

Summary
Background
HSD3B1(1245A>C) has been mechanistically linked to castration-resistant prostate cancer by encoding an altered enzyme that augments dihydrotestosterone synthesis. We hypothesized that men inheriting the HSD3B1(1245C) allele would exhibit resistance to androgen deprivation therapy (ADT).
Methods
We determined HSD3B1 genotype retrospectively in men treated with ADT for post-prostatectomy biochemical failure and correlated genotype with long-term clinical outcomes. Patients who received postoperative adjuvant or salvage radiotherapy were eligible, provided they had residual active disease as reflected by continued increase in their PSA after treatment. We analyzed progression-free survival (PFS; primary endpoint), distant metastasis-free survival (DMFS), and overall survival (OS) according to HSD3B1 genotype. Multivariable analyses were performed to assess the independent predictive value of HSD3B1 genotype on outcomes. Results were externally validated in two additional cohorts, including a second post-prostatectomy biochemical failure cohort as well as a metastatic cohort. There was no age limit for eligibility in the primary or validation cohorts.
Findings
The study included 443 patients: 118 in the primary cohort, 137 in the post-prostatectomy validation cohort, and 188 in the metastatic validation cohort. In the primary study cohort, median PFS diminished as a function of the number of variant alleles inherited: 6.6 years in homozygous wild-type men (95% CI, 3.8 to not reached); 4.1 years in heterozygotes (95% CI, 3.0 to 5.5); and 2.5 years in homozygous variant men (95% CI, 0.7 to not reached); P=0.011. Median DMFS likewise decreased according to the number of variant alleles inherited: 9.1 years (95% CI, 7.4 to not reached); 6.8 years (95% CI, 4.3 to 7.4); and 3.6 years (95% CI, 1.0 to 7.3), respectively; P=0.014. Finally, OS diminished with the number of variant alleles inherited: 5-year and 10-year OS 82% (95% CI, 69 to 94) and 55% (95% CI, 35 to 75) in homozygous wild-type men; 74% (95% CI, 62 to 85) and 35% (95% CI, 21 to 49) in heterozygotes; and 58% (95% CI, 30 to 86) and 0% in homozygous variant men; P=0.0064. On multivariable analysis, the hazard ratio (HR) for progression was 1.6 for men with at least one variant allele (95% CI, 1.0 to 2.7; P=0.074), which compared favorably with Gleason score (HR 1.3 for Gleason score 8–10 vs. 6–7; 95% CI 0.8 to 2.0; P=0.31), though neither factor reached statistical significance with the small sample size. The impact of homozygous variant genotype on metastasis (HR 2.8; 95% CI, 1.1 to 6.7; P=0.025) and death (HR 3.5; 95% CI 1.3 to 9.5; P=0.013) was maintained on multivariable analysis. Findings in the external cohorts independently validated the impact of HSD3B1(1245C) on outcomes.
Interpretation
Inheritance of the HSD3B1(1245C) allele that enhances dihydrotestosterone synthesis is associated with prostate cancer resistance to ADT. Our findings nominate HSD3B1 as a powerful genetic biomarker capable of distinguishing men who are a priori likely to fare favorably with androgen deprivation therapy from those who harbor disease liable to behave more aggressively, and who therefore may warrant early escalated therapy. Future studies should stratify by HSD3B1 genotype in light of the profound differences in outcomes according to the number of variant alleles present.
Funding
Prostate Cancer Foundation, National Institutes of Health, Department of Defense, Howard Hughes Medical Institute, American Cancer Society, Conquer Cancer Foundation of the American Society of Clinical Oncology, Cleveland Clinic Research Programs Committee and Department of Radiation Oncology, and Gail and Joseph Gassner Development Funds.
Introduction
Nearly all prostate cancers express the androgen receptor (AR), the importance of which is underscored by androgen deprivation therapy (ADT), the most effective and widely used prostate cancer systemic therapy for the past 70 years.1 ADT improves survival in combination with radiotherapy for selected patients.2,3 Likewise, ADT confers a survival advantage when given immediately after prostatectomy in node-positive disease.4 ADT represents the cornerstone of treatment in men with metastatic disease,5 and has shown benefit even in the setting of biochemical failure after local therapy.6 Indeed, the recently published TOAD trial demonstrated improved survival with early versus delayed ADT in non-metastatic men with increasing PSA, the vast majority of whom had biochemical failure after local therapy.7 Although nearly all men will demonstrate a response to ADT, most will eventually develop castration-resistant prostate cancer (CRPC). However, the duration of response to ADT varies widely.8,9
Evolution from castration-sensitive prostate cancer to CRPC hinges on AR reactivation, which can occur by several mechanisms.10,11 Since the underlying processes typically emerge under selection pressure from ADT, they generally cannot be used to determine a priori how patients will respond to ADT. A major advance in the last decade has been increased appreciation of intratumoral androgen synthesis.12,13 Prior to ADT, tumor androgen supply is dominated by gonadal testosterone. With gonadal suppression during ADT, the serum testosterone level is dramatically depleted, inhibiting tumor growth. However, proliferation can continue in the context of intratumoral androgen synthesis, likely in large part from adrenal precursor steroids and possibly in some part due to de novo synthesis from cholesterol. Strong evidence for the importance of this is found in the survival benefit from abiraterone, which depletes intratumoral androgens, and enzalutamide, which competes with intratumoral androgens.14–18 Additionally, transcripts for multiple steroidogenic enzymes, including AKR1C3, HSD3B1 and HSD3B2, are consistently upregulated in CRPC.19 Recently, a mutation in HSD3B1 was shown to furnish a novel mechanism of resistance to ADT.20 HSD3B1 encodes 3β-hydroxysteroid dehydrogenase-1 (3βHSD1), the isoenzyme predominantly expressed in peripheral tissues—including the prostate, skin, breast, placenta, and other tissues—that is responsible for catalyzing the rate-limiting step in the conversion of adrenal androgen precursors to dihydrotestosterone (DHT), and which is required for all pathways of DHT synthesis.21 HSD3B1(1245A>C) changes amino acid 367N→T and renders 3βHSD1 resistant to proteasomal degradation, causing profound accumulation and, effectively, gain-of-function. The resultant increased intratumoral metabolic flux of adrenal precursors to more potent androgens, including DHT (the most potent androgen), thus enhances androgen receptor activation and accelerates tumor proliferation, despite castration. Notably, while the HSD3B1(1245C) allele can be acquired by mutation, it is also heritable in the form of a single nucleotide polymorphism (SNP; rs1047303) with allelic frequency of 15–35% in most cohorts (varies by ethnicity; much lower among Asians and higher among Caucasians).22
Currently, the clinical relevance of HSD3B1(1245C) inheritance in prostate cancer is unknown. One study has linked this variant allele with increased progression to CRPC, though not with other endpoints.23 However, that study was limited by the extremely low prevalence of the HSD3B1(1245C) allele among Chinese men, and thus included zero men homozygous for the variant allele and very few who had even one allele. Additionally, the study was confounded by the fact that nearly twice as many men included in the homozygous wild-type cohort started ADT when they already had metastatic disease. As such, the potential clinical relevance of the variant allele remained undetermined.
The present study was designed to specifically test the mechanism-based hypothesis that men inheriting the variant HSD3B1(1245C) allele would demonstrate evidence of intrinsic resistance to ADT and that this would manifest as shorter time to progression, inferior distant-metastasis free survival, and potentially inferior overall survival. Moreover, based on the number of variant alleles inherited, we specifically hypothesized that men homozygous for the variant allele would have the worst outcomes, men homozygous for the wild-type allele would have the best outcomes, and heterozygotes would potentially have an intermediate course.
Methods
Study design and participants
We used large, prospectively-maintained prostate cancer registries at Cleveland Clinic and Mayo Clinic to correlate HSD3B1 genotype with long-term clinical outcomes. Biological samples and clinical data were obtained with individual written patient consent under informed consent protocols approved by local institutional review boards. Having performed extensive quality assurance and data verification, all authors vouch for the completeness and integrity of the data and statistical analysis. All patients were identified prior to determination of genotype, and tissue processing and genotyping were performed by investigators blinded to clinical data. Finally, data collection and analysis were uncoupled, as the study statistician (CAR) was not involved in collecting clinical data or genotyping.
The primary study cohort included all men who underwent prostatectomy at Cleveland Clinic on or before December 31, 2009 who were treated with ADT for biochemical failure. There was no age limit for eligibility. Biochemical failure was defined as a PSA value of ≥0.2 ng/mL followed by a higher value, or a single PSA value of ≥0.5 ng/mL.24 This definition was specified to reduce the likelihood of including men with low-level detectable PSA produced from residual benign tissue or biologically indolent cancer, since inclusion of such patients would undermine the study’s ability to test the mechanism-based hypothesis. Patients who received postoperative adjuvant or salvage radiotherapy were eligible, provided they had residual active disease as reflected by continued increase in their PSA after treatment.
The post-prostatectomy validation cohort was obtained from the National Cancer Institute-funded Sponsored Programs of Research Excellence (SPORE) database at Mayo Clinic. We identified all men who: (1) received ADT for biochemical and/or non-metastatic clinical failure following prostatectomy, and (2) had blood samples available for research. To ascertain the impact of HSD3B1 genotype among men with more advanced disease, we queried a separate Mayo database and identified men enrolled with metastatic CRPC who had blood samples banked for research. Time-to-event outcomes were measured from the date ADT was originally initiated. To minimize confounding, patients were excluded if they had received radiation therapy for initial local therapy, since use of ADT with RT was not adequately captured in this cohort. Patients with confirmed or probable neuroendocrine prostate cancer were excluded, given that such cancers are generally not dependent on the AR axis. There were no age limits in defining the validation cohorts.
Procedures
Prostatectomy specimens from the primary cohort were reviewed to obtain cores of non-neoplastic tissue for germline genotyping. Peripheral blood mononuclear cells were used for genotyping in the Mayo cohorts. Notably, HSD3B1 has one homolog (HSD3B2) and four non-processed pseudogenes, which have very closely related DNA sequences that may obscure detection of the variant sequence.21 We therefore developed a melting assay using an unlabeled locked nucleic acid oligonucleotide probe in an asymmetric polymerase chain reaction to perform targeted genotyping at this locus. See appendix page 1 and 7 for details.
Outcomes
Time-to-event outcomes, including the primary endpoint of progression-free survival and the secondary endpoints of distant metastasis-free survival and overall survival, were measured from the date ADT was initiated. Progression was an investigator-assessed composite endpoint defined as the first occurrence of: (1) a second increase in PSA on ADT (no absolute threshold); (2) radiographic or clinical progression; or (3) initiation of second-line therapy. Radiographic or clinical progression included development of a local recurrence (on physical exam, computed tomography [CT], or magnetic resonance imaging [MRI]), distant metastasis (bone scan and/or CT/MRI), or death from prostate cancer. Radiographic, clinical, and laboratory (PSA) assessment frequencies were at the discretion of the treating physicians, but were independent of genotype (genotype was unknown at the time of clinical decision-making).
Statistical analysis
Progression-free survival, distant metastasis-free survival, and overall survival were analyzed using Kaplan-Meier methods. The log-rank test for trend with one degree of freedom was used when assessing for potential gene-dosage effects across the three groups. Cox proportional hazards regression was performed to further assess potential allele-dosage effects. Demographic and treatment characteristics were compared across genotypes to assess for confounders using Fisher’s exact test and Kruskal-Wallis analysis of variance. Fisher’s exact test was utilized rather than the Chi-square test because the latter is less accurate when counts are low. Multivariable Cox proportional hazards models were used to account for potential confounders. Covariates included common clinicopathologic variables that are known to correlate with clinical outcomes, including American Joint Commission on Cancer T stage, N stage, PSA at ADT initiation, and Gleason score. We examined T-stage as a categorical variable using all T stages in two separate models. In the first model, stage T2 was the reference group, examining 3a versus 2 and 3b-4 versus 2. In the second model, stage T3a was the reference group, examining 3b-4 versus 3a and 2 versus 3a. Stage T4 was combined with stage T3b because there was only one T4 patient in our study population. Stage T3b-4 is typically more predictive of poor clinical outcomes than T3a, and thus we did not combine all of T3 into a single category. All tests were two-sided, and P-values less than or equal to 0.05 were interpreted as statistically significant. Analyses were performed with the use of SAS software, version 9.3 (SAS Institute, Cary, NC).
Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. JWDH, GA, CA Reichard, CA Reddy, CMG, RC, LR, MK, DT, and NS had access to raw data. The corresponding author had full access to all data and had the final responsibility for the decision to submit for publication.
Results
Determination of HSD3B1 Genotype
Our melting analysis technique was able to reliably distinguish genotypes (appendix page 2), and was validated with Sanger sequencing for 60 samples (20 for each genotype) with 100% concordance.
Patient Characteristics
We identified 177 men in the Cleveland Clinic registry who met study criteria and underwent prostatectomy between January 1, 1996 and December 31, 2009. Of these, 118 (67%) were successfully genotyped and therefore analyzable, reflecting the well-established challenge of genotyping using formalin-fixed paraffin-embedded tissue.25–27 HSD3B1(1245C) allelic frequency in this cohort was 36% (86/236 alleles). Genotype distribution and clinical characteristics are shown in Table 1. There were no statistically significant differences in demographic or treatment characteristics by genotype, although the crude proportions of T3b-4 tumors and Gleason score 8–10 disease were higher among homozygous variant men. Multivariable analyses were used to adjust accordingly.
Table 1.
Demographic and treatment characteristics of the primary cohort by HSD3B1 genotype
| Characteristic | Homozygous wild-type N = 44/118 (37%) |
Heterozygous N = 62/118 (52%) |
Homozygous variant N = 12/118 (10%) |
P-value |
|---|---|---|---|---|
| Follow-up (years) | 0.36 | |||
| Median | 7.1 | 6.7 | 6.2 | |
| Interquartile range | 4.0–9.0 | 4.1–9.0 | 3.6–6.8 | |
|
| ||||
| Age (years) | 0.36 | |||
| Median | 65 | 65 | 63 | |
| Interquartile range | 60–70 | 55–70 | 57–67 | |
|
| ||||
| Race – no. (%) | 0.16 | |||
| White | 34 (77) | 56 (90) | 11 (92) | |
| Black | 8 (18) | 4 (7) | 0 | |
| Hispanic | 0 | 1 (2) | 0 | |
| Asian | 1 (2) | 0 | 0 | |
| Other | 1 (2) | 1 (2) | 1 (8) | |
|
| ||||
| Pathologic T stage – no. (%) | 0.056 | |||
| T2 | 14 (32) | 9 (15) | 0 | |
| T3a | 13 (30) | 17 (27) | 4 (33) | |
| T3b-4 | 17 (39) | 36 (58) | 8 (67) | |
|
| ||||
| Pathologic N stage – no. (%) | 0.97 | |||
| N0 | 30 (68) | 39 (63) | 9 (75) | |
| N1 | 10 (23) | 16 (26) | 2 (17) | |
| NX | 4 (9) | 7 (11) | 1 (8) | |
|
| ||||
| PSA at ADT initiation (ng/mL) | 0.33 | |||
| Median | 3.3 | 1.5 | 1.9 | |
| Mean (standard deviation) | 8.4 (18.8) | 5.5 (11.5) | 5.5 (6.9) | |
| Interquartile range | 0.8–8.0 | 0.6–5.4 | 1.1–7.3 | |
|
| ||||
| Gleason score – no. (%) | 0.42 | |||
| 6 | 2 (5) | 0 | 0 | |
| 7 | 22 (50) | 35 (57) | 5 (42) | |
| 8–10 | 20 (46) | 27 (44) | 7 (58) | |
|
| ||||
| ADT type – no. (%) | 0.69 | |||
| GnRH agonist or orchiectomy | 41 (93) | 56 (90) | 12 (100) | |
| Androgen receptor antagonist | 3 (7) | 6 (10) | 0 | |
|
| ||||
| ADT use – no. (%) | 0.23 | |||
| Continuous | 25 (57) | 27 (44) | 8 (67) | |
| Intermittent | 19 (43) | 35 (57) | 4 (33) | |
|
| ||||
| Adjuvant/Salvage RT – no. (%) | 0.66 | |||
| No | 31 (71) | 38 (61) | 8 (67) | |
| Yes | 13 (30) | 24 (39) | 4 (33) | |
|
| ||||
| Neoadjuvant treatment – no. (%) | 0.97 | |||
| None | 31 (71) | 45 (73) | 9 (75) | |
| ADT | 10 (23) | 14 (23) | 2 (17) | |
| Chemotherapy or immunotherapy | 3 (7) | 3 (5) | 1 (8) | |
PSA denotes prostate-specific antigen level. Pathologic stages refer to the American Joint Commission on Cancer tumor–node– metastasis (TNM) staging system.28 ADT denotes androgen deprivation therapy. GnRH agonist denotes gonadotropin-releasing hormone agonist. RT denotes radiation therapy. P-values represent comparisons across the three genotypes.
Of 140 patients in the Mayo Clinic SPORE registry who met study criteria and underwent prostatectomy between January 1, 1987 and December 31, 2011, 98% were successfully genotyped, which yielded 137 analyzable patients. HSD3B1(1245C) allelic frequency was 26% (70/274 alleles), and the genotype distribution was 56% (77/137) homozygous wild-type, 36% (50/137) heterozygous, and 7% (10/137) homozygous variant. Of 204 patients enrolled in the Mayo Clinic metastatic prostate cancer registry between September 1, 2009 and July 31, 2013 who met study criteria, 92% were successfully genotyped, which yielded 188 analyzable patients. These men had initially been diagnosed between January 1, 1983 and July 31, 2012. HSD3B1(1245C) allelic frequency was 27% (101/376 alleles), and the genotype distribution was 52% (98/188) homozygous wild-type, 42% (79/188) heterozygous, and 6% (11/188) homozygous variant. Pooling all three cohorts, the HSD3B1(1245C) allelic frequency was 29% (257/886 alleles). The pooled genotype distribution was 49% (219/443) homozygous wild-type, 43% (191/443) heterozygous, and 7% (33/443) homozygous variant.
Demographic and treatment characteristics of the validation cohorts are listed in the appendix (pages 8 and 9). In both cohorts there were no significant differences according to genotype, with the exception of N-stage in the Mayo SPORE cohort. The distribution of N1 disease in this cohort was 5/10 (50%) in the homozygous variant group, as compared to 13% (10/77) and 16% (8/50) in the homozygous wild-type and heterozygous groups, respectively (P=0.025). This discrepancy was adjusted for during multivariable analysis. Median follow-up for the SPORE cohort (4.9 years; interquartile range [IQR] 2.6–7.6) was shorter than for the primary cohort (6.7 years; IQR 3.9–8.9), and was only 3.2 years (IQR 1.8–4.6) in the homozygous variant group. In view of the limited number of metastatic and death events during the short observation period (appendix page 3), the SPORE cohort was used to validate the impact of HSD3B1 genotype on progression-free survival only. In contrast, the median follow up from ADT initiation for the Mayo metastatic cohort was 6.0 years (IQR 3.1–9.2), which was sufficient to enable analysis of both progression-free survival and overall survival. Distant metastasis-free survival was not calculated in this cohort, since these were metastatic patients. Chemotherapy was commonly used for CRPC, and its usage was similar across genotypes: 57% (56/98) in homozygous wild-type men, 61% (48/79) in heterozygous men, and 55% (6/11) in homozygous variant men (P=0.83). Docetaxel was used in 96% of cases (106/110).
Progression-Free Survival
Progression-free survival (PFS) was significantly associated with HSD3B1 genotype in the primary study cohort (Figure 1A), and diminished as a function of the number of HSD3B1(1245C) alleles inherited: median 6.6 years in homozygous wild-type men (95% CI, 3.8 to not reached); 4.1 years in heterozygotes (95% CI, 3.0 to 5.5); and 2.5 years in homozygous variant men (95% CI, 0.7 to not reached); P=0.011. Relative to the homozygous wild-type genotype, inheritance of two copies of the variant allele was predictive of worse PFS (hazard ratio (HR) 2.4; 95% CI, 1.1 to 5.3; P=0.029). Inheritance of one copy of the variant allele was also predictive of worse PFS (HR 1.7; 95% CI, 1.0 to 2.9; P=0.041). On multivariable analysis (appendix page 10), the hazard ratio for progression was 1.6 for men with at least one variant allele (95% CI, 1.0 to 2.7; P=0.074), which compared favorably with Gleason score (HR 1.3 for Gleason score 8–10 vs. 6–7; 95% CI 0.8 to 2.0; P=0.31), though neither factor reached statistical significance with the small sample size. The distribution of each type of progression event is described in the appendix (page 11). In the Mayo SPORE post-prostatectomy cohort PFS was associated with HSD3B1 genotype (Figure 2A). Median PFS was 3.3 years in homozygous wild-type men (95% CI, 1.9 to 4.9); 2.8 years in heterozygotes (95% CI, 2.1 to 5.1); and 0.9 years in homozygous variant men (95% CI, 0.2 to 2.6; P=0.0022). Homozygous variant men had a 3.4-times risk of progression relative to homozygous wild-type men (95% CI, 1.6 to 7.0; P=0.0013), whereas heterozygous men did not differ significantly from the wild-type group (HR 1.0; 95% CI, 0.7 to 1.7; P=0.85). The impact of homozygous variant genotype on PFS persisted upon adjustment for lymph node status (HR 2.7; 95% CI 1.2 to 5.9; P=0.013), while heterozygotes did not differ significantly from wild-type men (HR 1.0; 95% CI, 0.6 to 1.6; P=0.98). Finally, median PFS in the Mayo metastatic cohort also correlated with HSD3B1 genotype (Figure 2B). As in the other two cohorts, the largest difference was seen between homozygous wild-type men (1.8 years; 95% CI, 1.2 to 2.5) and homozygous variant men (0.8 years; 95% CI, 0.3 to 1.6; P=0.024). The corresponding hazard ratio for progression was 2.0 (95% CI 1.1 to 3.8; P=0.027). Heterozygous men had an intermediate PFS (1.4 years; 95% CI, 1.0 to 1.8) and hazard ratio for progression of 1.1 (95% CI 0.8 to 1.5; P=0.38).
Figure 1. Progression-free survival, distant metastasis-free survival, and overall survival according to HSD3B1 genotype in the primary study cohort.
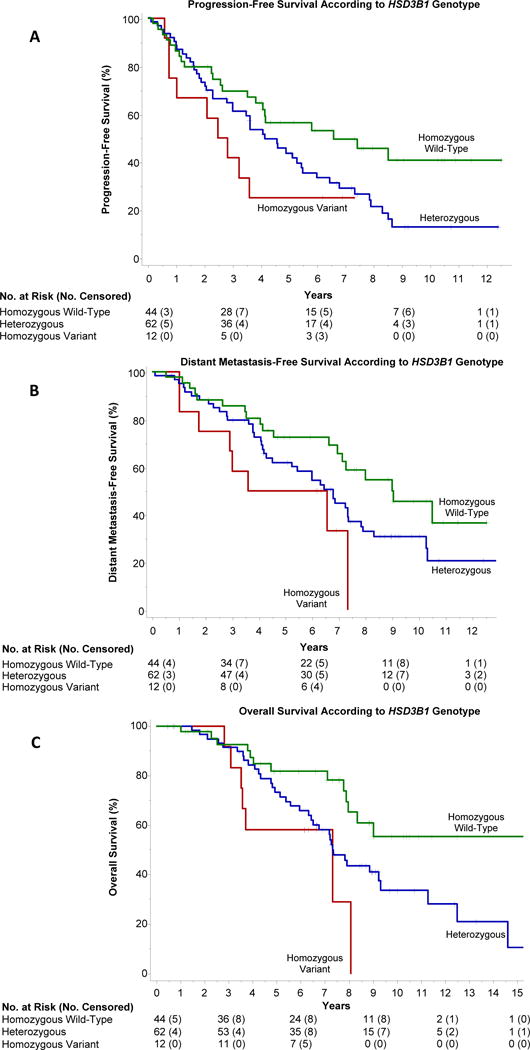
Progression-free survival (PFS), distant metastasis-free survival (DMFS), and overall survival (OS) diminished as a function of the number of variant alleles inherited. Median PFS from initiation of ADT (Panel A) was 6.6 years in homozygous wild-type men (95% CI, 3.8 to not reached); 4.1 years in heterozygotes (95% CI, 3.0 to 5.5); and 2.5 years in homozygous variant men (95% CI, 0.7 to not reached); P=0.011. Relative to the homozygous wild-type genotype, inheritance of two copies of the variant allele was predictive of worse PFS (HR 2.4; 95% CI, 1.1 to 5.3; P=0.029). Inheritance of one copy of the variant allele was also predictive of worse PFS (HR 1.7; 95% CI, 1.0 to 2.9; P=0.041. Median DMFS from initiation of ADT (Panel B) was 9.1 years in homozygous wild-type men (95% CI, 7.4 to not reached); 6.8 years in heterozygotes (95% CI, 4.3 to 7.4); and 3.6 years in homozygous variant men (95% CI, 1.0 to 7.3); P=0.014. Compared to the homozygous wild-type genotype, inheritance of two copies of the variant allele was predictive of worse DMFS (HR 2.7; 95% CI, 1.2 to 6.2; P=0.022). The corresponding hazard ratio for heterozygotes was 1.7 (95% CI, 1.0 to 2.8; P=0.074). Overall survival from initiation of ADT (Panel C) at 5 years and 10 years, respectively, was 82% (95% CI, 69 to 94) and 55% (95% CI, 35 to 75) in homozygous wild-type men; 74% (95% CI, 62 to 85) and 35% (95% CI, 21 to 49) in heterozygotes; and 58% (95% CI, 30 to 86) and 0% in homozygous variant men; P=0.0064. Relative to the homozygous wild-type genotype, inheritance of two copies of the variant allele was predictive of worse OS (HR 3.3; 95% CI, 1.3 to 8.3; P=0.013), as was one copy of the variant allele (HR 2.0; 95% CI, 1.1 to 3.7; P=0.036).
Figure 2. Progression-free survival in post-prostatectomy (A) and metastatic (B) validation cohorts according to HSD3B1 genotype.
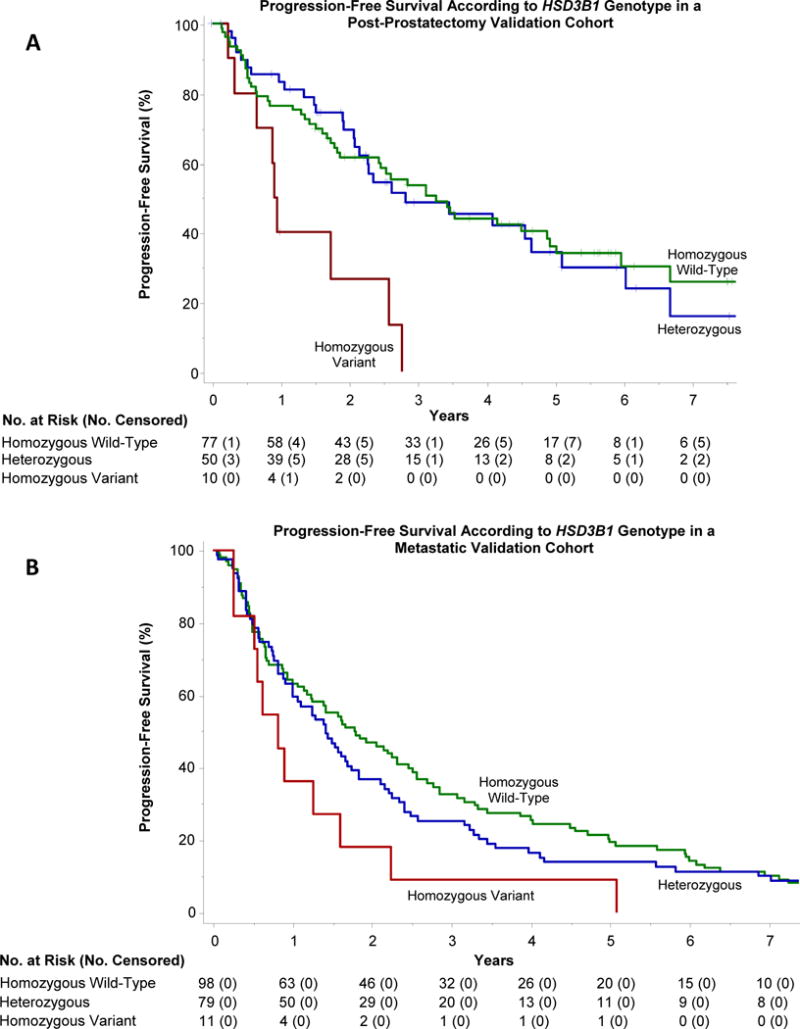
Progression-free survival (PFS) diminished according to HSD3B1 genotype in both validation cohorts. In the post-prostatectomy cohort (Panel A), median PFS was 3.3 years in homozygous wild-type men (95% CI, 1.9 to 4.9); 2.8 years in heterozygotes (95% CI, 2.1 to 5.1); and 0.9 years in homozygous variant men (95% CI, 0.2 to 2.6; P=0.0022). Homozygous variant men had a 3.4-times risk of progression relative to homozygous wild-type men (95% CI, 1.6 to 7.0; P=0.0013), while the corresponding hazard ratio for heterozygous men was 1.0 (95% CI, 0.7 to 1.7; P=0.85). In the metastatic cohort (Panel B), median PFS was likewise longest in homozygous wild-type men (1.8 years; 95% CI, 1.2 to 2.5) and shortest in homozygous variant men (0.8 years; 95% CI, 0.3 to 1.6; P=0.024). The corresponding hazard ratio for progression was 2.0 (95% CI 1.1 to 3.8; P=0.027). Heterozygous men had an intermediate PFS of 1.4 years (95% CI, 1.0 to 1.8) and hazard ratio for progression of 1.1 (95% CI 0.8 to 1.5; P=0.38).
Distant Metastasis-Free Survival
Distant metastasis-free survival (DMFS, i.e., the probability of surviving without distant metastases), which was assessable only in the primary study cohort due to length of follow up and the absence of metastatic disease at ADT initiation, was significantly associated with HSD3B1 genotype. DMFS decreased according to the number of variant alleles inherited: median 9.1 years in homozygous wild-type men (95% CI, 7.4 to not reached); 6.8 years in heterozygotes (95% CI, 4.3 to 7.4); and 3.6 years in homozygous variant men (95% CI, 1.0 to 7.3; P=0.014) (Figure 1B). Compared to the homozygous wild-type genotype, inheritance of two copies of the variant allele was predictive of worse DMFS (HR 2.7; 95% CI, 1.2 to 6.2; P=0.022). The corresponding hazard ratio for heterozygous men was 1.7 (95% CI, 1.0 to 2.8; P=0.074). The impact of homozygous variant genotype on DMFS was confirmed on multivariable analysis (HR 2.8; 95% CI, 1.1 to 6.7; P=0.025). For heterozygous men the corresponding hazard ratio was 1.8 (95% CI, 1.0 to 3.3; P=0.050).
Overall Survival
Overall survival (OS) was significantly associated with HSD3B1 genotype in both cohorts with sufficiently mature follow up to enable its analysis. Figure 1C demonstrates a stepwise reduction in OS as a function of the number of variant alleles inherited in the primary cohort (P=0.0064). The 5-year and 10-year point estimates of OS, respectively, were 82% (95% CI, 69 to 94) and 55% (95% CI, 35 to 75) in homozygous wild-type men; 74% (95% CI, 62 to 85) and 35% (95% CI, 21 to 49) in heterozygotes; and 58% (95% CI, 30 to 86) and 0% in homozygous variant men. Relative to the homozygous wild-type genotype, inheritance of two copies of the variant allele was predictive of worse OS (HR 3.3; 95% CI, 1.3 to 8.3; P=0.013). Inheritance of one copy of the variant allele was also predictive of worse OS (HR 2.0; 95% CI, 1.1 to 3.7; P=0.036). Multivariable analysis confirmed the impact of HSD3B1 genotype on survival, with a hazard ratio of 3.5 in homozygous variant men (95% CI 1.3 to 9.5; P=0.013), and a hazard ratio of 2.0 in heterozygotes (95% CI, 1.0 to 3.9; P=0.054). Similarly, in the Mayo metastatic cohort median OS from the time of ADT initiation was reduced according to the number of variant alleles present: median 9.7 years (95% CI 6.7 to 12.1) in homozygous wild-type men; 6.8 years (95% CI 5.2 to 8.0) in heterozygotes; and 4.6 years (95% CI 1.6 to 7.5) in homozygous variant men; P=0.0042 (Figure 3). Compared with homozygous wild-type men, the hazard ratio for death was 2.5 (95% CI 1.2 to 5.0; P=0.013) in homozygous variant men and 1.5 (95% CI 1.0 to 2.1; P=0.036) among heterozygotes. Outcomes for each cohort with all genotypes combined are included in the appendix (pages 4–6). Prostate cancer-specific survival for the primary cohort (which had cause of death data) is also depicted in the appendix (page 6). As with overall survival, prostate cancer-specific survival diminished according to HSD3B1 genotype (P=0.029).
Figure 3. Overall survival in a metastatic validation cohort according to HSD3B1 genotype.
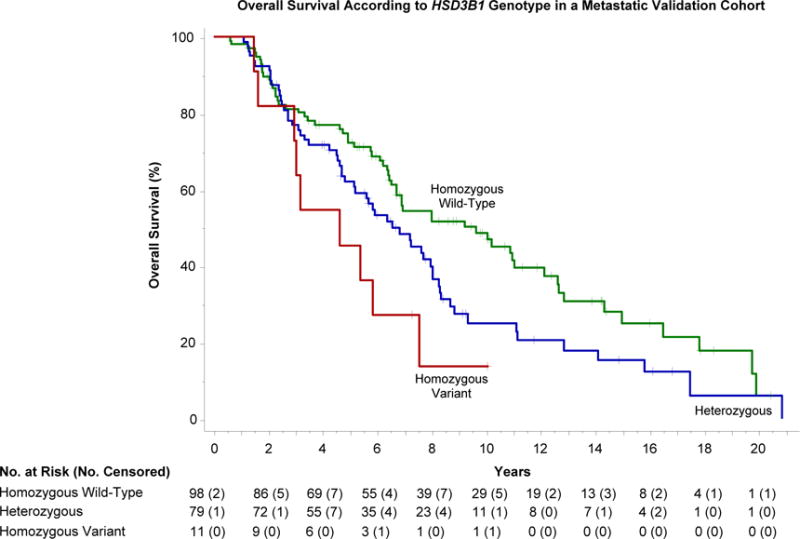
Overall survival diminished according to the number of variant HSD3B1 alleles inherited: median, 9.7 years (95% CI 6.7 to 12.1) in homozygous wild-type men; 6.8 years (95% CI 5.2 to 8.0) in heterozygotes; and 4.6 years (95% CI 1.6 to 7.5) in homozygous variant men; P=0.0042. Compared with homozygous wild-type men, the hazard ratio for death was 2.5 (95% CI 1.2 to 5.0; P=0.013) in homozygous variant men and 1.5 (95% CI 1.0 to 2.1; P=0.036) among heterozygotes.
Discussion
In our study, designed specifically to assess the possibility that HSD3B1(1245C) might be predictive of resistance to ADT, we found that inheritance of this allele was associated with decreased progression-free survival, distant metastasis-free survival, and overall survival, with differences measured in years. These results are biologically credible, since tumor cells carrying the HSD3B1(1245C) allele are able to more efficiently produce their own DHT. The observed stepwise decrement in all three endpoints would be exceedingly unlikely to occur by chance, and strongly suggests allele dose-dependence. Finally, validation of our initial findings in two independent cohorts confirms the impact of HSD3B1(1245C) inheritance. Genotype may also be prognostic regardless of ADT, although the mechanistic underpinnings suggest the associated growth advantage would be most pronounced with ADT.
As an extension of our preclinical work identifying a link between HSD3B1(1245C) and increased 3βHSD1 enzyme levels, we previously evaluated tumors in men with CRPC.20 In homozygous variant tumors we measured a robust increase in 3βHSD1 enzyme levels, whereas in heterozygous variant tumors we did not see any clear increase in enzyme level. However, expression level was measured in only two such tumors in the latter group, and thus no definitive conclusion regarding the significance of heterozygous inheritance could be drawn. In contrast, in the current study we analyze 191 heterozygotes, with the majority of findings suggesting heterozygous inheritance is biologically relevant. Figure 2A is the only set of curves among the six Kaplan-Meier plots in Figures 1–3 that does not suggest a difference between heterozygotes and homozygous wild-type men, and this is the cohort with the shortest follow-up. Apart from the question of whether there is an intermediate increase in enzyme level associated with a single variant allele, our prior data show that loss of heterozygosity (LOH) of the wild-type allele occurs under selective pressure from ADT. Three of eleven (27%) patients with germline heterozygous inheritance had developed CRPC tumors with LOH of the wild-type allele, whereas none had lost the variant allele. It is probable that LOH of the wild-type allele in tumors could contribute to the difference in outcomes between heterozygous men and homozygous wild-type men. On a more fundamental level, the discordance between germline inheritance and tumor DNA reflects somatic alterations, which are known to increase as tumors evolve, particularly under selective pressure from ADT. Our prior work revealed that even men who are homozygous wild-type can eventually acquire the variant allele (3/25 men evaluated [12%]). Tumor DNA from CRPC is challenging to obtain in large numbers of patients and is unavailable from these cohorts. Nonetheless, we would anticipate that future studies taking somatic alterations in HSD3B1 into account by analyzing tumor DNA would demonstrate an even stronger association between genotype and clinical outcomes.
Since Huggins and Hodges published their seminal work on the therapeutic effects of castration in 1941,1 ADT has been the cornerstone of systemic therapy for prostate cancer. To date, it has been challenging to predict innate resistance to ADT. A previous study evaluated 109 SNPs in several steroidogenic genes, including HSD3B1, and found no clear association with biochemical recurrence among men with resected prostate cancer.29 However, it is important to note that the majority of patients in the primary cohort analyzed in that study were not treated with ADT. This is a crucial difference relative to the current study, since the growth advantage of the variant allele would be most pronounced under conditions of castration, as noted above. Furthermore, as also mentioned earlier, HSD3B1 has one homolog (HSD3B2) and four non-processed pseudogenes, which have very closely related DNA sequences that can obscure detection of the variant sequence. The accuracy of high-throughput genotyping techniques, including the methods used in multi-SNP analyses, can be adversely impacted by such similar sequences and is highly dependent on optimal primer design.30,31 Such concerns provided the motivation for our development and validation of the specific high-resolution melting analysis technique we used herein, inasmuch as accurate genotyping was crucial to undertaking the present study.
Another prior study associated a noncoding SNP 13 kb upstream of HSD3B1 (rs1856888) with development of CRPC.32 This SNP might influence the expression of HSD3B1 or have an independent regulatory role. Alternatively, the correlation between this SNP and CRPC may be due to its proximity to HSD3B1 and the biochemical activity the latter confers. Several other studies have shown an association between germline variants in transmembrane steroid transporters and the development of CRPC.33–35 Therefore, other germline variants may confer additional information in combination with HSD3B1(1245C).
The ramifications of a biomarker able to predict ADT resistance are far-reaching. Our findings suggest HSD3B1 genotype could a priori distinguish men with disease likely to respond favorably to ADT from those who harbor disease prone to behave more aggressively, and who therefore may merit escalated therapy. Combined androgen blockade (CAB) with the upfront addition of an AR antagonist to castration has long been debated, but analyses of multiple trials suggest that the clinical benefit of CAB in unselected patients is small at best.36 Although speculative, it is possible that there might be differential benefit from CAB, with little incremental gain for homozygous wild-type HSD3B1(1245A) men, but meaningful utility in men possessing the variant allele. HSD3B1(1245C) genotype could also guide future studies with regard to selective early incorporation of highly-potent inhibitors of the AR axis, such as enzalutamide or abiraterone acetate. It is mechanistically plausible that men who possess the HSD3B1(1245C) allele, especially two copies, could considerably benefit if one of these agents were started with ADT rather than waiting until development of CRPC. With respect to the possible use of such agents for men who inherit the HSD3B1(1245C) allele, it is notable that abiraterone is clinically converted by 3βHSD to the more potent anti-androgen metabolite, D4A37. D4A is further converted to other metabolites in patients, including 3-keto-5α-abiraterone, which stimulates AR.38 Nonetheless, the ultimate clinical effect of these steroidal metabolites of abiraterone remains to be determined.
Similar reasoning would suggest HSD3B1 genotype could be informative in decisions regarding chemohormonal therapy. Two landmark trials, Eastern Cooperative Oncology Group E3805 (CHAARTED) and STAMPEDE, have demonstrated that chemohormonal therapy prior to development of CRPC substantially improves survival relative to ADT alone.39,40 It is probable that cytotoxic therapy would be most beneficial in patients least likely to have a durable response to ADT, whereas men who are apt to have a sustained response to ADT may benefit less. HSD3B1 genotype could therefore help guide management, particularly for men whose ability to tolerate chemotherapy is marginal.
There are multiple other domains in which HSD3B1 genotype might be influential, one of which is the combination of ADT with radiotherapy. There are also potential implications for refining identification of men for active surveillance. Active surveillance might be riskier in men with homozygous variant HSD3B1(1245C) inheritance, since they are not likely to respond durably to ADT, which would be their recourse if the window for curative local therapy were missed. In contrast, homozygous wild-type inheritance may reinforce a recommendation for active surveillance. Similarly, HSD3B1 genotype could be informative when considering salvage radiation therapy. Finally, our results provide insight concerning the potential impact of pharmacologic inhibition of 3βHSD1. Due to the relatively low prevalence of the homozygous variant genotype, HSD3B1(1245C) as a biomarker could conceivably be used in the clinic as a binary factor: no variant alleles versus one-or-more variant alleles. We felt it was informative to analyze the three groups separately in this report, given that homozygous variant patients appear to fare particularly poorly, although we acknowledge that the clinical implications may be similar for one vs. two variant alleles in some clinical situations.
With respect to limitations, each of our individual cohorts has a relatively modest sample size, although the similarity of results in the three cohorts and the evidence of allele dose-dependence strongly support the influence of HSD3B1(1245C) inheritance. Nonetheless, future work prospectively evaluating the role of HSD3B1 will be a high priority, given the retrospective design of our study. Correlation of serum and tissue steroid profiles with genotype will likewise be informative. There is some variation in the prevalence of each genotype across the three cohorts we studied (homozygous wild-type 37–56%, heterozygous 36–52%, and homozygous variant 6–10%). Aside from random variation, the allelic distribution may be somewhat different in the populations sampled, even among the Caucasian subsets. Future studies will provide additional information to refine prevalence estimates. Further analysis of non-Caucasian patients would be of value as well, given the underrepresentation of such men in our cohorts, though this may be limited by the lower allelic frequency in non-Caucasian populations. In the present study we evaluated two biochemical-failure cohorts and found HSD3B1 genotype to be associated with outcomes in this setting. We also explored whether men on the opposite end of the disease spectrum (i.e. the cohort of men who ultimately developed metastatic CRPC) had different outcomes according to HSD3B1 status, and our data demonstrate that indeed they did. However, the more general question of whether the same is true of an unselected population of men with metastatic prostate cancer warrants further investigation.
One other potential limitation of our study is that there was not a uniform follow-up schedule. Systematic differences in assessment intervals can lead to spurious results (as for example when comparing progression-free survival across trials with different assessment schedules).41,42 However, in the current study, assessment frequency was at the discretion of the treating physicians for all patients without regard to genotype (genotype was unknown at the time of clinical decision-making). Thus, while the lack of a uniform interval for assessments should be considered when comparing our cohorts with one another or with additional cohorts, it would not reduce the validity of comparing men by genotype within each of our study cohorts, which was our objective.
In summary, our findings in three independent cohorts nominate germline HSD3B1 genotype as a genetic biomarker of resistance to ADT for prostate cancer, and are concordant with what would be predicted from the underlying molecular mechanism.
Supplementary Material
Research in context.
Evidence before this study
We queried PubMed for biomarkers of resistance to androgen deprivation therapy (ADT) in prostate cancer. We search human studies published between January 1, 1980 and February 1, 2016 with the search terms “androgen deprivation therapy” AND (“resistance” or “efficacy”) AND “prostate cancer” AND “biomarker.” We identified 144 studies, common limitations of which included dependence on information not available at ADT initiation, emphasis on associations between polymorphisms with no known mechanistic correlate, lack of association with endpoints other than time to progression, and absence of validation.
Added value of this study
We tested the pre-specified, mechanism-based hypothesis that men inheriting the HSD3B1(1245C) allele would exhibit innate resistance to ADT, given that this allele has recently been established to increase intratumoral conversion of androgen precursor steroids to more potent androgens that can drive disease progression, despite castration.
We demonstrated that HSD3B1 genotype has a profound impact on long-term clinical endpoints, including progression-free survival, distant metastasis-free survival, and overall survival. Moreover, there is a stepwise difference in outcomes according to the number of HSD3B1(1245C) alleles inherited, with differences measured in years. Finally, we have validated the influence of HSD3B1 genotype by analyzing three independent cohorts, representing both the post-prostatectomy and metastatic disease contexts.
Implications of all the available evidence
Our data demonstrate that HSD3B1 genotype is a powerful genetic biomarker of resistance to ADT, and can identify men who may benefit a priori from escalated therapy. This may have ramifications for selective early incorporation of highly-potent inhibitors of the androgen receptor axis, and could potentially inform selection of patients for chemohormonal therapy. Future studies should stratify by HSD3B1 genotype in light of the profound differences in outcomes according to the number of variant alleles present.
Acknowledgments
This work was supported by a Cleveland Clinic Research Programs Committee Grant with matching funds from the Cleveland Clinic Department of Radiation Oncology (to J.H.), a grant from the U.S Department of Defense Congressionally Directed Medical Research Programs (to J.H.), a Merit Award from the Conquer Cancer Foundation of the American Society of Clinical Oncology (to J.H.), funding from the Prostate Specialized Program of Research Excellence (P50-CA091956; to D.T.), the Gail and Joseph Gassner Development Funds (to M.K.), a Howard Hughes Medical Institute Physician-Scientist Early Career Award (to N.S.), a grant from the Prostate Cancer Foundation (to N.S.), an American Cancer Society Research Scholar Award (to N.S.), a grant from the U.S. Army Medical Research and Materiel Command (W81XWH-09-1-0301; to N.S.), and additional grants from the National Cancer Institute (R01CA172382, R01CA190289 and R01CA168899; to N.S.).
We thank Drs. John Suh and Rahul Tendulkar for providing key organizational support; Ms. Patricia White for assistance with the Cleveland Clinic prostate cancer registry; Ms. Paula Carver and Ms. Karen Streator Smith for assistance with the tissue samples; and all of the patients and families who made this study possible.
Footnotes
Contributions
JWDH, GA, K-HC, and NS conceived and designed the study and developed the methodology. JWDH, GA, CA Reichard, CM-G, RC, LR, MK, DT, EAK, and NS acquired data. JWDH, CA Reddy, and NS analyzed and interpreted the data. KR, BJD, RJK, MK, DT, EAK, and NS provided administrative, technical, or material support. JWDH, CA Reddy, MK, DT, EAK and NS provided study supervision. All authors wrote and provided final approval of the manuscript.
Declaration of Interests
A patent for 3β-hydroxysteroid dehydrogenase in steroid-dependent disease has been filed by Cleveland Clinic. All grant support and other funding is listed in the Acknowledgments section.
References
- 1.Huggins C, Hodges CV. Studies on Prostatic Cancer. I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Research. 1941 Apr 1;1(4):293–7. doi: 10.3322/canjclin.22.4.232. 1941. [DOI] [PubMed] [Google Scholar]
- 2.Bolla M, Gonzalez D, Warde P, et al. Improved survival in patients with locally advanced prostate cancer treated with radiotherapy and goserelin. N Engl J Med. 1997 Jul 31;337(5):295–300. doi: 10.1056/NEJM199707313370502. Epub 1997/07/31. eng. [DOI] [PubMed] [Google Scholar]
- 3.D’Amico AV, Chen MH, Renshaw AA, Loffredo M, Kantoff PW. Androgen suppression and radiation vs radiation alone for prostate cancer: a randomized trial. JAMA. 2008 Jan 23;299(3):289–95. doi: 10.1001/jama.299.3.289. Epub 2008/01/24. eng. [DOI] [PubMed] [Google Scholar]
- 4.Messing EM, Manola J, Sarosdy M, Wilding G, Crawford ED, Trump D. Immediate hormonal therapy compared with observation after radical prostatectomy and pelvic lymphadenectomy in men with node-positive prostate cancer. N Engl J Med. 1999 Dec 9;341(24):1781–8. doi: 10.1056/NEJM199912093412401. Epub 1999/12/10. eng. [DOI] [PubMed] [Google Scholar]
- 5.Hussain M, Tangen CM, Berry DL, et al. Intermittent versus continuous androgen deprivation in prostate cancer. N Engl J Med. 2013 Apr 4;368(14):1314–25. doi: 10.1056/NEJMoa1212299. Epub 2013/04/05. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moul JW, Wu H, Sun L, et al. Early versus delayed hormonal therapy for prostate specific antigen only recurrence of prostate cancer after radical prostatectomy. J Urol. 2004 Mar;171(3):1141–7. doi: 10.1097/01.ju.0000113794.34810.d0. Epub 2004/02/10. eng. [DOI] [PubMed] [Google Scholar]
- 7.Duchesne GM, Woo HH, Bassett JK, et al. Timing of androgen-deprivation therapy in patients with prostate cancer with a rising PSA (TROG 03.06 and VCOG PR 01-03 [TOAD]): a randomised, multicentre, non-blinded, phase 3 trial. Lancet Oncol. 2016 May 4; doi: 10.1016/S1470-2045(16)00107-8. Epub 2016/05/09. Eng. [DOI] [PubMed] [Google Scholar]
- 8.Crawford ED, Eisenberger MA, McLeod DG, et al. A controlled trial of leuprolide with and without flutamide in prostatic carcinoma. N Engl J Med. 1989 Aug 17;321(7):419–24. doi: 10.1056/NEJM198908173210702. Epub 1989/08/17. eng. [DOI] [PubMed] [Google Scholar]
- 9.Eisenberger MA, Blumenstein BA, Crawford ED, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998 Oct 8;339(15):1036–42. doi: 10.1056/NEJM199810083391504. Epub 1998/10/08. eng. [DOI] [PubMed] [Google Scholar]
- 10.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005 Nov 10;23(32):8253–61. doi: 10.1200/JCO.2005.03.4777. Epub 2005/11/10. eng. [DOI] [PubMed] [Google Scholar]
- 11.Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014 Sep 11;371(11):1028–38. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohler JL, Gregory CW, Ford OH, 3rd, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004 Jan 15;10(2):440–8. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 13.Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008 Jun 1;68(11):4447–54. doi: 10.1158/0008-5472.CAN-08-0249. Epub 2008/06/04. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011 May 26;364(21):1995–2005. doi: 10.1056/NEJMoa1014618. Epub 2011/05/27. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013 Jan 10;368(2):138–48. doi: 10.1056/NEJMoa1209096. Epub 2012/12/12. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012 Sep 27;367(13):1187–97. doi: 10.1056/NEJMoa1207506. Epub 2012/08/17. eng. [DOI] [PubMed] [Google Scholar]
- 17.Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014 Jul 31;371(5):424–33. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryan CJ, Smith MR, Fizazi K, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015 Feb;16(2):152–60. doi: 10.1016/S1470-2045(14)71205-7. [DOI] [PubMed] [Google Scholar]
- 19.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006 Mar 1;66(5):2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 20.Chang KH, Li R, Kuri B, et al. A Gain-of-Function Mutation in DHT Synthesis in Castration-Resistant Prostate Cancer. Cell. 2013 Aug 29;154(5):1074–84. doi: 10.1016/j.cell.2013.07.029. Epub 2013/09/03. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, Melner MH. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocr Rev. 2005 Jun;26(4):525–82. doi: 10.1210/er.2002-0050. Epub 2005/01/06. eng. [DOI] [PubMed] [Google Scholar]
- 22.NCBI dbSNP database [Last accessed January 9, 2016]. Available from: http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=1047303.
- 23.Wu G, Huang S, Nastiuk KL, et al. Variant allele of HSD3B1 increases progression to castration-resistant prostate cancer. Prostate. 2015 May;75(7):777–82. doi: 10.1002/pros.22967. Epub 2015/03/04. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephenson AJ, Kattan MW, Eastham JA, et al. Defining biochemical recurrence of prostate cancer after radical prostatectomy: a proposal for a standardized definition. J Clin Oncol. 2006 Aug 20;24(24):3973–8. doi: 10.1200/JCO.2005.04.0756. Epub 2006/08/22. eng. [DOI] [PubMed] [Google Scholar]
- 25.Baak-Pablo R, Dezentje V, Guchelaar HJ, van der Straaten T. Genotyping of DNA samples isolated from formalin-fixed paraffin-embedded tissues using preamplification. J Mol Diagn. 2010 Nov;12(6):746–9. doi: 10.2353/jmoldx.2010.100047. Epub 2010/09/18. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann U, Kreipe H. Real-time PCR analysis of DNA and RNA extracted from formalin-fixed and paraffin-embedded biopsies. Methods. 2001 Dec;25(4):409–18. doi: 10.1006/meth.2001.1263. Epub 2002/02/16. eng. [DOI] [PubMed] [Google Scholar]
- 27.Okello JB, Zurek J, Devault AM, et al. Comparison of methods in the recovery of nucleic acids from archival formalin-fixed paraffin-embedded autopsy tissues. Anal Biochem. 2010 May 1;400(1):110–7. doi: 10.1016/j.ab.2010.01.014. Epub 2010/01/19. eng. [DOI] [PubMed] [Google Scholar]
- 28.Greene FL, American Joint Committee on Cancer . AJCC cancer staging manual. 6th. New York: Springer-Verlag; 2002. p. xiv, 421. [Google Scholar]
- 29.Levesque E, Huang SP, Audet-Walsh E, et al. Molecular markers in key steroidogenic pathways, circulating steroid levels, and prostate cancer progression. Clin Cancer Res. 2013 Feb 1;19(3):699–709. doi: 10.1158/1078-0432.CCR-12-2812. Epub 2012/11/29. eng. [DOI] [PubMed] [Google Scholar]
- 30.van den Boom D, Wjst M, Everts RE. MALDI-TOF mass spectrometry. Methods Mol Biol. 2013;1015:71–85. doi: 10.1007/978-1-62703-435-7_4. Epub 2013/07/05. eng. [DOI] [PubMed] [Google Scholar]
- 31.Syrmis MW, Moser RJ, Whiley DM, et al. Comparison of a multiplexed MassARRAY system with real-time allele-specific PCR technology for genotyping of methicillin-resistant Staphylococcus aureus. Clin Microbiol Infect. 2011 Dec;17(12):1804–10. doi: 10.1111/j.1469-0691.2011.03521.x. Epub 2011/05/21. eng. [DOI] [PubMed] [Google Scholar]
- 32.Ross RW, Oh WK, Xie W, et al. Inherited variation in the androgen pathway is associated with the efficacy of androgen-deprivation therapy in men with prostate cancer. J Clin Oncol. 2008 Feb 20;26(6):842–7. doi: 10.1200/JCO.2007.13.6804. Epub 2008/02/19. eng. [DOI] [PubMed] [Google Scholar]
- 33.Yang M, Xie W, Mostaghel E, et al. SLCO2B1 and SLCO1B3 may determine time to progression for patients receiving androgen deprivation therapy for prostate cancer. J Clin Oncol. 2011 Jun 20;29(18):2565–73. doi: 10.1200/JCO.2010.31.2405. Epub 2011/05/25. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wright JL, Kwon EM, Ostrander EA, et al. Expression of SLCO transport genes in castration-resistant prostate cancer and impact of genetic variation in SLCO1B3 and SLCO2B1 on prostate cancer outcomes. Cancer Epidemiol Biomarkers Prev. 2011 Apr;20(4):619–27. doi: 10.1158/1055-9965.EPI-10-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Harshman LC, Xie W, et al. Association of SLCO2B1 Genotypes With Time to Progression and Overall Survival in Patients Receiving Androgen-Deprivation Therapy for Prostate Cancer. J Clin Oncol. 2015 Dec 14; doi: 10.1200/JCO.2015.62.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Prostate Cancer Trialists’ Collaborative Group. Lancet. 2000 Apr 29;355(9214):1491–8. Epub 2000/05/09. eng. [PubMed] [Google Scholar]
- 37.Li Z, Bishop AC, Alyamani M, et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. 2015 Jul 16;523(7560):347–51. doi: 10.1038/nature14406. Epub 2015/06/02. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z, Alyamani M, Li J, Upadhyay U, Balk SP, Taplin M-E, Auchus RJ, Sharifi N. Redirecting abiraterone metabolism to biochemically fine-tune prostate cancer anti-androgen therapy. Nature. 2016;533:547–51. doi: 10.1038/nature17954. Epub 2016/05/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sweeney CJ, Chen YH, Carducci M, et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N Engl J Med. 2015 Aug 20;373(8):737–46. doi: 10.1056/NEJMoa1503747. Epub 2015/08/06. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.James ND, Sydes MR, Clarke NW, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. 2016 Mar 19;387(10024):1163–77. doi: 10.1016/S0140-6736(15)01037-5. Epub 2016/01/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panageas KS, Ben-Porat L, Dickler MN, Chapman PB, Schrag D. When you look matters: the effect of assessment schedule on progression-free survival. J Natl Cancer Inst. 2007 Mar 21;99(6):428–32. doi: 10.1093/jnci/djk091. Epub 2007/03/22. eng. [DOI] [PubMed] [Google Scholar]
- 42.Gignac GA, Morris MJ, Heller G, Schwartz LH, Scher HI. Assessing outcomes in prostate cancer clinical trials: a twenty-first century tower of Babel. Cancer. 2008 Sep 1;113(5):966–74. doi: 10.1002/cncr.23719. Epub 2008/07/29. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.