other Ca+2 channels. Then we asked how this TRPA-1-dependent Ca+2 flux is transduced to activate SKN-1: we examined the involvement of candidate Ca+2-sensitive kinases that modulate C. elegans behavior and lifespan [28–30]. Knockdown of unc-43 (Ca2+/calmodulin-dependent kinase/CaMKII), but not cmk-1 (another CaMK) or pkc-2 (Protein kinase C), reduced expression of the SKN-1 reporter gst-4p∷gfp in glod-4 animals (Figure 3I; Figure S3E). Corroborating this, knockdown of glod-4 significantly shortened lifespan of unc-43(n498,n1186), but not cmk-1(oy21) or pkc-2(ok328) mutants (Figure 3J; Figure S3F), thus specifically implicating the role of UNC-43 in this pathway.
Next, we investigated how this TRPA-1/SKN-1 signaling cascade downstream provides physiological protection under elevated α-DC conditions. We found that the TRPA-1/SKN-1 network regulates the expression of GLOD-4, a fundamental α-DC detoxification enzyme: glod-4p∷gfp reporter showed a trpa-1- and skn-1-dependent increase in expression upon MGO treatment (Figure 4A; Figure S4A, see also Figure 4C). Consistent with the exacerbated pathogenic phenotypes, we found that trpa-1 or skn-1 knockdown results in increase of MGO and GO levels, both in N2 and glod-4 (Figure 4B; Figure S4B). In particular, the increase in MGO and GO due to skn-1 or trpa-1 knockdown in glod-4 background suggests the existence of additional α-DC detoxification pathway(s). We then examined whether these could involve the conserved co-factor-independent glyoxalase enzyme(s) [31], DJR-1.1 and -1.2 (human DJ1) and how they may complement GLOD-4 for α-DC detoxification. We found that the expression of all the glyoxalases (glod-4, djr-1.1, and djr-1.2) are strongly regulated by trpa-1 and skn-1 (Figure 4C). Interestingly, while djr-1.1 expression is glod-4-dependent, djr-1.2 expression is not, suggesting a co-option in the trpa-1/skn-1-mediated α-DC detoxification network. Further, changes to MGO and GO levels due to djr-.1.1 and djr-1.2 knockdown are comparable to that of glod-4 knockdown (Figure 4D; Figure S4C). Accordingly, djr-1.1 and djr-1.2 knockdown result in increased touch sensitivity and reduced lifespan phenotypes similar to glod-4 (Figures 4E and 4F). Interestingly, prior studies indicate that the specific activity of DJ1 as a glyoxalase may be much lower than GLO1 [31, 32]. However, these studies were conducted only in in vitro systems, which entails that enzymes may not be correctly folded and/or the setup could be missing an unidentified co-factor, thus limiting the predictability of the enzymes’ in vivo function(s).
Figure 4. Downstream glyoxalases mediate α-DC detoxification in response to TRPA-1/SKN-1 activation.
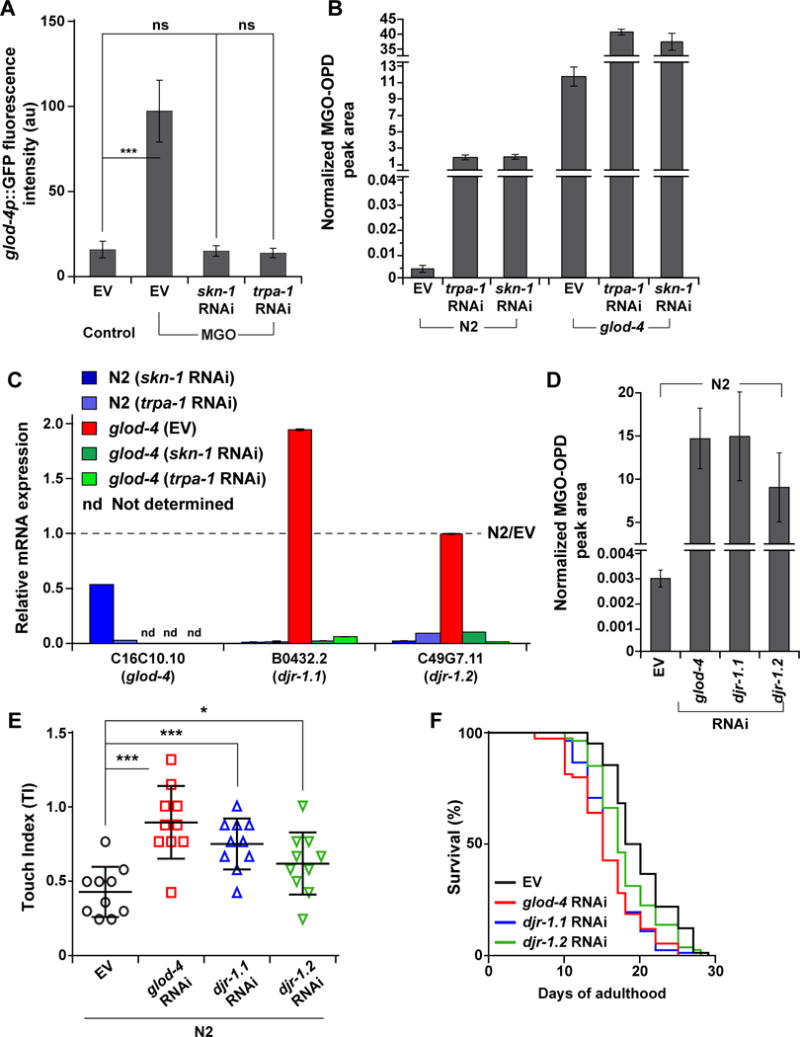
(A) Quantification of GFP intensity of glod-4p∷gfp reporter strain, reared on empty vector (EV, L4440), skn-1, or trpa-1 RNAi, supplemented with water (control) or MGO (7 mM). n = 10.
(B) Levels of MGO in N2 and glod-4 animals, reared on EV, trpa-1, or skn-1 RNAi.
(C) qPCR analysis of conserved glyoxalases, glod-4, djr-1.1, and djr-1.2, in N2 and glod-4, reared on EV, skn-1, or trpa-1 RNAi. The data is normalized to the corresponding expression levels in N2 animals reared on EV (represented by the dotted line).
(D) Levels of MGO in N2, reared on EV, glod-4, djr-1.1, or djr-1.2 RNAi.
(E) Touch indices during young adult stage for N2, reared on EV, glod-4, djr-1.1, and djr-1.2 RNAi.
(F) Survival curves for N2, reared on EV, glod-4, djr-1.1, and djr-1.2 RNAi.
Data are represented as mean ± SD. Significance: *P<0.05, **P<0.005 and ***P<0.0005.
C. elegans glod-4 model for drug development
With a firm understanding of the biochemical regulation of α-DC stress, we set out to answer one of the more contemporary and practical questions: can we use the glod-4 model to identify novel therapies for treating pathologies associated with α-DC stress? Taking advantage of the ease of experimental setup in C. elegans [33], we screened a library of natural products (TimTec Inc. NPL-640), first to ameliorate the hyperesthesia phenotype exhibited by glod-4 animals. Among the hits, podocarpic acid (Figure 5A), a natural product isolated from the New Zealand conifer Dacrydium cupressinum [34], had the best all-round positive effect. Podocarpic acid (PA) was able to alleviate the glod-4 touch sensitivity phenotypes (Figure 5B; Figure S5A), prevent neuronal damages in aging glod-4 animals, and extend glod-4 lifespan (Figures 5C and 5D; Figure S5B). Interestingly, PA did not prove to be a lifespan extending compound for N2 animals (Figure S5C).
Figure 5. Podocarpic acid (PA) is a TRPA-1 agonist and SKN-1 activator that ameliorates pathogenic phenotypes of C. elegans glod-4.
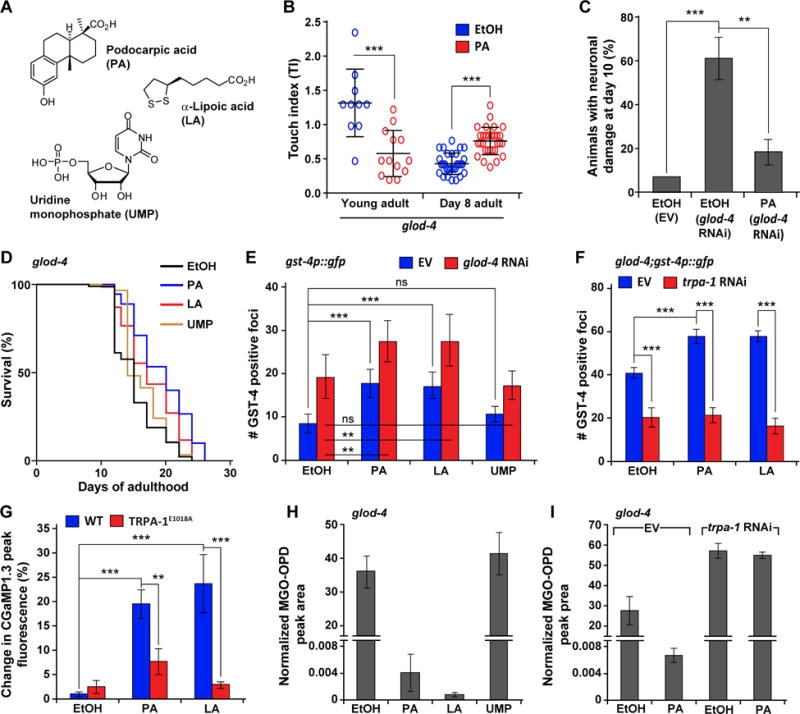
(A) Structures of podocarpic acid (PA), α-lipoic acid (LA), and uridine monophosphate (UMP).
(B)Touch indices in glod-4, supplemented with EtOH (control) or PA (20 μM) during young adult stage or day 8 of adulthood.
(C) Neuronal damage in pan-neuronal GFP animals (unc-33p∷gfp) at day 10 of adulthood, reared on empty vector (EV, L4440) or glod-4 RNAi, supplemented with EtOH (control) or PA (20 μM). n = 45.
(D) Survival curves for glod-4 mutant animals supplemented with EtOH (control), PA, LA, or UMP (20 μM).
(E) Quantification of GFP foci in gst-4p∷gfp animals reared on EV or glod-4 RNAi, supplemented with EtOH (control), PA, LA, or UMP (20 μM). n = 10.
(F) Quantification of GFP foci in glod-4;gst-4p∷gfp animals reared on EV or trpa-1 RNAi, supplemented with EtOH (control), PA or LA (20 μM). n = 15.
(G) Percentage change in peak fluorescence intensity observed in transgenic animals expressing intestinal GCaMP1.3 (Ca2+ sensor) and a wild-type TRPA-1 or TRPA-1E1018A (Ca2+ impermeable) mutant channel, in response to EtOH (control), PA or LA (20 μM). n = 10.
Levels of MGO in
(H) glod-4, supplemented with EtOH (control), PA, LA, or UMP (20 μM) or in
(I) glod-4, reared on EV or trpa-1 RNAi, supplemented with EtOH (control) or PA.
Data are represented as mean ± SD. Significance: *P<0.05, **P<0.005 and ***P<0.0005.
Next, we checked if this new compound utilizes the TRPA-1/SKN-1-controlled α-DC detoxification pathway for ameliorating the pathogenic phenotypes associated with glod-4. We found that PA activates SKN-1 in C. elegans, similar to known Nrf2 activators such as α-lipoic acid (LA) (Figures 5A and 5E). LA is currently used as a dietary supplement for diabetic complications [35] and performed similar to PA in glod-4 lifespan assays (Figure 5D). Interestingly, we found that the PA or LA-induced SKN-1 activation also requires TRPA-1: trpa-1 knockdown in glod-4;gst-4p∷gfp animals, reduced expression of gst-4 to wild-type levels (Figure 5F). More importantly, this PA and LA-mediated SKN-1 activation was observed to a similar extent in both N2 and glod-4 backgrounds (Figures 5E and 5F). This suggests that TRPA-1/SKN-1 activation via α-DCs and via PA or LA happens largely through distinct mechanisms. Further, PA and LA supplementation results in a robust Ca+2 flux, which is significantly reduced when the Ca+2-impermeable TRPA-1E1018A channel is present (Figure 5G; Figure S5D), suggesting that TRPA-1 activation is key for these drugs’ function. Finally, PA and LA alleviated the pathogenic phenotypes of glod-4 animals by reverting the high endogenous MGO and GO to almost wild-type-like levels (Figures 1B and 5H; Figure S5E). Consistent with action of PA to be dependent on trpa-1, under trpa-1 knockdown, PA has no effect on MGO levels (Figure 5I). Thus, in the absence of glod-4, activation of trpa-1 can ameliorate α-DC stress, perhaps by engaging DJR-1.1 and -1.2. Uridine monophosphate (UMP) was used as a negative control in these assays. While UMP, a hit from our drug screen, results in reduced touch sensitivity (Figure S5A), it does not ameliorate any of the other deleterious phenotypes associated with glod-4 (Figures 5D and 5H; Figure S5E), nor does it activate SKN-1 (Figure 5E).
Conservation of MGO-induced neurotoxicity and its rescue
MGO and TRP channels, specifically TRPA1 have been previously implicated in neuropathic pain [25]. In C. elegans, we found TRPA-1 to evoke a robust Ca2+ flux in response to elevated MGO. Hence, we questioned if this response is conserved across taxa. To that end, we expressed worm and rat TRPA1 in mammalian HEK293 cells and examined MGO-induced Ca+2 flux response. We found that MGO was able to induce a similar Ca2+ flux through both rat and worm TRPA1 channels (Figure 6A and 6B). To check if this response is specific to TRPA1, we used native HEK293 cells (expressing GFP sensor only) or HEK293 cells expressing mouse TRPM8, a channel known to result in Ca2+ flux when activated by menthol [36]. Our results show that menthol but not MGO, triggers a Ca+2 flux through the TRPM8 channel (Figure 6C), whereas neither compounds resulted in Ca+2 flux in the native HEK293 cells (Figure 6A–6C); suggesting specificity of MGO for TRPA1 activation. In contrast, allyl isothiocyanate (AITC), the compound responsible for the pungent smell in wasabi and mustard oil, known to activate mammalian TRPA1, failed to activate the worm TRPA1 channel (Figures S6A and S6B), suggesting that MGO and AITC may have distinct activation mechanisms.
Figure 6. Methylglyoxal (MGO)-induced neurotoxicity is sensed and rescued through a conserved mechanism.
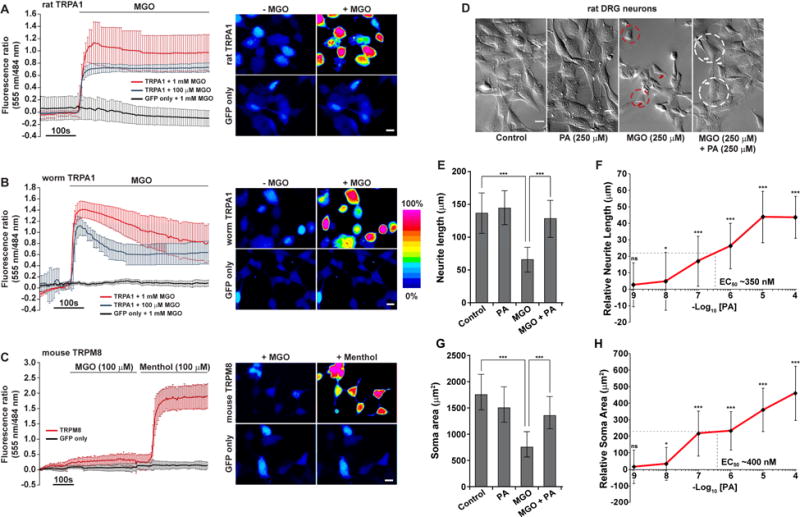
Fluorescence ratio (555/484 nm) changes for the membrane-permeable Ca+2 indicator Rhod-3 AM (left) and representative Rhod-3 AM images (Scale bar is 10 μm) in pseudo color scale (right) for HEK293 cells transfected with
(A) TRPA1 (RAT) and GFP or GFP only. Cells were treated with 100 μM or 1 mM MGO and images captured before (−MGO) or 100 s after 100 μM MGO application. n ≥6.
(B) TRPA1 (worm) and GFP or GFP only, treated and imaged as in Fig. 6a. n ≥5.
(C) TRPM8 (mouse) and GFP or GFP only. Cells were treated with 100 μM MGO first and then switched to 100 μM menthol. Images were captured after 100 s of incubation with MGO and menthol. n=9.
(D) DIC images for differentiated 50B11 cells (immortalized rat DRG neuronal cells) treated with ethanol (control), podocarpic acid/PA (250 μM), MGO (250 μM), or a combination of PA and MGO (each at 250 μM). Shrinkage in cell bodies (red dotted circles), retraction in neurite outgrowth (red arrows) and diminished neuronal networking is visible in MGO (only)-treated cells. Amelioration of size of the cell bodies (white dotted circle) and length of neurite outgrowth emerging from the edge of the soma (white arrows) due to PA treatment. Scale bar 50 μm.
(E–F) Neurite length and
(G–H) soma size quantification in rat DRG neuronal cells, treated with water (control), PA (various concentrations), 250 μM MGO, and 250 μM of MGO and PA (various concentrations). For dose response figures, the individual data points are presented relative to MGO (250 μM) measurements. PA concentration is 250 μM in (E) and (G).
Data are represented as mean ± SD. Significance: *P<0.05, **P<0.005 and ***P<0.0005.
See also Figure S6.
Next, we examined whether podocarpic acid (PA) rescues MGO-mediated neurotoxicity. We used the 50B11 cell line (Figure S6C) [37], an immortalized rat dorsal root ganglion (DRG) neuronal cell line that natively expresses Trpa1 [25]. In differentiated DRG neurons, exposure to MGO resulted in significant neuronal damage evident by shrinkage in cell bodies and significant retraction of neurite outgrowths (Figure 6D). We found that PA was able to ameliorate these MGO-induced neurotoxic phenotypes (Figures 6D–6H) at EC50 ~400 nM. These results further argue the validity of C. elegans as a model for identifying therapeutic leads against α-DC-induced pathologies.
DISCUSSION
Accumulation of reactive α-DCs, e.g. GO, MGO, 3DG and derived AGEs have been implicated as the root-cause for multiple diabetic complications [38]. Additionally, α-DC and AGE stress have been associated with neurodegenerative disorders, for which diabetes is an additional risk factor, such as Alzheimer’s disease, Parkinson’s disease, and ATTR amyloidosis [6]. Hence, the C. elegans glod-4 model established in this work may have far-reaching clinical relevance. More importantly, all pathogenic phenotypes in glod-4 mutant occur within a couple of weeks that can take years to develop in humans, significantly fast-tracking biochemical discovery and drug development. As the first step, we have uncovered a conserved regulatory network that mediates endogenous α-DC detoxification (Figure 7) based on TRPA-1/TRPA1, flux of Ca+2 ions relayed by UNC-43/CaMKII, and the p38/MAPK kinases SEK-1 and PMK-1 to SKN-1. In response, SKN-1 initiates a transcription program geared towards activation of a multi-faceted α-DC detoxification (Figure 7). Interestingly, it has been shown that the TRPA-1 mediates the lifespan extension upon cold sensation in C. elegans [28]. Even though cold-mediated TRPA-1 activation results in a similar Ca+2 flux, downstream signaling involves distinct kinases PKC-2 and SGK-1 and result in DAF-16/FOXO activation [28]. The existence of such a TRPA-1 signaling plasticity suggests that other yet-to-be identified signaling components are involved in deciding the course of the organism’s response to an endogenous (α-DCs) vs. an exogenous (cold) stress. Further, it has been shown that a wide range of TRPA1 agonists such as allyl isothiocyanate (AITC), to activate TRPA1 through covalent modification of specific cysteine residues [39]. Interestingly, C. elegans do not have these specific Cys residues and is therefore refractory to AITC activation [28]. However, we observe that the MGO-induced TRPA1 activation is conserved from C. elegans to mammals, suggesting a previously unreported mode of TRPA1 activation by MGO, which could result in the aforementioned plasticity.
Figure 7. Model for α-DC (endogenous stress)- or cold (exogenous stress)-induced TRPA-1 activation and subsequent divergence of downstream signaling in C. elegans.
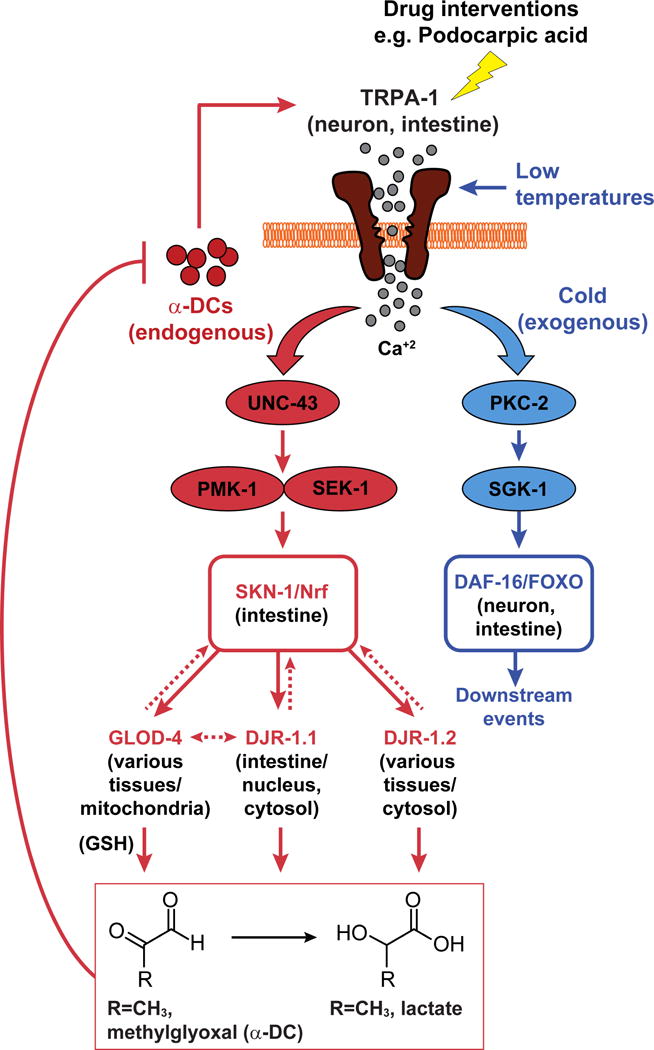
TRPA-1/TRPA1 activation via α-DCs is relayed through UNC-43 (CaMKII), PMK-1, and SEK-1 (MAPK) to SKN-1/Nrf resulting in the expression of various downstream glyoxalases to achieve organism-wide α-DC detoxification. In contrast, the effects of cold-induced TRPA-1 activation is mediated through DAF-16/FOXO regulation via PKC-2 (protein kinase C) and SGK-1 (serum- and glucocorticoid-inducible kinase). Drug-induced activation of TRPA1-Nrf2 ameliorates pathologies associated with elevated α-DC buildup.
Downstream, the activation of the TRPA1-Nrf pathway ultimately results in the expression of evolutionarily conserved glutathione-dependent glyoxalase glod-4, and co-factor–independent glyoxalases djr-1.1, and djr-1.2 that convert reactive α-DCs to significantly less reactive metabolites, e.g. MGO to D-lactate (Figure 7). While Nrf2-dependent regulation of GLO1 has previously been shown in mammalian cell lines [40], C. elegans studies that looked for SKN-1 downstream targets did not feature these glyoxalases [41]. Moreover, the mammalian functional ortholog of the glutathione-independent glyoxalase has only recently been identified as DJ1, an enzyme implicated in early onset Parkinson’s disease [31]. DJR-1.1 and DJR-1.2 are C. elegans ortholog of DJ1 [31], and our data shows that along with GLOD-4, both these enzymes are responsible for α-DC detoxification. The redundancy of the glyoxalases perhaps arise to eradicate deleterious reactive α-DCs across the organism, as well as all relevant organelles as each of the glyoxalases are expressed in complementary cellular compartments (Figure 7) [11, 31]. Interestingly, individuals suffering from long-term diabetes are at a higher risk of Parkinson’s disease [42]. Our results thus take a step closer towards understanding the biochemical ink between diabetes and Parkinson’s disease.
Our drug screen and concomitant identification of podocarpic acid suggests that amelioration of α-DC stress represents a viable option for mitigating related pathologies, which remains underutilized from a clinical perspective. Such rapid phenotypic drug screens have the potential to offer several advantages over target-based screens; generally by overcoming problems faced in target-based approaches such as metabolic instability and toxicity due to off-target effects [43]. Our results with α-lipoic acid (LA), which is documented to ameliorate diabetic complications in mice and humans [44], argues for the validity of using the worm model for studying diabetes-like pathologies. However, LA is extensively metabolized in mammals, limiting this compound’s potential as a drug [45]. Thus, the identification of podocarpic acid (PA) as a novel TRPA1 activator is quite exciting. The rescue of α-DC-induced phenotypes using PA in C. elegans as well as rat DRG neurons, suggests TRPA1 activators are viable candidates for treating diabetic pathologies, even with a basal Nrf activation due to accumulated α-DCs. Additionally, most enzymes featured in our TRPA1-Nrf pathway, represent viable druggable targets (Figure 7), particularly Nrf2 [46]. However, attempts at direct Nrf2 activation in vivo has resulted in a multitude of unwanted side-effets [47, 48], e.g. promotion of cancerous tumours, development of atheroschlerosis, etc, often outweighing its potential health benefits. Our results suggest that we may be able to circumvent this by indirectly activating Nrfs via TRPA1. Future studies in mammalian in vivo models will corroborate the utility of such indirect Nrf activation and its clinical significance. However, our results contrast some previous findings that suggest TRPA1 channel antagonists in the amelioration of hypersesthesia [49]. As noted earlier, results with such antagonist treatment should be intrepreted with caution [25], as there may be confounding off-target effects of these compounds. Moreover, while TRPA1 antagonism can provide temporary relief by numbing neuropathic pain, our results show that the presence of a functional TRPA1 is ultimately neuroprotective, beneficial to organismal healthspan. Thus, drug-induced TRPA1 activation can be a viable strategy for ameliorating α-DC stress as observed in diabetes, and neurodegenrative conditions such as Parkinson’s and Alzeimer’s disease.
EXPERIMENTAL PROCEDURES
Strains
Nematode stocks were maintained on Nematode Growth Medium (NGM) plates made with Bacto agar (BD Biosciences) and seeded with bacteria (Escherichia coli strain OP50-1 unless otherwise specified) at 20 °C (http://www.wormbook.org/). The following C. elegans strains were used: wild type (N2, Bristol), VC343: glod-4(gk189), BC15643: sEx15643 [rCesC16C10.10∷gfp + pCeh361], OH438: otIs117 [unc-33p∷gfp + unc-4(+)], CL2166: dvIs19 [(pAF15)gst-4p∷gfp∷nls], LD1171: IdIs3 [gcs-1p∷gfp+pRF4(rol-6(su1006))], EU31: skn-1(zu135) IV/nT1 [unc-?(n754) let-?], LG348: skn-1(zu135)/nT1[qIs51], geIs9[gpa-4p∷skn-1b∷gfp+rol-6(su1006)], LG357: skn-1(zu135)/nT1[qIs51], geIs10[ges-1p∷skn-1c∷gfp+rol-6(su1006)], LD1: Idls7 [skn-1b/c∷gfp + pRF4(rol-6(su1006))], KU25: pmk-1(km25), KU4: sek-1(km4), VC345: sgk-1(ok538), CX10: osm-9(ky10), CX4544: ocr-2(ak47), TQ225: trp-1(sy690), TQ194: trp-2(sy691), TQ296: trp-4(sy695), TQ233: trpa-1(ok999), TQ2772: xuEx866[pges-1∷trpa-1(E1018A) ∷sl2∷mcherry2];xuEx19[plfe-2b∷GCaMP1.3+plfe-2b∷dsred];trpa-1(ok999), TQ1996: unc-43(n498, n1186), TQ2746: cmk-1(oy21), TQ2571: pkc-2(ok328), TQ3056: xuIs180[plfe2b∷GCaMP1.3+plfe-2b∷dsred];N2, TJ356: zIs356[daf-16p∷daf-16a/b∷GFP + rol-6], BC15643: sEx15643[rCes C16C10.10∷gfp + pCeh361], CF1553: muIs84 [(pAD76) sod-3p∷GFP + rol-6(su1006)], glod-4;dvIs19 [(pAF15)gst-4p∷gfp∷nls], glod-4;otIs117 [unc-33p∷gfp + unc-4(+)], glod-4;skn-1(zu135) IV/nT1 [unc-?(n754) let-?]. Compound mutants were constructed using standard techniques. Bacterial clones for RNAi feeding protocol were obtained from either the Ahringer library or the ORFeome RNAi v1.1 library (Open Biosystems, GE Healthcare, CO).
Touch assay
Mechanosensory responsiveness to gentle touch was adapted from a previously described protocol [50]. Briefly, each animal was touched alternately in the anterior (head and nose) region and in the posterior (tail) region with an eyelash with a ~4–5 s delay between individual touches. Typically, an omega turn or diversion in head direction resulting from anterior touch was counted as a positive response. Posterior touch response (not counted towards positive response) functioned towards resetting of the response to anterior touch. Touch index (TI) scores were generated by dividing the total number of positive responses over the number of negative responses per animal.
Calcium imaging of HEK293 cells
Appropriate HEK293-derived cells were seeded on collagen-coated glass bottom culture dishes (MatTek Corporation). Cells were loaded with 10 μM of Rhod-3 AM (Life Technology) for 30 min at 37 °C. After 30 min they were washed twice with standard Tyrode’s solution (135 mM NaCl, 4 mM KCl, 10 mM glucose, 10 mM HEPES, 2mM CaCl2, and 1 mM MgCl2 at pH=7.4) at room temperature. Calcium imaging was performed on an Olympus BX51WI Axiovert microscope under a 60× objective. Fluorescent images were documented upon sequential excitation with 555 nm followed by 484 nm with a Roper CoolSnap CCD camera. After establishing a baseline 555/484 ratio, methylglyoxal or other agonists were diluted with Tyrode’s solution (100 μM or 1 mM final concentration) and were perfused into cells. Images were processed with the MetaFlour (Olympus) software.
Statistical analyses
All data analyses for lifespan were performed using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA). Survival curves were plotted using Kaplan-Meier method and comparison between survival curves to measure significance (P values) was performed using Log-rank (Mantel-Cox) test. All remaining pairwise comparisons for the quantification data were done using two-tailed Student’s t-test. P values from the significance testing were designated as follows: *P<0.05, **P<0.005 and ***P<0.0005.
Supplementary Material
Acknowledgments
The 50B11 cell line was a gift from Dr. Ahmet Höke (Johns Hopkins University). We thank Dr. Akos Gerencser (Buck Institute Morphology and Imaging Core) for help with DIC imaging. We thank Profs. Julie Anderson, Richmond Sarpong, Simon Melov, Bradford Gibson; Drs. Subhash Katewa, Marc Lucanic, Kathleen Dumas and Amit Khanna for their suggestions. This work was supported by grants from the American Federation of Aging Research (to P.K.), Larry L. Hillblom Foundation (to P.K.), the Griswold Family Fund, the 2016 Buck Institute Impact Circle (to P.K.) and the NIH (T32 AG000266 to J.C.; R01AG038688, AG038012, AG045835 to P.K.; and R01AG048072 to X.Z.S.X.). We thank Prof. Keith Blackwell for the LD1 strain. Some strains were provided by the CGC (funded by NIH Office of Research Infrastructure Programs (P40 OD010440)) and by the C. elegans Gene-Knockout Consortium. The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
J.C., N.B., and P.K. conceived the study and designed experiments. N.B., D.H., and A.Ri. processed the biological samples for LC-MS/MS analyses. N.B. and A.Ra. analyzed the LC-MS/MS data. J.C. performed the C. elegans lifespan, touch assays, fluorescent microscopy, genetic expression studies, and Ca+2 channel-based studies. J.C. and A.Ri. performed the C. elegans motility-related assays. G.L. supervised and D.H. performed the touch assay-based natural product library screen. T.H.P. performed the C. elegans lifespan experiments for glyoxalase RNAi. J.G., J.L., and X.Z.S.X. designed, performed, and analyzed the HEK293-based channel studies. D.B. and C.L. performed the DRG cell line-based assays with the assistance of J.C. and A.Ri. J.C. and M.C. constructed the C. elegans compound mutant strains. J.C., N.B., and P.K. analyzed the results from all biological experiments. J.C., N.B., and P.K. wrote the manuscript.
References
- 1.Thornalley PJ. Methylglyoxal, glyoxalases and the development of diabetic complications. Amino acids. 1994;6:15–23. doi: 10.1007/BF00808119. [DOI] [PubMed] [Google Scholar]
- 2.Peppa M, Vlassara H. Advanced glycation end products and diabetic complications: a general overview. Hormones. 2005;4:28–37. doi: 10.14310/horm.2002.11140. [DOI] [PubMed] [Google Scholar]
- 3.Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic complications. The Korean journal of physiology & pharmacology: official journal of the Korean Physiological Society and the Korean Society of Pharmacology. 2014;18:1–14. doi: 10.4196/kjpp.2014.18.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monnier VM, Sell DR, Genuth S. Glycation products as markers and predictors of the progression of diabetic complications. Annals of the New York Academy of Sciences. 2005;1043:567–581. doi: 10.1196/annals.1333.065. [DOI] [PubMed] [Google Scholar]
- 5.Brownlee M. Advanced protein glycosylation in diabetes and aging. Annual review of medicine. 1995;46:223–234. doi: 10.1146/annurev.med.46.1.223. [DOI] [PubMed] [Google Scholar]
- 6.Sousa Silva M, Gomes RA, Ferreira AE, Ponces Freire A, Cordeiro C. The glyoxalase pathway: the first hundred years… and beyond. The Biochemical journal. 2013;453:1–15. doi: 10.1042/BJ20121743. [DOI] [PubMed] [Google Scholar]
- 7.Islam MS, Koya D, Portha B. Animal models of diabetes and its associated complications. Journal of diabetes research. 2013;2013:593204. doi: 10.1155/2013/593204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giacco F, Du X, D’Agati VD, Milne R, Sui G, Geoffrion M, Brownlee M. Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes. 2014;63:291–299. doi: 10.2337/db13-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Distler MG, Palmer AA. Role of Glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: recent advances and mechanistic insights. Frontiers in genetics. 2012;3:250. doi: 10.3389/fgene.2012.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, Sauer SK, Eberhardt M, Schnolzer M, Lasitschka F, et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nature medicine. 2012;18:926–933. doi: 10.1038/nm.2750. [DOI] [PubMed] [Google Scholar]
- 11.Morcos M, Du X, Pfisterer F, Hutter H, Sayed AA, Thornalley P, Ahmed N, Baynes J, Thorpe S, Kukudov G, et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging cell. 2008;7:260–269. doi: 10.1111/j.1474-9726.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 12.Riddle DL, Blumenthal T, Meyer BJ, Priess JR. In: Introduction to C elegans In C elegans II. 2nd. Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. Cold Spring Harbor; NY: 1997. [Google Scholar]
- 13.Henning C, Liehr K, Girndt M, Ulrich C, Glomb MA. Extending the spectrum of alpha-dicarbonyl compounds in vivo. The Journal of biological chemistry. 2014;289:28676–28688. doi: 10.1074/jbc.M114.563593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rabbani N, Thornalley PJ. Measurement of methylglyoxal by stable isotopic dilution analysis LC-MS/MS with corroborative prediction in physiological samples. Nature protocols. 2014;9:1969–1979. doi: 10.1038/nprot.2014.129. [DOI] [PubMed] [Google Scholar]
- 15.Takekuma K, Ando F, Niino N, Shimokata H. Prevalence of hyperesthesia detected by current perception threshold test in subjects with glucose metabolic impairments in a community. Internal medicine. 2002;41:1124–1129. doi: 10.2169/internalmedicine.41.1124. [DOI] [PubMed] [Google Scholar]
- 16.Mendler M, Schlotterer A, Ibrahim Y, Kukudov G, Fleming T, Bierhaus A, Riedinger C, Schwenger V, Herzig S, Hecker M, et al. daf-16/FOXO and glod-4/glyoxalase-1 are required for the life-prolonging effect of human insulin under high glucose conditions in Caenorhabditis elegans. Diabetologia. 2015;58:393–401. doi: 10.1007/s00125-014-3415-5. [DOI] [PubMed] [Google Scholar]
- 17.Rabbani N, Thornalley PJ. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochemical and biophysical research communications. 2015;458:221–226. doi: 10.1016/j.bbrc.2015.01.140. [DOI] [PubMed] [Google Scholar]
- 18.Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PloS one. 2010;5:e8758. doi: 10.1371/journal.pone.0008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlotterer A, Kukudov G, Bozorgmehr F, Hutter H, Du X, Oikonomou D, Ibrahim Y, Pfisterer F, Rabbani N, Thornalley P, et al. C. elegans as model for the study of high glucose- mediated life span reduction. Diabetes. 2009;58:2450–2456. doi: 10.2337/db09-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jimenez-Osorio AS, Gonzalez-Reyes S, Pedraza-Chaverri J. Natural Nrf2 activators in diabetes. Clinica chimica acta; international journal of clinical chemistry. 2015;448:182–192. doi: 10.1016/j.cca.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 21.An JH, Blackwell TK. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes & development. 2003;17:1882–1893. doi: 10.1101/gad.1107803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staab TA, Griffen TC, Corcoran C, Evgrafov O, Knowles JA, Sieburth D. The conserved SKN-1/Nrf2 stress response pathway regulates synaptic function in Caenorhabditis elegans. PLoS genetics. 2013;9:e1003354. doi: 10.1371/journal.pgen.1003354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.An JH, Vranas K, Lucke M, Inoue H, Hisamoto N, Matsumoto K, Blackwell TK. Regulation of the Caenorhabditis elegans oxidative stress defense protein SKN-1 by glycogen synthase kinase-3. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16275–16280. doi: 10.1073/pnas.0508105102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hertweck M, Gobel C, Baumeister R. C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Developmental cell. 2004;6:577–588. doi: 10.1016/s1534-5807(04)00095-4. [DOI] [PubMed] [Google Scholar]
- 25.Andersson DA, Gentry C, Light E, Vastani N, Vallortigara J, Bierhaus A, Fleming T, Bevan S. Methylglyoxal evokes pain by stimulating TRPA1. PloS one. 2013;8:e77986. doi: 10.1371/journal.pone.0077986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Julius D. TRP channels and pain. Annual review of cell and developmental biology. 2013;29:355–384. doi: 10.1146/annurev-cellbio-101011-155833. [DOI] [PubMed] [Google Scholar]
- 27.Kindt KS, Viswanath V, Macpherson L, Quast K, Hu H, Patapoutian A, Schafer WR. Caenorhabditis elegans TRPA-1 functions in mechanosensation. Nature neuroscience. 2007;10:568–577. doi: 10.1038/nn1886. [DOI] [PubMed] [Google Scholar]
- 28.Xiao R, Zhang B, Dong Y, Gong J, Xu T, Liu J, Xu XZ. A genetic program promotes C. elegans longevity at cold temperatures via a thermosensitive TRP channel. Cell. 2013;152:806–817. doi: 10.1016/j.cell.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reiner DJ, Newton EM, Tian H, Thomas JH. Diverse behavioural defects caused by mutations in Caenorhabditis elegans unc-43 CaM kinase II. Nature. 1999;402:199–203. doi: 10.1038/46072. [DOI] [PubMed] [Google Scholar]
- 30.Robatzek M, Thomas JH. Calcium/calmodulin-dependent protein kinase II regulates Caenorhabditis elegans locomotion in concert with a G(o)/G(q) signaling network. Genetics. 2000;156:1069–1082. doi: 10.1093/genetics/156.3.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JY, Song J, Kwon K, Jang S, Kim C, Baek K, Kim J, Park C. Human DJ-1 and its homologs are novel glyoxalases. Human molecular genetics. 2012;21:3215–3225. doi: 10.1093/hmg/dds155. [DOI] [PubMed] [Google Scholar]
- 32.Rabbani N, Xue M, Thornalley PJ. Activity, regulation, copy number and function in the glyoxalase system. Biochemical Society transactions. 2014;42:419–424. doi: 10.1042/BST20140008. [DOI] [PubMed] [Google Scholar]
- 33.Petrascheck M, Ye X, Buck LB. An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature. 2007;450:553–556. doi: 10.1038/nature05991. [DOI] [PubMed] [Google Scholar]
- 34.Cui YM, Yasutomi E, Otani Y, Yoshinaga T, Ido K, Sawada K, Ohwada T. Design, synthesis and characterization of podocarpate derivatives as openers of BK channels. Bioorganic & medicinal chemistry letters. 2008;18:5197–5200. doi: 10.1016/j.bmcl.2008.08.081. [DOI] [PubMed] [Google Scholar]
- 35.Vallianou N, Evangelopoulos A, Koutalas P. Alpha-lipoic Acid and diabetic neuropathy. The review of diabetic studies: RDS. 2009;6:230–236. doi: 10.1900/RDS.2009.6.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu B, Qin F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:1674–1681. doi: 10.1523/JNEUROSCI.3632-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen W, Mi R, Haughey N, Oz M, Hoke A. Immortalization and characterization of a nociceptive dorsal root ganglion sensory neuronal line. Journal of the peripheral nervous system: JPNS. 2007;12:121–130. doi: 10.1111/j.1529-8027.2007.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabbani N, Thornalley PJ. Glyoxalase in diabetes, obesity and related disorders. Seminars in cell & developmental biology. 2011;22:309–317. doi: 10.1016/j.semcdb.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541–545. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- 40.Xue M, Rabbani N, Momiji H, Imbasi P, Anwar MM, Kitteringham N, Park BK, Souma T, Moriguchi T, Yamamoto M, et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. The Biochemical journal. 2012;443:213–222. doi: 10.1042/BJ20111648. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Robida-Stubbs S, Tullet JM, Rual JF, Vidal M, Blackwell TK. RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS genetics. 2010;6 doi: 10.1371/journal.pgen.1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santiago JA, Potashkin JA. System-based approaches to decode the molecular links in Parkinson’s disease and diabetes. Neurobiology of disease. 2014;72(Pt A):84–91. doi: 10.1016/j.nbd.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 43.Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacological reviews. 2011;63:411–436. doi: 10.1124/pr.110.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gomes MB, Negrato CA. Alpha-lipoic acid as a pleiotropic compound with potential therapeutic use in diabetes and other chronic diseases. Diabetology & metabolic syndrome. 2014;6:80. doi: 10.1186/1758-5996-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teichert J, Hermann R, Ruus P, Preiss R. Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers. Journal of clinical pharmacology. 2003;43:1257–1267. doi: 10.1177/0091270003258654. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends in pharmacological sciences. 2013;34:340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 47.Barajas B, Che N, Yin F, Rowshanrad A, Orozco LD, Gong KW, Wang X, Castellani LW, Reue K, Lusis AJ, et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:58–66. doi: 10.1161/ATVBAHA.110.210906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei H, Hamalainen MM, Saarnilehto M, Koivisto A, Pertovaara A. Attenuation of mechanical hypersensitivity by an antagonist of the TRPA1 ion channel in diabetic animals. Anesthesiology. 2009;111:147–154. doi: 10.1097/ALN.0b013e3181a1642b. [DOI] [PubMed] [Google Scholar]
- 50.Hobert O, Moerman DG, Clark KA, Beckerle MC, Ruvkun G. A conserved LIM protein that affects muscular adherens junction integrity and mechanosensory function in Caenorhabditis elegans. The Journal of cell biology. 1999;144:45–57. doi: 10.1083/jcb.144.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.