

Abstract
Transforming growth factor (TGF)-β–induced epithelial-mesenchymal transition (EMT) leads to the formation of ocular fibrotic pathologies, such as anterior subcapsular cataract and posterior capsule opacification. Remodeling of the actin cytoskeleton, mediated by the Rho family of GTPases, plays a key role in EMT. However, how actin dynamics affect downstream markers of EMT has not been fully determined. Our previous work suggests that myocardin-related transcription factor A (MRTF-A), an actin-binding protein, might be an important mediator of TGFβ-induced EMT in lens epithelial cells. The aim of the current study was to determine the requirement of RhoA/ROCK signaling in mediating TGFβ-induced nuclear accumulation of MRTF-A and ultimate expression of α-smooth muscle actin (αSMA), a marker of a contractile myofibroblast phenotype. Using rat lens epithelial explants, we demonstrate that ROCK inhibition using Y-27632 prevents TGFβ-induced nuclear accumulation of MRTF-A, E-cadherin/β-catenin complex disassembly, and αSMA expression. Using a novel inhibitor specifically targeting MRTF-A signaling, CCG-203971, we further demonstrate the requirement of MRTF-A nuclear localization and activity in the induction of αSMA expression. Overall, our findings suggest that TGFβ-induced cytoskeletal reorganization through RhoA/ROCK/MRTF-A signaling is critical to EMT of lens epithelial cells.
INTRODUCTION
Cataract is an opacification of the ocular lens that results in obstruction of light and gradual loss of vision, and it continues to be the leading cause of blindness worldwide, affecting nearly 20 million people (1,2). Cataract surgery in developed countries is the most common operative procedure performed, while effective treatment is not readily available in economically developing countries. Consequently, in reaching the target of reducing avoidable visual impairment by 25% by the year 2019, the World Health Organization has outlined a global eye health action plan that prioritizes offering cataract surgery to those in need, further increasing the number of cataract surgeries performed. Cataract surgery, however, is not without its problems and can lead to secondary loss of vision, also known as posterior capsule opacification (PCO) (for review; 3,4). PCO involves a fibroproliferative response in which remnant lens epithelial cells found in the capsular bag after surgery proliferate and migrate to the posterior capsule, where they undergo epithelial-mesenchymal transition (EMT). Ocular trauma (5,6), implantable contact lenses (7,8), and diseases such as atopic dermatitis (9,10) and retinitis pigmentosa (11) can also trigger a similar fibrotic wound-healing response in the lens that results in a primary cataract, known as anterior subcapsular cataract (ASC) (12,13).
Transforming growth factor (TGF)-β has been implicated in ASC and PCO, and both its active and latent forms have been detected in the aqueous humor of the anterior chamber of the eye (14,15) in response to surgical or accidental injury (16–20). A role for TGFβ in ASC and PCO has also been shown experimentally in a number of animal models. For example, treatment of excised whole rat lenses with TGFβ disrupts the normal architecture of the anterior lens and results in characteristic changes associated with the conversion of lens epithelial cells into mesenchymal cells, indicative of ASC formation (21,22). Similarly, treating ex vivo rat lens explants with TGFβ triggers EMT and the formation of spindle-shaped cells immunoreactive to αSMA, a marker of a contractile myofibroblast phenotype (15,23,24). During EMT, TGFβ induces dramatic cytoskeletal reorganization, implicating the Rho family of GTPases, which are involved in regulating actin polymerization. Activated RhoA, bound to GTP, signals through its two effectors, Rho-associated coiled-coil–forming protein serine/threonine kinase (ROCK1 and 2) and mammalian diaphanous homologs (mDia1 and 2), which are both required for assembly of actin stress fibers and focal adhesions in the promotion of actomyosin contractile force generation (25). The involvement of RhoA signaling through ROCK in TGFβ-induced EMT has been demonstrated in a number of in vitro and in vivo animal systems (26–31). In lens epithelial cells, TGFβ induces RhoA activation with a corresponding increase in actin stress fibers and focal adhesions (32). Further, RhoA/ROCK signaling has been implicated in TGFβ-induced αSMA expression in mouse lens epithelial cells (27); however, the mechanism by which RhoA/ROCK signaling is linked to the expression of EMT-related markers has not been fully defined.
Rho-induced actin polymerization is monitored and then relayed to the nucleus through actin-binding proteins, specifically myocardin-related transcription factors (MRTFs). These transcription factors have been implicated in EMT (33–38); however, little is known about their role in the lens (39). The two isoforms, MRTF-A and -B, are widely expressed in numerous tissues, where they are normally bound to monomeric actin (G-actin). Upon actin polymerization, G-actin is incorporated into stress fibers as filamentous actin (F-actin), leading to free MRTF accumulating in the nucleus. Nuclear MRTF forms a complex with serum response factor (SRF) activating transcription of cytoskeletal genes associated with EMT, including αSMA (34,40–42). Indeed, TGFβ has been shown to stimulate nuclear accumulation of MRTF-A in several organ fibrosis models (38,43,44), including in recent work from our group showing TGFβ-induced nuclear localization of MRTF-A in lens epithelial cells (39). However, whether TGFβ-induced changes in MTRF-A in the lens are ROCK-dependent is not known.
Understanding the mechanisms involved in TGFβ-induced EMT is critical in the development of therapeutic strategies for various fibrotic diseases, including ocular fibrotic pathologies such as ASC and PCO. Therefore, using lens epithelial explants, the current study examines RhoA/ROCK signaling in the context of TGFβ-induced cytoskeletal remodeling. In particular, we seek to determine whether TGFβ-induced nuclear accumulation of MRTF-A is ROCK-dependent and required for expression of αSMA.
MATERIALS AND METHODS
Reagents
Recombinant human TGFβ2 and inhibitor CCG-203971 were obtained from R&D Systems. Y-27632 was purchased from Calbiochem. Primary antibodies were: E-cadherin from BD Transduction Laboratories; active β-catenin (clone 8E7) from Millipore; myosin light chain (phospho S20), pan actin [2Q1055], and GAPDH from Abcam; MRTF-A from Santa Cruz Biotechnology; and αSMA fluorescein isothiocyanate (FITC) conjugated and unconjugated from Sigma-Aldrich. All secondary antibodies for immunofluorescence staining were purchased from Invitrogen, while secondary antibodies for Western blots were obtained from LI-COR Biosciences.
Ex Vivo Rat Lens Epithelial Explants: Preparation and Treatment
Lens epithelial explants were obtained from 17- to 19-d-old Wistar rats (Charles River Laboratories), as previously described (45). Briefly, rat lenses were isolated and placed in 35 mm culture dishes containing prewarmed serum-free medium M199, supplemented with antibiotics (all from Invitrogen). The posterior suture of the lens was located and a small incision was made to remove the posterior capsule. This revealed the fiber mass, which was then gently peeled off the anterior epithelium. Once separated, the epithelium was then pinned to the culture dish with a blunt tool, exposing lens epithelial cells to culture medium. After 24 h, confluent epithelial explants were left untreated in serum-free M199 or treated with TGFβ2. For ROCK inhibitor studies, lens explants were pretreated with Y-27632 (20 μM) (46) for one hour, followed by cotreatment with TGFβ2 (4 ng/mL) for 48 and 72 h. For MRTF-A inhibition studies, CCG-203971 was dissolved in dimethyl sulfoxide and lens explants were pretreated with CCG-203971 (5 μM) for one hour, followed by cotreatment with TGFβ2 (4 ng/mL) for 48 h. Untreated explants for CCG-203971 studies were incubated in dimethyl sulfoxide (0.1%). All animal studies were performed according to the Canadian Council on Animal Care Guidelines (Guide to the Care and Use of Experimental Animals, second edition, 1993) and the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
Immunofluorescence Staining
Lens explants were fixed with 10% neutral buffered formalin for 15 min and washed with phosphate-buffered saline (Invitrogen). Explants were then detached from their culture dish and immersed in permeabilizing buffer (0.1% Triton X-100, 0.5% sodium dodecyl sulphate, and 5% donkey serum) for one hour at the bottom of their own 12 × 75 mm glass culture tube. Primary (overnight at 4°C) and secondary (1.5 h at room temperature) antibody incubation, as well as phosphate-buffered saline washes were carried out in these tubes under gentle agitation, ensuring that explants were always immersed in a small volume of solution at the bottom of the tube. Following the last set of washes, explants were flattened onto a slide and mounted in Prolong Gold antifade reagent with DAPI (Invitrogen) to visualize nuclei. Immunostained explants were analyzed using a Zeiss Apotome inverted microscope or a Leica DMRA2 fluorescence microscope equipped with a Q-Imaging RETIGA 1300i FAST digital camera. All immunofluorescence images were taken from the center of the explant and were representative of the entire explant. All images in figures labeled “Control” are representative of both untreated and inhibitor-only–treated explants. Images were captured using OpenLab software 4.0, and minor adjustments in brightness/contrast were made using Adobe Photoshop CS3. MRTF-A nuclear localization was analyzed using Fiji image processing software (47), where nuclear MRTF-A fluorescence intensity was measured and normalized to cytoplasmic fluorescence. Nuclear/cytoplasmic ratios were reported relative to untreated control ratios.
Western Blot
Lens explants (n ≥ 5 per sample per experiment) were pooled and lysed in Triton lysis buffer (50mM Tris, pH 8.0, 150mM NaCl, 1% Triton X-100) with cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail (Roche). Equal amounts of total protein were loaded on a gel, as determined by DC Protein Assay (Bio-Rad), and sodium dodecyl sulfate polyacrylamide gel electrophoresis was performed. Resolved bands were transferred onto a nitrocellulose membrane. Membranes were blocked with Odyssey Blocking Buffer (LI-COR Biosciences) for one hour and incubated with unconjugated αSMA or E-cadherin and GAPDH primary antibodies overnight at 4°C. Blots were washed in Tris-buffered saline with Tween-20 (1%) and probed with corresponding secondary antibodies. Densitometry on visualized bands was performed using Fiji image processing software, where each band was normalized to its corresponding GAPDH band. GAPDH-normalized values were reported as a fold reduction in αSMA or E-cadherin expression relative to TGFβ-treated controls (equal to 1).
Statistical Analysis
Data are expressed as means ± standard error of the mean (SEM), and statistical significance was determined by two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test performed post hoc (GraphPad Prism, version 6.0). Exact p values are reported and considered significant when p <.05.
RESULTS
Inhibition of RhoA/ROCK Signaling Prevents TGFβ-Induced Actin Polymerization and Expression of αSMA
To investigate the role of RhoA/ROCK signaling in TGFβ-induced EMT in the lens, rat lens explants (n ≥ 5 per treatment) were treated with TGFβ for 72 h in the presence and absence of a ROCK inhibitor, Y-27632. This compound targets the ATP-dependent kinase domain of ROCK1 and 2. One of the primary roles for ROCK, and the role of interest, is its regulation of actin-myosin cytoskeletal organization mediated through phosphorylation/activation of myosin regulatory light chains (MLCs). Immunofluorescence staining revealed that TGFβ-treated lens explants were rich in cytoplasmic actin filaments that stained positively for phosphorylated MLC, a mark of actomyosin contractility (Figure 1). In contrast, control and TGFβ + Y-27632–treated explants showed a distinct absence of actin filaments, instead exhibiting both diffuse cortical actin and phosphorylated MLC staining, indicating an inhibition of ROCK signaling.
Figure 1.

Y-27632 prevents TGFβ-induced actomyosin contractility. Lens explants were treated with TGFβ (4 ng/mL) in the presence and absence of Y-27632 (20 μM) for 48 h. Fixed explants were costained for pan-actin and phosphorylated myosin light chain (pMLC), and mounted in medium with DAPI to colocalize nuclei. Scale bar = 100 μm.
Next, explants were immunostained for rhodamine-phalloidin, which revealed that TGFβ treatment triggered polymerization of actin into F-actin–containing stress fibers positive for αSMA (Figure 2A; n ≥ 5 per treatment). Not all F-actin fibers within lens cells colocalized with αSMA; however, the large proportion that did are suggestive of a contractile, myofibroblast phenotype. In the presence of Y-27632, these effects of TGFβ were prevented, where lens epithelial cells did not exhibit detectable F-actin or αSMA staining. To quantify the previous finding, Western blot analysis was carried out on pooled explant lysates (n ≥ 5 per sample per experiment) following a 72 h treatment period. A significant 5.81 ± 0.07–fold reduction in αSMA protein was confirmed in the TGFβ + Y-27632–treated explant cells, as compared with explant cells treated with TGFβ alone (p <.0001; n = 4; Figures 2B and C). Similar results were obtained with a 48 h time course (data not shown).
Figure 2.

TGFβ-induced actin polymerization and induction in αSMA expression are ROCK-dependent. (A) Lens explants were treated with TGFβ (4 ng/mL) in the presence and absence of Y-27632 (20 μM) for 72 h. Fixed explants were costained for F-actin and αSMA and mounted in medium with DAPI to colocalize nuclei. (B) Western blots of pooled lens explant lysates (n ≥ 5 explants per sample) were probed for αSMA, with GAPDH as loading control. (C) Densitometric quantification of αSMA/GAPDH protein indicating the fold reduction in αSMA expression normalized to TGFβ-treated explants. Data expressed as the means ± SEM from four separate experiments (** p <.0001). Scale bar = 100 μm (A).
TGFβ-Induced Disruption to E-Cadherin–Based Adherens Junctions Is ROCK-Dependent
The E-cadherin/β-catenin complex provides a direct link between cell-cell adhesion complexes and the actin cytoskeleton, therefore E-cadherin and β-catenin localization were assessed in our lens explants. It was found that TGFβ treatment resulted in a loss of E-cadherin staining from cell borders (Figure 3A), exhibiting a disorganized patterning that was associated with an increase in αSMA expression (data not shown). In comparison, untreated and Y-27632–treated explants exhibited E-cadherin staining that was localized to the cell membrane. Treating explants with TGFβ in the presence of Y-27632 prevented membranous E-cadherin loss and disorganization, resembling control explants (n ≥ 5 per treatment). Accordingly, TGFβ treatment also led to dissociation of β-catenin from the membrane, resulting in its cytosolic/nuclear accumulation (Figure 3B), coinciding with induction in αSMA expression (data not shown). In contrast, in the presence of Y-27632, TGFβ treatment was unable to alter the localization of β-catenin, where it remained at the cell membrane, similar to control explants (n ≥ 5 per treatment).
Figure 3.

Y-27632 prevents TGFβ-induced E-cadherin and β-catenin delocalization from the cell membrane. Lens explants (n ≥ 5 per treatment) were fixed and costained for (A) E-cadherin and αSMA or (B) β-catenin and αSMA following TGFβ (4 ng/mL) treatment in the presence and absence of Y-27632 after 48 h. All explants were mounted in medium with DAPI to colocalize nuclei. Scale bar = 100 μm (A).
MRTF-A Is a Downstream Target of RhoA/ROCK Signaling in Rat Lens Explants
Our previous work in rat lens explants demonstrated expression and nuclear accumulation of MRTF-A in response to TGFβ treatment (39). Therefore, we next sought to determine whether TGFβ-induced MRTF-A nuclear localization in lens explants is ROCK-dependent. In control explants, MRTF-A was predominantly localized in the cytoplasm with no detectable αSMA expression (Figure 4A). Treatment with TGFβ triggered distinct nuclear accumulation of MRTF-A (Figure 4A, inset), which was associated with increased expression of αSMA. In contrast, lens explants treated with TGFβ in the presence of Y-27632 exhibited cytoplasmic MRTF-A staining corresponding to an absence of αSMA, similar to controls (n ≥ 5 per treatment). Next, we quantified the ratio of nuclear-to-cytoplasmic fluorescence intensity of MRTF-A across the lens explant treatment groups. Accordingly, TGFβ-treated explants exhibited a nuclear-to-cytoplasmic ratio 20.7 ± 3.2–fold higher than untreated controls (p = .0012; Figure 4B). In contrast, MRTF-A nuclear-to-cytoplasmic fluorescence intensity with TGFβ treatment in the presence of Y-27632 was 39.8 ± 3.2–fold lower than explants treated with TGFβ alone (p = .001), not differing from controls.
Figure 4.
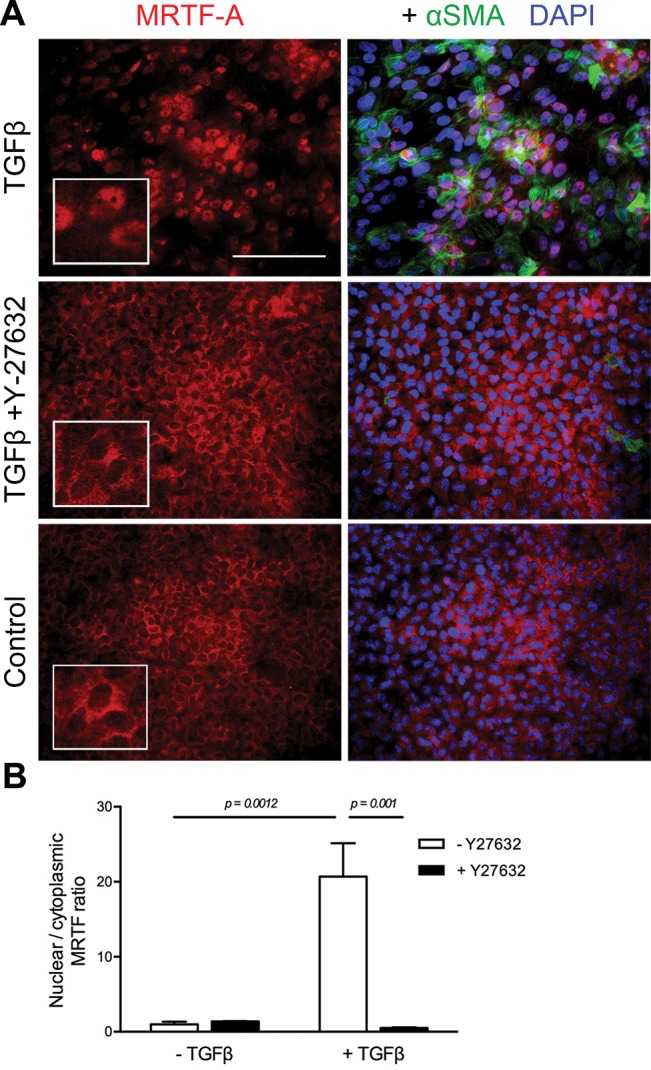
TGFβ-induced MRTF-A nuclear localization is ROCK-dependent. (A) The presence of Y-27632 (20 μM) prevented nuclear localization of MRTF-A in response to TGFβ (4 ng/mL) treatment for 48 h. Fixed explants were costained for MRTF-A and αSMA and mounted in medium with DAPI to colocalize nuclei. (B) Quantification of the ratio of nuclear-to-cytoplasmic MRTF-A signal by measuring intensity of fluorescence in images as described in Materials and Methods. Data expressed as means ± SEM from three separate experiments. Scale bar and insets = 100 μm (A).
Having established that TGFβ triggers nuclear accumulation of MRTF-A through RhoA/ROCK signaling, we next specifically targeted MRTF-A–mediated signaling using a novel inhibitor, CCG-203971, a derivative of CCG-1423. The advantages of CCG-203971 over the original compound are a significant reduction in cytotoxicity, with increased potency and selectivity (48). To better understand how CCG-203971 affects actin dynamics, lens explants were stained with DNaseI, which selectively binds unpolymerized G-actin monomers (Figure 5). Under untreated and CCG-203971–treated conditions, the majority of G-actin appears to be localized in the lens cell nuclei. In contrast, TGFβ leads to a near depletion of G-actin pools from the nuclei, with some nonselective staining of G-actin incorporating into filaments. Interestingly, treating explants with TGFβ in the presence of CCG-203971 prevents G-actin pool depletion, and staining resembles that of control explants (n ≥ 5 per treatment).
Figure 5.
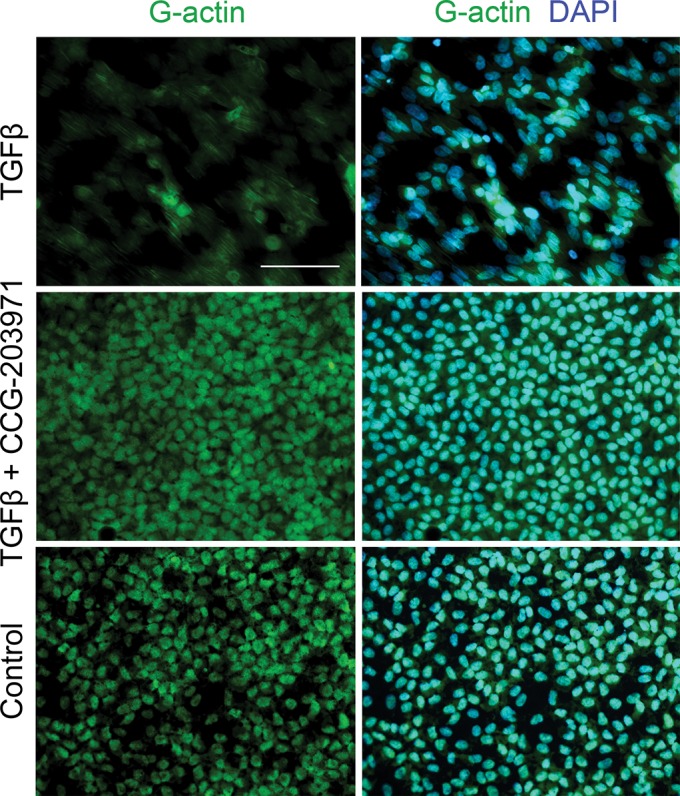
CCG-203971 prevents TGFβ-induced nuclear G-actin depletion. Lens explants were treated with TGFβ (4 ng/mL) in the presence and absence of CCG-203971 (5 μM). After 48 h, explants were fixed and stained with DNase I, which selectively binds G-actin monomers, then mounted in medium with DAPI to colocalize nuclei. Scale bar = 100 μm.
Next, the effects of CCG-203971 on lens EMT were analyzed. Explants treated with TGFβ in the presence of CCG-203971 revealed cytoplasmic localization of MRTF-A with an absence of αSMA (Figure 6A). This was in contrast to explants treated with TGFβ alone, which, as seen in the above experiments, exhibited nuclear accumulation of MRTF-A and expression of αSMA. Western blot analysis on pooled explant lysates (n ≥ 5 per sample per experiment) confirmed a significant 6.85 ± 0.05–fold reduction in αSMA protein in the TGFβ + CCG-203971–treated explant cells, as compared with explant cells treated with TGFβ alone (p < .0001; n = 3; Figures 6B and C).
Figure 6.

CCG-203971 prevents TGFβ-induced actin polymerization and αSMA expression. (A) Lens explants were treated with TGFβ (4 ng/mL) in the presence and absence of CCG-203971 (5 μM). Fixed explants were costained for MRTF-A and αSMA and mounted in medium with DAPI to colocalize nuclei. (B) Pooled lens explant lysates (n ≥ 5 explants per sample) were probed for αSMA and GAPDH as loading control. (C) Densitometric quantification of αSMA/GAPDH indicating the fold reduction in αSMA expression normalized to TGFβ-treated explants. Data are expressed as means ± SEM from three separate experiments (** p <.0001). Scale bar = 100 μm (A).
Further, the effects of CCG-203971 on E-cadherin and β-catenin localization were examined. It was found that treating explants with TGFβ in the presence of CCG-203971 prevented TGFβ-induced delocalization of E-cadherin (Figure 7A) and β-catenin (Figure 7B) from the cell membrane, where both adherens junction constituents exhibited distinct staining at sites of cell-cell contact (n ≥ 5 per treatment). Corresponding Western blot analysis on pooled explant lysates (n ≥ 5 per sample per experiment) using an antibody that recognizes the cytoplasmic domain of E-cadherin showed a 3.2 ± 0.9–fold induction in E-cadherin in the TGFβ + CCG-203971–treated group compared with TGFβ-treated explants (n = 3; Figures 7C and D).
Figure 7.

CCG-203971 prevents TGFβ-induced E-cadherin and β-catenin delocalization from the cell membrane. Lens explants (n ≥ 5 per treatment) were fixed and costained for (A) E-cadherin and αSMA or (B) β-catenin and αSMA following TGFβ (4 ng/mL) treatment in the presence and absence of CCG-203971 (5 μM) after 48 h. All explants were mounted in medium with DAPI to colocalize nuclei. (C) Pooled lens explant lysates (n ≥ 5 explants per sample) were probed for the cytoplasmic domain of E-cadherin and GAPDH as loading control. (D) Densitometric quantification of E-cadherin indicating the fold reduction in E-cadherin/GAPDH expression normalized to TGFβ-treated explants. Data are expressed as means ± SEM from three separate experiments. Scale bar = 100 μm (A).
DISCUSSION
Actin cytoskeletal reorganization is a phenotypic change widely associated with TGFβ-induced EMT. Understanding how actin dynamics affect downstream expression of EMT-related targets is critical to furthering our knowledge of the pathophysiology of fibrosis of the lens and other tissues. In this paper, we have identified a RhoA/ROCK signaling pathway that is required for TGFβ–induced EMT of lens epithelial cells using the ROCK inhibitor Y-27632. This compound has been widely used in the study of ROCK-dependent signaling as a potential therapeutic target in a number of diseases, including cancer (49), asthma (50), hypertension (51), corneal endothelial dysfunction (52), and glaucoma (53–55). In our model of lens EMT, as occurs in PCO and ASC, TGFβ stimulation of lens epithelial explants induced assembly of actin into pMLC-positive stress fibers, which along with coexpression of αSMA, indicates a transition to a myofibroblast phenotype. In the presence of Y-27632, TGFβ-induced stress fiber formation and actomyosin contractility were prevented and corresponded with an absence of αSMA expression. This finding suggests that polymerization of actin is required in order for αSMA to be incorporated into filaments. Our findings are in line with studies carried out in other epithelial systems reporting that RhoA signaling was indispensable to TGFβ-induced cytoskeletal remodeling (26,28) and αSMA expression (28), as well as in mouse lens epithelial cell culture demonstrating that αSMA expression is ROCK-dependent (27). Importantly, we have further extended these findings to show that ROCK-mediated stimulation of αSMA expression occurs via the actin binding protein MRTF-A.
Transcriptional activation and expression of αSMA is regulated by SRF along with its coactivators, the MRTFs. The subcellular localization of MRTF-A is tightly regulated by the ratio of G- to F-actin in the cell, as determined by the state of Rho-mediated actin polymerization. G-actin binds to the RPEL motifs on MRTF-A and, when bound, G-actin has been proposed to inhibit nuclear import, enhance nuclear export, and repress transcription of MRTF-A target genes (56). Thus far, work in proximal tubular epithelial cells has been pivotal in determining the involvement of MRTF-A subcellular localization in the context of TGFβ-induced EMT (34,37,57). Morita et al. (2007) demonstrated that the presence of constitutively active MRTF-A was sufficient to induce expression of αSMA, in contrast to dominant negative MRTF-A, which prevented the TGFβ-induced decrease in expression of epithelial markers, including E-cadherin and β-catenin. Here we demonstrate that the presence of Y-27632 prevented TGFβ-induced nuclear accumulation of MRTF-A, which corresponded with the absence of αSMA, suggesting that in our lens explants, TGFβ signals through Rho/ROCK/MRTF-A to induce expression of αSMA. Furthermore, subcellular localization of MRTF-A may act as an indicator of EMT.
To further corroborate our findings, a novel specific inhibitor of the MRTF-A pathway, CCG-203971, was used. This inhibitor is a derivative of CCG-1423, which was identified to inhibit an MRTF-A/SRF transcriptional reporter downstream of RhoA (58). CCG-1423 has been shown to decrease mesenchymal protein expression, including αSMA in podocyte EMT in diabetic nephropathy (59), while both CCG-1423 and CCG-203971 blocked matrix-stiffness and TGFβ-induced αSMA expression in intestinal fibroblasts (60). The precise mechanism of action of these inhibitors has not been definitively established; however, it is suggested that they retain MRTF-A in the cytoplasm by: (A) binding the nuclear localization signal of MRTF-A, preventing nuclear import (61), or (B) inhibiting depletion of G-actin pools in the nucleus, resulting in nuclear export of MRTF-A (62). The observed cytoplasmic MRTF-A staining in our explants in the presence of TGFβ and CCG-203971 coincides with a substantially higher level of G-actin localized to the nucleus when compared with the near depletion of G-actin pools observed in explant cells exclusively treated with TGFβ. This finding suggests that one of the ways in which CCG-203971 acts in our system is by retaining G-actin in lens cell nuclei, preventing the emergence of polymerized cytosolic filaments, and thus nuclear accumulation of MRTF-A, similar to mechanism (B) outlined above. The presence of CCG-203971 prevented TGFβ-induced αSMA expression, which demonstrates the critical nature of the balance of G- to F-actin in MRTF-A–mediated expression of αSMA. By preventing nuclear accumulation of MRTF-A, CCG-203971 is preventing transcriptional activation of MRTF-A/SRF–regulated target genes.
In an earlier study in mouse lens epithelial cell explants, we demonstrated that TGFβ leads to delocalization of adherens junction constituents E-cadherin and β-catenin from the cell membrane, resulting in their disorganized patterning (45). The current work demonstrates that this TGFβ-induced E-cadherin and β-catenin delocalization is ROCK/MRTF-A-dependent, where both remained intact at the cell membrane in the presence of Y-27632 as well as CCG-203971. Similarly, in renal epithelial cells, the presence of Y-27632 following TGFβ treatment prevented TGFβ-induced adherens junction disassembly, including β-catenin delocalization and phosphorylation (30). As well, activated RhoA has been shown to mediate disassembly of epithelial cell contacts, preferentially signaling through ROCK rather than mDia (63). Based on the emerging role of E-cadherin–based adherens junctions as mechanosensors, it is possible that a RhoA/ROCK-mediated increase in cytoskeletal tension is sensed through cadherin-based contacts (64). A compromise in cell-cell adhesion integrity can then lead to dissociation of β-catenin, which is involved in regulating the transcription of EMT-related genes (65). Indeed, novel concurrent work in our lab demonstrated that β-catenin–mediated signaling, specifically when in complex with CREB-binding protein, is required for TGFβ-induced EMT, outlining a manner in which the effects of EMT are propagated in a feed-forward loop (66). Therefore, our findings demonstrate a key role for RhoA/ROCK and MRTF-A signaling in regulating the integrity of adhesion complexes in response to TGFβ, essential in determining cell phenotype.
Overall, the current study in lens epithelial explants demonstrates that the progression of TGFβ-induced EMT, culminating in the expression of αSMA, is regulated by actin dynamics through RhoA/ROCK/MRTF-A signaling. Specifically, TGFβ-mediated disassembly of the E-cadherin/β-catenin complex and subcellular localization of MRTF-A are ROCK-dependent and critical to TGFβ-induced αSMA expression. Our findings suggest that targeting MRTF-A activity downstream of ROCK may serve as a promising means of preventing TGFβ-induced EMT involved in fibrosis of the lens and other tissues.
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health, R01-017146 (PI: JW-M). AK acknowledges the Natural Sciences and Engineering Research Council of Canada for financial support. We thank S Bowman for his invaluable suggestions and thoughts on the manuscript.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Korol A, Taiyab A, West-Mays JA. (2016) RhoA/ROCK signaling regulates TGFβ-induced epithelial-mesenchymal transition of lens epithelial cells through MRTF-A. Mol. Med. 22:713–23.
REFERENCES
- Pascolini D, Mariotti SP. Global estimates of visual impairment: 2010. Br J Ophthalmol. 2012;96:614–18. doi: 10.1136/bjophthalmol-2011-300539. [DOI] [PubMed] [Google Scholar]
- World Health Organization Visual Impairment and Blindness. Fact Sheet No. 282. 2012 [Google Scholar]
- West-Mays JA, Sheardown H. Posterior capsule opacification. In: Levin LA, Albert DM, editors. Ocular Disease: Mechanisms and Management. Saunders, Philadelphia: 2010. pp. 277–82. [Google Scholar]
- Wormstone IM, Wang L, Liu CS. Posterior capsule opacification. Exp Eye Res. 2009;88:257–69. doi: 10.1016/j.exer.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Chinwattanakul S, Prabhasawat P, Kongsap P. Corneal injury by bee sting with retained stinger—a case report. J Med Assoc Thai. 2006;89:1766–9. [PubMed] [Google Scholar]
- Schnaidt AG, et al. Delayed loss of vision after hammer and chisel injury. Ophthalmologe. 2009;106:1017–21. doi: 10.1007/s00347-009-1955-8. [DOI] [PubMed] [Google Scholar]
- Khalifa YM, et al. Cataract development associated with collagen copolymer posterior chamber phakic intraocular lenses: clinicopathological correlation. J Cataract Refract Surg. 2010;36:1768–74. doi: 10.1016/j.jcrs.2010.04.039. [DOI] [PubMed] [Google Scholar]
- Schmidinger G, Lackner B, Pieh S, Skorpik C. Long-term changes in posterior chamber phakic intraocular collamer lens vaulting in myopic patients. Ophthalmology. 2010;117:1506–11. doi: 10.1016/j.ophtha.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Brandonisio TM, Bachman JA, Sears JM. Atopic dermatitis: a case report and current clinical review of systemic and ocular manifestations. Optometry. 2001;72:94–102. [PubMed] [Google Scholar]
- Katsushima H, Miyazaki I, Sekine N, Nishio C, Matsuda M. [Incidence of cataract and retinal detachment associated with atopic dermatitis] Nippon Ganka Gakkai Zasshi. 1994;98:495–500. [PubMed] [Google Scholar]
- Cohen S. Ocular histopathologic studies of neonatal and childhood adrenoleukodystrophy. Am J Ophthalmol. 1983;95:82–96. doi: 10.1016/0002-9394(83)90336-7. [DOI] [PubMed] [Google Scholar]
- Lovicu FJ, Steven P, Saika S, McAvoy JW. Aberrant lens fiber differentiation in anterior subcapsular cataract formation: a process dependent on reduced levels of Pax6. Invest Ophthalmol Vis Sci. 2004;45:1946–53. doi: 10.1167/iovs.03-1206. [DOI] [PubMed] [Google Scholar]
- Novotny GE. Formation of cytoplasm-containing vesicles from double-walled coated invaginations containing oligodendrocytic cytoplasm at the axon-myelin sheath interface in adult mammalian central nervous system. Acta Anat (Basel) 1984;119:106–12. doi: 10.1159/000145869. [DOI] [PubMed] [Google Scholar]
- de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW. Transforming growth factor-beta: induced epithelial-mesenchymal transition in the lens: a model for cataract formation. Cells Tissues Organs. 2005;179:43–55. doi: 10.1159/000084508. [DOI] [PubMed] [Google Scholar]
- Gordon-Thomson C, de Iongh RU, Hales AM, Chamberlain CG, McAvoy JW. Differential cataractogenic potency of TGF-beta1, -beta2, and -beta3 and their expression in the postnatal rat eye. Invest Ophthalmol Vis Sci. 1998;39:1399–1409. [PubMed] [Google Scholar]
- Cousins SW, McCabe MM, Danielpour D, Streilein JW. Identification of transforming growth factor-beta as an immunosuppressive factor in aqueous humor. Invest Ophthalmol Vis Sci. 1991;32:2201–11. [PubMed] [Google Scholar]
- Lovicu FJ, et al. TGFbeta induces morphological and molecular changes similar to human anterior subcapsular cataract. Br J Ophthalmol. 2002;86:220–6. doi: 10.1136/bjo.86.2.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JV, et al. Adenoviral gene transfer of bioactive TGFbeta1 to the rodent eye as a novel model for anterior subcapsular cataract. Mol Vis. 2007;13:457–69. [PMC free article] [PubMed] [Google Scholar]
- Saika S, et al. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am J Pathol. 2004;164:651–63. doi: 10.1016/S0002-9440(10)63153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallentin N, Wickstrom K, Lundberg C. Effect of cataract surgery on aqueous TGF-beta and lens epithelial cell proliferation. Invest Ophthalmol Vis Sci. 1998;39:1410–18. [PubMed] [Google Scholar]
- Dwivedi DJ, et al. Matrix Metalloproteinase Inhibitors Suppress Transforming Growth Factor-β-Induced Subcapsular Cataract Formation. Am J Pathol. 2006;168:69–79. doi: 10.2353/ajpath.2006.041089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales AM, Chamberlain CG, McAvoy JW. Cataract induction in lenses cultured with transforming growth factor-beta. Invest Ophthalmol Vis Sci. 1995;36:1709–13. [PubMed] [Google Scholar]
- Banh A. Lens-Specific Expression of TGF Induces Anterior Subcapsular Cataract Formation in the Absence of Smad3. Invest Ophthalmol Vis Sci. 2006;47:3450–60. doi: 10.1167/iovs.05-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales AM, Schulz MW, Chamberlain CG, McAvoy JW. TGF-beta 1 induces lens cells to accumulate alpha-smooth muscle actin, a marker for subcapsular cataracts. Curr Eye Res. 1994;13:885–90. doi: 10.3109/02713689409015091. [DOI] [PubMed] [Google Scholar]
- Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348 Pt 2:241–55. [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HJ, Yoo J. Rho activation is required for transforming growth factor-beta–induced epithelial-mesenchymal transition in lens epithelial cells. Cell Biol Int. 2007;31:1225–30. doi: 10.1016/j.cellbi.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Masszi A, et al. Central role for Rho in TGF-beta1–induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol. 2003;284:F911–24. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- Tavares AL, Mercado-Pimentel ME, Runyan RB, Kitten GT. TGF beta: mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev Dyn. 2006;235:1589–98. doi: 10.1002/dvdy.20771. [DOI] [PubMed] [Google Scholar]
- Tian YC, Fraser D, Attisano L, Phillips AO. TGF-beta1–mediated alterations of renal proximal tubular epithelial cell phenotype. Am J Physiol Renal Physiol. 2003;285:F130–42. doi: 10.1152/ajprenal.00408.2002. [DOI] [PubMed] [Google Scholar]
- Zhang K, et al. TGF-beta1 induces the dissolution of tight junctions in human renal proximal tubular cells: role of the RhoA/ROCK signaling pathway. Int J Mol Med. 2013;32:464–8. doi: 10.3892/ijmm.2013.1396. [DOI] [PubMed] [Google Scholar]
- Maddala R, Reddy VN, Epstein DL, Rao V. Growth factor induced activation of Rho and Rac GTPases and actin cytoskeletal reorganization in human lens epithelial cells. Mol Vis. 2003;9:329–36. [PubMed] [Google Scholar]
- Busche S, Descot A, Julien S, Genth H, Posern G. Epithelial cell-cell contacts regulate SRF-mediated transcription via Rac-actin-MAL signalling. J Cell Sci. 2008;121:1025–35. doi: 10.1242/jcs.014456. [DOI] [PubMed] [Google Scholar]
- Fan L, et al. Cell contact–dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol Biol Cell. 2007;18:1083–97. doi: 10.1091/mbc.E06-07-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fintha A, et al. Characterization and role of SCAI during renal fibrosis and epithelial- to-mesenchymal transition. Am J Pathol. 2013;182:388–400. doi: 10.1016/j.ajpath.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–42. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- Morita T, Mayanagi T, Sobue K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J Cell Biol. 2007;179:1027–42. doi: 10.1083/jcb.200708174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JW, Riley PN, Nalluri SM, Ashar PK, Gomez EW. Matrix Rigidity Mediates TGFbeta1-Induced Epithelial-Myofibroblast Transition by Controlling Cytoskeletal Organization and MRTF-A Localization. J Cell Physiol. 2015;230:1829–39. doi: 10.1002/jcp.24895. [DOI] [PubMed] [Google Scholar]
- Gupta M, Korol A, West-Mays JA. Nuclear translocation of myocardin-related transcription factor-A during transforming growth factor beta-induced epithelial to mesenchymal transition of lens epithelial cells. Mol Vis. 2013;19:1017–28. [PMC free article] [PubMed] [Google Scholar]
- Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol. 2007;292:C70–81. doi: 10.1152/ajpcell.00386.2006. [DOI] [PubMed] [Google Scholar]
- Sebe A, et al. Rac, PAK and p38 regulate cell contact-dependent nuclear translocation of myocardin-related transcription factor. FEBS Lett. >2008;582:291–8. doi: 10.1016/j.febslet.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XH, et al. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci. 2007;120:1801–9. doi: 10.1242/jcs.001586. [DOI] [PubMed] [Google Scholar]
- Fan WH, Karnovsky MJ. Increased MMP-2 expression in connective tissue growth factor over-expression vascular smooth muscle cells. J Biol Chem. 2002;277:9800–5. doi: 10.1074/jbc.M111213200. [DOI] [PubMed] [Google Scholar]
- Hinson JS, Medlin MD, Lockman K, Taylor JM, Mack CP. Smooth muscle cell-specific transcription is regulated by nuclear localization of the myocardin-related transcription factors. Am J Physiol Heart Circ Physiol. 2007;292:H1170–80. doi: 10.1152/ajpheart.00864.2006. [DOI] [PubMed] [Google Scholar]
- Korol A, et al. Matrix metalloproteinase- 9-null mice are resistant to TGF-beta-induced anterior subcapsular cataract formation. Am J Pathol. 2014;184:2001–12. doi: 10.1016/j.ajpath.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehata M, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev. 2015;82:518–29. doi: 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JL, et al. Design and synthesis of tag-free photoprobes for the identification of the molecular target for CCG-1423, a novel inhibitor of the Rho/MKL1/SRF signaling pathway. Beilstein J Org Chem. 2013;9:966–73. doi: 10.3762/bjoc.9.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath N, Olson MF. Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012;13:900–8. doi: 10.1038/embor.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume H. RhoA/Rho-kinase as a therapeutic target in asthma. Curr Med Chem. 2008;15:2876–85. doi: 10.2174/092986708786242831. [DOI] [PubMed] [Google Scholar]
- Duong-Quy S, Bei Y, Liu Z, Dinh-Xuan AT. Role of Rho-kinase and its inhibitors in pulmonary hypertension. Pharmacol Ther. 2013;137:352–64. doi: 10.1016/j.pharmthera.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Koizumi N, Okumura N, Ueno M, Kinoshita S. New therapeutic modality for corneal endothelial disease using Rho-associated kinase inhibitor eye drops. Cornea. 2014;33 Suppl 11:S25–31. doi: 10.1097/ICO.0000000000000240. [DOI] [PubMed] [Google Scholar]
- Murphy KC, et al. The formation of cortical actin arrays in human trabecular meshwork cells in response to cytoskeletal disruption. Exp Cell Res. 2014;328:164–71. doi: 10.1016/j.yexcr.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PV, Deng PF, Kumar J, Epstein DL. Modulation of aqueous humor outflow facility by the Rho kinase-specific inhibitor Y-27632. Invest Ophthalmol Vis Sci. 2001;42:1029–37. [PubMed] [Google Scholar]
- Waki M, Yoshida Y, Oka T, Azuma M. Reduction of intraocular pressure by topical administration of an inhibitor of the Rho-associated protein kinase. Curr Eye Res. 2001;22:470–4. doi: 10.1076/ceyr.22.6.470.5489. [DOI] [PubMed] [Google Scholar]
- O’Connor JW, Gomez EW. Cell adhesion and shape regulate TGF-beta1–induced epithelial-myofibroblast transition via MRTF-A signaling. PLoS One. 2013;8:e83188. doi: 10.1371/journal.pone.0083188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elberg G, et al. MKL1 mediates TGF-beta1–induced alpha-smooth muscle actin expression in human renal epithelial cells. Am J Physiol Renal Physiol. 2008;294:F1116–28. doi: 10.1152/ajprenal.00142.2007. [DOI] [PubMed] [Google Scholar]
- Evelyn CR, et al. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6:2249–60. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- Zhao L, et al. Critical role of serum response factor in podocyte epithelial-mesenchymal transition of diabetic nephropathy. Diab Vasc Dis Res. 2016;13:81–92. doi: 10.1177/1479164115588545. [DOI] [PubMed] [Google Scholar]
- Johnson LA, et al. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta–induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis. 2014;20:154–65. doi: 10.1097/01.MIB.0000437615.98881.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Watanabe B, Nakagawa Y, Minami S, Morita T. RPEL proteins are the molecular targets for CCG-1423, an inhibitor of Rho signaling. PLoS One. 2014;9:e89016. doi: 10.1371/journal.pone.0089016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist MR, et al. Redox modification of nuclear actin by MICAL-2 regulates SRF signaling. Cell. 2014;156:563–76. doi: 10.1016/j.cell.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol. 2002;4:408–15. doi: 10.1038/ncb796. [DOI] [PubMed] [Google Scholar]
- le Duc Q, et al. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol. 2010;189:1107–15. doi: 10.1083/jcb.201001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoux B, Nelson WJ, Yan J, Mege RM. The mechanotransduction machinery at work at adherens junctions. Integr Biol (Camb) 2015;7:1109–19. doi: 10.1039/c5ib00070j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiyab A, Korol A, Deschamps PA, West-Mays JA. β-catenin/CBP-dependent signaling regulates TGF-β-induced epithelial to mesenchymal transition in lens epithelial cells. Invest Ophthalmol Vis Sci. 2016 doi: 10.1167/iovs.16-20162. [In press] [DOI] [PMC free article] [PubMed] [Google Scholar]