

Abstract
Atrial fibrillation (AF) is the most common age-related cardiac arrhythmia. It is a progressive disease, which makes treatment difficult. The progression of AF is caused by the accumulation of damage in cardiomyocytes which makes the atria more vulnerable for AF. Especially structural remodeling and electrical remodeling, together called electropathology are sustainable in the atria and impair functional recovery to sinus rhythm after cardioversion.
The exact electropathological mechanisms underlying persistence of AF are at present unknown. High resolution wavemapping studies in patients with different types of AF showed that longitudinal dissociation in conduction and epicardial breakthrough were the key elements of the substrate of longstanding persistent AF. A double layer of electrically dissociated waves propagating transmurally can explain persistence of AF (Double Layer Hypothesis) but the molecular mechanism is unknown. Derailment of proteasis –defined as the homeostasis in protein synthesis, folding, assembly, trafficking, guided by chaperones, and clearance by protein degradation systems – may play an important role in remodeling of the cardiomyocyte. As current therapies are not effective in attenuating AF progression, step-by-step analysis of this process, in order to identify potential targets for drug therapy, is essential. In addition, novel mapping approaches enabling assessment of the degree of electropathology in the individual patient are mandatory to develop patient-tailored therapies. The aims of this review are to
1) summarize current knowledge of the electrical and molecular mechanisms underlying AF
2) discuss the shortcomings of present diagnostic instruments and therapeutic options and
3) to present potential novel diagnostic tools and therapeutic targets.
Keywords: Atrial Fibrillation, Heat Shock Protein, Diagnosis, Therapy
Introduction
The first electrocardiogram (ECG) of atrial fibrillation (AF) was recorded by Einthoven in 1906.[1] Nowadays, AF is one of the most common arrhythmias with a prevalence varying from <0.1% to >12% in the elderly which is expected to be doubled in patients over 55 years by 2060.[2,3] AF is originally known as a disease of the ageing population. However, an increasing prevalence is seen in young adults, especially in endurance athletes[4] and patients with congenital heart disease.[5] Hence, a continuous rise in the number of AF associated hospitalizations and healthcare costs is to be expected.[6] Several treatment modalities have been developed, but all are associated with high recurrence rates or negative side effects. The aims of this review are to
summarize current knowledge of the electrical and molecular mechanisms underlying AF,
discuss the shortcomings of present diagnostic instruments and therapeutic options and
to present potential novel diagnostic tools and targets for future therapy.
Deficiencies in Diagnostic Tools of Atrial Fibrillation
AF is usually diagnosed by a surface ECG or Holter recording.However, diagnosis of new onset, paroxysmal or asymptomatic AF can be challenging. An ECG only captures several seconds of the heart rhythm and episodes of AF can therefore be easily missed. The use of long-term ambulatory electrocardiography devices or implantable loop recorders increases the chance of detecting AF paroxysms. In addition, these devices also allow determination of the total duration of all AF episodes within a specific time frame, the so-called AF burden. However, electrocardiographic recordings do not provide any information on the mechanism underlying AF. Recent studies[7]-[10] suggest that body surface mapping arrays, containing 252 electrodes, may be useful to identify driver regions in patients with AF. Yet, none of the currently available recording techniques can determine the degree and extensiveness of atrial electropathology. Hence, when a patient presents with AF, we have no diagnostic tool available for evaluating the mechanism underlying AF and determining the stage of the disease at any time in the process.
Mechanisms Of Atrial Fibrillation: From Past To The Present
Experiments performed by Gordon Moe[11] nearly 60 years ago, provided the basis for the ongoing debate on the underlying cause for AF. In isolated canine atria, he showed that AF could be due to either fibrillatory conduction (AF caused by an ectopic focus with a high frequency discharge resulting in non-uniform excitation of the atria) or true fibrillation (AF persists independently from the site where it was initiated). In 1959, Moe[11] introduced the so-called Multiple Wavelet Hypothesis which further described the features of true fibrillation. In this hypothesis, Moe postulated that persistence of AF depended on the average number of wavelets. With the total number of wavelets being increased, the probability of extinguishment and thus termination of AF would become smaller. Twenty-six years later, Allessie et al.[12] performed the first experimental evaluation of Moe’s multiple wavelet hypothesis. In a canine right atrium, during 0.5 second of acutely induced AF, he demonstrated in series of consecutive excitation maps that there was a continuous beat-tobeat change in activation pattern. The critical number of wavelets in both right and left atria necessary to perpetuate AF was estimated to be between three and six. Ever since, numerous experimental and clinical mapping studies,[11,13-21] reporting on perpetuation of AF, are supportive on either a focal (repetitive ectopic discharges) or reentrant mechanism (mother-wave, rotor, multiple wavelets). In the past years, most clinical studies reported on the presence of rotors in patients with various types of AF.[20]
Electropathology Associated With Persistence Of Atrial Fibrillation
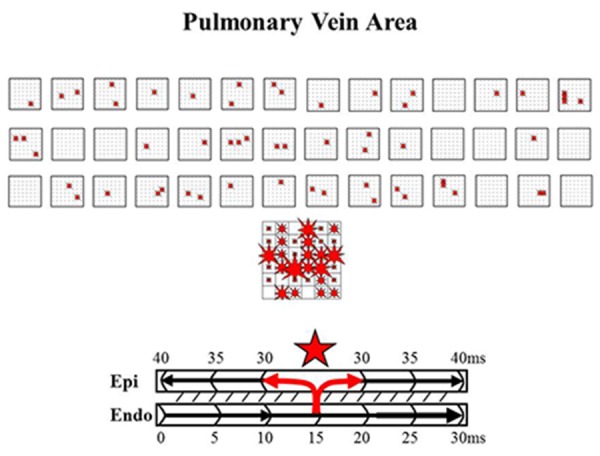
High-resolution wavemapping studies[22] of AF in patients with valvular heart disease and longlasting persistent AF, demonstrated that a large proportion of fibrillation waves were so-called focal waves. These waves appeared in the middle of the mapping area and could not be explained by fibrillation waves propagating in the epicardial plane. Focal fibrillation waves appeared scattered throughout the mapping area and were not repetitive (Figure 1). The coupling interval was longer than the dominant AF cycle length, and unipolar electrograms at the epicardial origin of these waves exhibited R-waves.[22] Hence, characteristics of these focal fibrillation waves strongly suggest that they originated from endo-epicardial breakthrough. These findings were supported by a report from Lee et al.[23] who observed that more than one third of the fibrillation waves in patients with persistent AF were of ‘focal’ origin without any area sustaining focal activity. Based on our observations, we recently introduced a new mechanism explaining persistence of AF independently of the presence of foci or re-entrant circuits in our Double Layer Hypothesis.[22,24] The “Double Layer Hypothesis” states that the substrate of longstanding persistent AF in humans is caused by progressive endo-epicardial dissociation, transforming the atria into an electrical double layer of dissociated waves that constantly ‘feed’ each other (Figure 1) Whereas in patients with short-lasting episodes of AF, the endo- and epicardial layers are still activated synchronously, in patients with longstanding persistent AF, the endo- and epicardial layers of the atrial wall are activated asynchronously. Over time, due to electrical and structural remodeling of the atria, the atrial wall is gradually transformed into a double layer of narrow anatomically delineated pathways. The exact molecular mechanisms underlying electrical dissociation are, however, unknown.
Figure 1. Epicardial breakthrough Upper panel: beat-to-beat variation in spatiotemporal distribution of epicardial breakthrough waves (‘focal waves’) during 6 seconds of persistent AF in a small area of 1.25 X 1.25cm between the pulmonary veins. Each asterisk indicates a breakthrough site. The large map shows all 55 epicardial breakthroughs sites. The size of the asterisk is proportional to the number of epicardial breakthroughs occurring at that site. The breakthrough map demonstrates a wide distribution of these focal waves; none of these breakthrough waves occurred, however, repetitively. Lower panel: schematic presentation of excitation of the endoand epicardial layer explaining how transmural conduction from the endocardium to the epicardium gives rise to an epicardial breakthrough wave. Hence, the endocardial layer serves in this case as a source for ‘new’ fibrillation waves in the epicardial layer.[22].
Molecular Mechanisms Underlying Electropathology AF
As mentioned above, AF is a progressive disease, which can be explained by the fact that AF itself induces alterations in both function and structure of the cardiomyocyte. These alterations induce an arrhythmogenic substrate which facilitates perpetuation of AF episodes.[25]
During the last decennia, various researchers aimed to identify the molecular mechanisms that underlie cardiomyocyte remodeling and AF progression. Although several pathways, especially related to ion channel remodeling, have been described, the exact molecular mechanisms driving AF remodeling and progression are still unidentified. The general concept is that during AF, cardiomyocytes are subjected to rapid and irregular excitation causing calcium overload in the cells which leads to fast and reversible electrical remodeling and slower, irreversible structural remodeling (Figure 2). The cardiomyocyte responds to a calcium overload by the functional downregulation of L-type Ca2+ current channels, which causes the shortening of action potential duration (APD) and electrical remodeling, thereby providing a further substrate for AF.[26-30] Also, several other ion channel currents are affected either on the expression level or phosphorylation and redox status.[31-33] In addition, various kinases and phosphatases become activated and regulate the function of ion channels and other downstream target proteins, for example transcriptions factors, various calcium handling proteins (such as RyR2, Sarcoplasmic Reticulum Ca2+ ATPases (SERCA), or Na+/Ca2+ exchanger) and the actin cytoskeleton.[34-38]
Figure 2. Overview of AF-induced cardiomyocyte remodeling AF induces time-related progressive remodeling. First, AF causes a stressful cellular Ca2+ overload, which results in a direct inhibition of the L-type Ca2+ channel, shortening of action potential duration and contractile dysfunction. These changes have an early onset and are reversible. The early processes protect the cardiomyocyte against Ca2+ overload but at the expense of creating a substrate for persistent AF. When AF persists derailment of proteostasis occurs, which results in microtubule disruption, cytoskeletal changes and degradation of proteins. The targets involved in proteostasis are RhoA/ROCK, HDAC6 and calpain. In addition, HSP induction has been found to counteract these targets. Derailment of proteostasis results in structural remodeling, myolysis/ hibernation, and consequently impaired contractile function and AF persistence. Thus drugs that normalize proteostasis via inhibition of RhoA/ROCK, calpain, and HDAC6, but also via induction of cardioprotective HSPs are of therapeutic interest for future treatment of clinical AF.
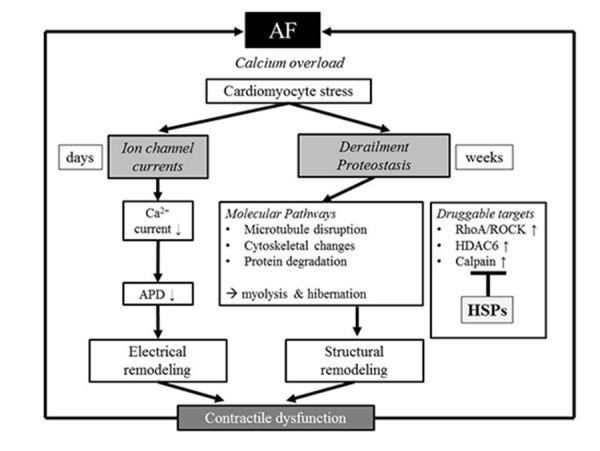
When AF persists beyond a few days, irreversible structural remodeling occurs, especially hibernation[39] (Figure 2). Various research groups[39-41] showed that hibernation is a form of tissue adaptation. It is defined as the ability of the cardiomyocytes to turn into a non-functional phenotype featuring irreversible degradation of the myofibril structure (myolysis), which leads to loss of atrial contraction.
While the early electrical remodeling is reversible30 a ‘second factor’ underlies the persistence of AF, having a time course comparable to AF-induced structural changes (hibernation/myolysis) in the atrial cardiomyocytes.[42] Thus, the prevention of structural remodeling represents a key target to attenuate cardiomyocyte remodeling and dysfunction and may improve the outcome of (electrical) cardioversion to normal sinus rhythm. We have strong indications that derailment of proteostasis represents this ‘second factor’ that underlies AF progression.[38,39,43-46]
Derailed Proteostasis Novel Concept Of Cardiomyocyte Remodeling
Proteostasis is defined as the homeostasis in protein synthesis, folding, assembly, trafficking, guided by chaperones, and clearance by protein degradation systems.[47-50] Healthy proteostasis is controlled by an exquisitely regulated network of molecular components and cellular pathways, the protein quality control (PQC) system.[47,51] Cells, including cardiomyocytes, are very sensitive to changes in the intra- and extracellular environment, induced by stressors, including AF. Stressors can cause derailment in the proteostasis by altering the stability of proteins, leading to protein damage, unfolding and breakdown, as observed for cardiac troponins and structural proteins.[38, 43] In the heart, various chaperones, especially Heat Shock Proteins (HSPs), are expressed to ensure a healthy cardiomyocyte proteostasis and optimal function of the heart. For example HSP27,cvHSP, HSP20 and HSP22 are important members of the PQC system and attenuate derailment of proteostasis in AF by assisting in the refolding of unfolded proteins,[38,51] prevention of AF-induced damage to contractile proteins[44,52] and attenuation of protein breakdown.[43] In this way, HSPs normalize the proteostasis and protect the cardiomyocyte against remodeling and AF progression.
Molecular Pathways Underlying Derailed Proteostasis
Recently, several molecular pathways were found to induce derailment of proteostasis. These pathways include the persistent activation of calpain, activation of RhoA/ROCK pathway and the activation of HDAC6.
Investigators found proof for a role of persistent activation of the calcium overload-induced protease calpain to underlie impairment of proteostasis and AF progression in experimental cardiomyocyte, and Drosophila model systems for AF,[43,52,53] but also in human permanent AF.[39] In experimental studies it was observed that calpain activation causes the degradation of contractile and structural proteins, and subsequently contributes to structural cardiomyocyte remodeling (myolysis) and dysfunction and AF progression.[43,53] The role of calpain was confirmed in human AF. Here, a significant induction in calpain activation was observed in patients with permanent AF, compared to patients with paroxysmal AF and controls in sinus rhythm.[39] Furthermore, patients with permanent AF revealed induced amounts of myolysis which correlated significantly with calpain activity levels, suggesting a role for calpain in derailment of cardiomyocyte proteostasis, structural remodeling and AF progression.
Also, during AF, RhoA-GTPases are activated. RhoA-GTPases represent a family of small GTP-binding proteins that are involved in cell cytoskeleton organization, migration, transcription and proliferation. They have an important role as regulators of the actin cytoskeleton in cardiomyocytes[54] and trigger the initiation of AF.[55,56] RhoA-GTPases activation results in conduction disturbances and cardiac dysfunction.[57,58] Recent studies[38] revealed that in AF, RhoAGTPase become activated resulting in the activation of its downstream effector ROCK and thereby stimulate the polymerization of G-actin to filamentous F-actin stress bundles. These stress bundles impair calcium homeostasis and contribute to contractile dysfunction, cardiomyocyte remodeling and AF progression.[38]
Furthermore, recently it was found that histone deacetylases (HDACs), such as HDAC6, are implicated in AF-induced cardiomyocyte remodeling.[43] HDACs affect cardiomyocyte proteostasis by epigenetically regulating protein expression and modulating various cytoplasmic proteins, including α-tubulin, a structural protein from the microtubule network.[59-61] By using mutant constructs, AF-induced contractile dysfunction and structural remodeling was proven to be driven by HDAC6 via deacetylation of α-tubulin and finally breakdown of microtubules by calpain. This effect of HDAC6 was observed in tachypaced HL-1 atrial cardiomyocytes, Drosophila, dogs and confirmed in patients with permanent AF.[43] HDAC6 inhibition by tubacin conserved the microtubule homeostasis and prevented depolymerized α-tubulin from calpain-mediated degradation. These results indicate a key role for HDAC6 in the derailment of cardiomyocyte proteostasis in experimental and clinical AF.
So, three key pathways in AF-induced structural and functional remodeling have been identified, and all these pathways impair a healthy proteostasis of the cardiomyocyte.
Induction Of HSPs Normalize Proteostasis
To maintain a good functioning PQC system, numerous chaperones are expressed to ensure a healthy cardiomyocyte proteostasis.[38] HSPs are under the control of heat shock transcription factor 1 (HSF1), and represent important chaperones in proteostatic control.[47,62] During excessive stress situations such as AF, HSP levels were found to become exhausted.[44] This finding suggests that upregulation of HSP levels might normalize proteostasis and improve cardiomyocyte function in AF. In clinical studies, induced HSP levels showed protection against AF initiation and progression. HSP70 atrial expression levels were found to correlate with reduced incidence of post-operative AF in patients in sinus rhythm undergoing cardiac surgery.[633,64] In another clinical study,[65] a potent Heat Shock Response (HSR) and high HSP27 levels have been associated with restoration of normal sinus rhythm in patients with permanent AF after mitral valve surgery. Higher atrial HSP27 levels were found to be related to shorter AF duration and less myolysis when comparing paroxysmal versus persistent AF and sinus rhythm.[44,66] These findings suggest that HSPs become activated after AF episodes, and exhaust in time in a stress related manner.[44] Consequently, PQC is lost and incorrect/damaged proteins accumulate in cardiomyocytes, inducing or accelerating remodeling, in turn resulting in AF progression and recurrence. Next to AF, also a loss of PQC is recognized to contribute to the deterioration of heart function, reduction of stress tolerance, and the possibility of reducing the threshold for manifestation of cardiac disease.[67]
Various in vitro and in vivo models for tachypacing-induced AF identified HSPs to protect against AF initiation and against the derailment of proteostasis and cardiomyocyte remodeling. HSPs increase SERCA activity and stimulate both the reuptake of Ca2+ into the sarcoplasmic reticulum and the removal of Ca2+ out of the cardiomyocyte via Na+/Ca2+ exchanger,[68] suggesting that HSPs attenuate AF progression by protecting against (tachypacing-induced) changes in calcium handling proteins. Several HSPs (including HSP27) were shown to reduce oxidative stress, thereby potentially preventing or restoring the redox status of the ion channels[69] and preventing damage to the actin cytoskeleton. This protective effect of HSP27 was found via direct binding to actin filaments and indirectly by preserving the redox status.[43,44,70-73] Reducing oxidative stress preserves proteostasis and electrophysiological and contractile function of the cardiomyocyte in AF. Moreover, HSPs prevent calpain activation[39,53] and thereby attenuate contractile protein degradation and conctractile dysfunction.
Deficiencies Of Present Therapy Of Atrial Fibrillation
Therapy of AF is aimed at either rhythm or rate control. Since AF induces electrical, structural, and contractile remodeling, therapy aimed at prevention or restoration of remodeling and consequently restoration of sinus rhythm should be the strategy of first choice.[74] The different AF treatment modalities include pharmacological therapy, electrical cardioversion (ECV), pacemaker implantation combined with His bundle ablation or surgical isolation of the pulmonary veins with or without additional linear lesions/substrate modification (endovascular or surgical). According to the Multiple Wavelet Theory, the stability of the fibrillatory process is determined by the number of simultaneously circulating wavelets. Anti-fibrillatory effects of class IA, IC and III drugs are based on widening of the excitable period (difference between AF cycle length and refractory period). When the excitable period widens, it is less likely that a fibrillation wave encounters atrial tissue, which is still refractory. This in turn decreases the degree of fractionation of fibrillation waves and subsequently also the number of fibrillation waves. It is most likely that when patients with AF have a variable degree of remodeling due to e.g. dissimilar underlying heart diseases or AF episodes of different durations anti-arrhythmic drugs will also widen the excitable gap to a variable degree. This in turn may explain differences in interindividual responses to anti-arrhythmic drugs. The acute success rate of intravenous chemical cardioversion (CCV) using various drugs including amiodarone and flecainide is 58-75%[75,76] for patients with paroxysmal or persistent AF and is highest when performed in AF 48 hours.[76] Immediate (prior to discharge) AF recurrences were observed in 3%[76] and AF relapsed in 30-40% of patients within one year with continuation of anti-arrhythmic drugs.[176] When CCV is unsuccessful, ECV is next treatment in line. Immediate restoration of sinus rhythm is achieved in 88-97%.[76-78] Comparable to CCV, AF recurrences are common; sinus rhythm is maintained for one year in only 40-60% of the patients.
Circumferential Pulmonary Vein Isolation (PVI), endovascular or surgical, is aimed isolating ectopic foci within the myocardial sleeves of the pulmonary veins. Endovascular PVI can be achieved with radiofrequency current, laser or cryothermal energy. Navigation of the ablation catheters can be performed either manually guided by fluoroscopy or electroanatomical mapping systems, or robotically using remote (non-) magnetic navigation systems.[79-81] Despite the promising acute success rates, one year AF free survival is approximately 40-50% and redo ablations are frequently performed.[79-82] This data is confirmed in a large meta-analysis by Ganesan et al.[83] In this study, the long-term success rate increased to 79,8%, however only after multiple ablation procedures. The overall complication rate associated with endovascular AF ablation is 5% including phrenic nerve palsy, pulmonary vein stenosis, pericardial effusion and cardiac tamponade.[82,84] From a theoretical point of view, PVI should be an effective treatment modality for patients with paroxysms of AF triggered by ectopic foci within the pulmonary veins. Recurrences of AF after pulmonary vein isolation can be due to incompleteness of circular lesions, conduction or an arrhythmogenic substrate located outside the pulmonary veins.[85] In addition, an arrhythmogenic substrate may also develop over time as a result of a progressive cardiomyopathy. Different ablation approaches targeting the assumed substrate of AF have therefore been developed in the past years[85] including ablation of ganglionated autonomic plexuses in epicardial fat pads or disruption of dominant rotors in the left or right atrium as recognized by high-frequency Complex Fractionated Atrial Electrograms (CFAE).[86] Wu et al.[87] concluded in a meta-analysis that CFAE ablation could reduce the recurrence of atrial tachycardia in patients with nonparoxysmal AF after a single procedure. This effect was not observed in patients with paroxysmal AF. The reported one year AF free survival after the first CFAE ablation is only 29% when performed as a standalone procedure[86] and 74% in CFAE ablation additional to PVI.[86,88] Endovascular ablation of the ganglionic plexi as a standalone procedure in patients with paroxysmal AF is associated with a significantly lower arrhythmia free survival when compared to the PVI.[89], [90] When performed additionally to (repeat) PVI in patients with persistent AF, 16 months success rate rises to 59%.[90] The recurrence rates of these (concomitant) substrate modifications are thus high, indicating that the arrhythmogenic substrate underlying persistence of AF was still not fully understood. Our Double Layer Hypothesis[22,24] provides the explanation why, in case the endo- and epicardial layers are electrically dissociated, ablative therapy is not successful anymore.
Future Diagnostic Tools
As large numbers of disorders are associated with AF and patients with AF reveal AF episodes of variable duration, it is most likely that there is a large degree of variation in the degree of atrial remodeling. In addition to this, within a patient, it is also likely that there is intraatrial variation in the degree of remodeling. Examples of regional differences in morphology of unipolar fibrillation potentials are shown in Figure 3. Hence, knowledge of the degree and extensiveness of the arrhythmogenic substrate in the individual patient is essential in order to evaluate a patient-tailored therapy for AF. For this purpose, we developed custom made mapping software (‘wave mapping’) which enabled visualization of the individual fibrillation waves and quantification of the fibrillatory process. By using this software, we compared electrophysiological properties of fibrillation waves recorded during induced AF in patients with normal atria (physiological AF) with persistent AF in patients with valvular heart disease (pathological AF) and demonstrated that electrical dissociation of atrial muscle bundles and epicardial breakthrough of fibrillation waves play a key role in development of the substrate of persistent AF (Figure 4).[24] In order to diagnose the arrhythmogenic substrate of AF in individual patients, we are currently evaluating a real-time, high resolution, multi-site epicardial mapping approach of the entire atria (Figure 5) as a novel diagnostic tool which can be applied as a routine procedure during cardiac surgery. An approach like this allows quantification of electrophysiological properties of the entire atria. In such manner, we study electropathology throughout the entire atria in patients with and without AF and with a diversity of underlying structural heart diseases. This novel mapping approach will not only be used to gain further insights into the arrhythmogenic substrate of AF, but will also be used to develop novel therapies or to improve existing treatment modalities. For example, it may guide ablative therapy when the arrhythmogenic substrate is confined to a circumscribed region. In addition, data acquired with this mapping approach will also provide the basis for development of less- or noninvasive mapping techniques.
Figure 3. Intra-individual variation in electrogram morphology. Typical examples of unipolar fibrillation electrograms recorded from the middle of respectively the right atrial appendage (RA), Bachmann’s Bundle (BB) and the pulmonary vein area (PV) obtained from a patient with mitral valve disease and persistent AF. In the right atrium, the fibrillation potentials contain a single deflection whereas fibrillation potentials recorded from Bachmann’s Bundle and the pulmonary vein area contain multiple deflections.

Figure 4. Inter-individual variation in characteristics of fibrillation waves. Examples of six consecutive wavemaps obtained from the right atrial free wall constructed during acute AF (upper panel) and persistent AF (lower panel); unipolar fibrillation electrograms recorded in the middle of the mapping area are shown on top. The mapping area activated by each individual fibrillation is represented by a color; every color indicates the moment of entrance in the mapping area (from red to purple); the arrows indicate the main trajectory of the fibrillation wave (black: peripheral fibrillation wave, white epicardial breakthrough wave). During acute AF, there are a fewer number of fibrillation waves and the patterns of activation are less complex, compared to persistent AF. In addition, ‘focal fibrillation waves’ occur more frequently during persistent AF.
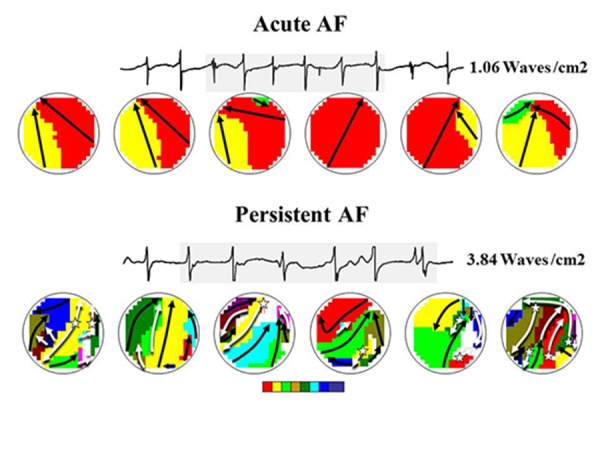
Figure 5. Atrial epicardial mapping Activation -, conduction block- and voltage maps constructed from Bachmann’s Bundle, right atrium, crista terminalis, pulmonary vein area, left atrioventricular groove, left atrial appendage during sinus rhythm, obtained from a patient with coronary artery disease. Electrograms recorded from the middle of the mapping area are shown on top. Arrows in the color-coded activation maps show the main trajectory of the excitation wave. Areas of slow conduction (<18cm/s) and conduction block (<30cm/s) are represented by respectively blue and red lines. Voltage maps show the peak-topeak amplitude of the atrial potentials.

The Future Novel Therapeutic Targets
Current therapies are directed at suppression of AF symptoms, but are not effective in attenuating AF remodeling , therefore there is a high need to identify novel therapeutic targets which will improve the clinical outcome. Novel targets include RhoA, calpain and HDAC6 inhibition, but also HSP induction. Recent studies revealed the important role of the RhoA/ROCK pathway activation in structural remodeling of cardiomyocytes during AF.[38] To maintain proper cardiac function, RhoA/ROCK inhibitors might be of therapeutic interest. Several RhoA and ROCK inhibitors have been developed. RhoA inhibitors CCG-1423 and Rhosin are studied in the preclinical phase.[91,92] Fasudil, Ezetimibe and AR-12286 are ROCK inhibitors currently studies in Phase II-IV trials for Raynaud’s phenomenon, vascular function study, atherosclerosis and glaucoma (Table 1).
Table 1. Novel therapeutic targets.
| Drug | Target | Phase | Indication | Ref (clinical trials.gov identifier) |
|---|---|---|---|---|
| GGA | HSP induction | Phase IV | Gastric ulcers Gastritis Gastric lesion | NCT01190657 NCT01547559 NCT01284647 NCT01397448 |
| NYK9354 | HSP induction | Pre-clinical | Atrial Fibrillation | (HoogstraBerends et al., 2012) |
| Leupeptin ALLN MDL-28170 A-705239 A-705253 | Calpain induction | Pre-clinical | reviewed in Cardiovascular Research (2012) 96, 23–31 | |
| Tubastatin | HDAC6 | Pre-clinical | Arthritis Anti-inflammatory | (Vishwakarma et al., 2013) |
| ACY-1215 | HDAC6 | Phase I/II | Myeloma | NCT01323751 NCT01583283 |
| Fasudil Ezetimibe AR-12286 | ROCK | Phase III Phase II Phase II Phase IV Phase II | Raynaud’s Phenomenon Vascular function study Atherosclerosis Atherosclerosis Glaucoma | NCT00498615 NCT00120718 NCT00670202 NCT00560170 NCT01936389 |
| CCG-1423 Rhosin | Rho | Pre-clinical | (Evelyn, et al., 2007) (Shang, et al.,2012) |
Calpain activation during AF causes the degradation of contractile and structural proteins, resulting in myolysis.[38,39,43] In vitro studies showed that inhibitors of calpain conserve the cardiomyocyte structure and function and therefore might have beneficial effects in the treatment of AF.[1,53] Various calpain inhibitors have been developed and preclinically studied. Disadvantages of the current developed inhibitors are that they show poor selectivity for subtypes of calpain and often have a high LogP value and therefore are hard to dissolve in aqueous solutions.[93]
HDAC6 inhibition, by tubacin, conserves-tubulin proteostasis, and prevents its degradation by calpain 1 and thereby protects against loss of calcium transient and cardiac remodeling in experimental model systems for AF. As tubacin is not suitable for in vivo studies due to low drug-likeness,[94] other promising HDAC6 inhibitors, such as tubastatin A and ACY-1215 have been recently developed[94-96] (Table 1). Interestingly, tubastatin A showed to protect against tachypacinginduced cardiac remodeling in a canine model for AF,[52] supporting the use of HDAC6 inhibitors as a novel therapeutic approach in AF.
Promoting maintenance of proteostasis by revitalization of the PQC system may prevent the derailment of proteostasis and structural and functional remodeling in AF. Interestingly, the heat shock response as part of the PQC system can be pharmacologically boosted, and consequently cardiac remodeling may be prevented, halted or even be restored. Indeed, as depicted earlier, increasing HSP expression, by either pharmacologic compounds or molecular biological means, displays cardioprotective effects in various models for AF and in patients. HSP induction provided protection against loss of actin proteostasis by reducing RhoA-GTPase-induced remodeling[38] and against activation of calpain.[38,43,44,46,52,53] Furthermore, in canine models for AF progression, treatment with geranylgeranylacetone induced HSP expression and prevented AF initiation and progression by inhibition of the prolongation of the effective refractory period (ERP), shortening of APD and reductions in L-type + current and it revealed protective effects against atrial conduction abnormalities.[44,97]
Whether HSP induction also protects via HDAC inhibition is currently unknown. Of all HSP inducing compounds, GGA represents the most efficacious compound for the pharmacological induction of HSPs. GGA has already been applied clinically in Japan since 1984 as an antiulcer drug with no reported serious adverse reactions.[98-102] Due to high LogP value for GGA, high dosages might be needed, therefore, GGA derivatives are developed with improve pharmaco-chemical properties[103] (table 1). Induction of HSP is suggested to be the most promising therapeutic approach with pleiotropic protective effects
HSPs As Biomarkers
Following stress, HSPs get expressed intracellular, but can also be presented on the cell surface or released to the surroundings.[104] HSPs in serum may act as a biomarker to reveal the stage of AF. Elevated serum HSP60 levels were found in patients with acute myocardial infarction and seemed to be predictive for post-AMI adverse events.[105] Elevated serum HSP70 and HSP60 have found to correlate to the severity of metabolic syndrome-associated factors in postmenopausal women.[106] HSP60 and HSP70 were found to positively associate with severity of cardiovascular disease.[107-112] Patients with coronary artery disease (CAD) have revealed antibodies to HSP27 in serum,[113] but a correlation between antibody titers to HSP27 and the extent of CAD could not be found. Several studies have reported increased serum levels for HSP27 several hours after myocardial infarction[114,115] In another study, anti-HSP27 levels were found to be higher in patients with more advanced cardiac artery disease, making the authors to conclude that serum anti-HSP27 titers may be associated with the presence and severity of cardiac artery disease.[116] Anti-HSP27 titers measured in patients with stroke were found significantly elevated.[117] These findings suggest that the measurement of HSP levels in serum, may be useful as biomarkers of disease initiation, and progression.
Conclusions
AF naturally tends to progress from trigger dependent paroxysmal AF to a more substrate mediated (longstanding) persistent or permanent AF. Trigger focused treatments (endovascular or surgical PVI) might be successful in patients with paroxysmal AF, however this approach will not be sufficient for patients suffering from more advanced types of AF, who require substrate modification. Even treatments aimed at substrate modification, such as CFAE ablation, Cox maze III and ganglion ablation, are associated with AF recurrences. This implies insufficient understanding of the electrophysiological and structural changes which form a substrate underlying AF. Hence, as long as the electropathological substrate remains poorly understood, and the stage of electropathology cannot be evaluated, it is challenging to define the optimal approach per individual patient. Therefore, research is focused on the dissection of molecular mechanisms underlying electropathology. New findings indicate a role for derailment of cardiomyocyte proteostasis in AF progression and identified novel innovative targets for drug therapy. These targets are directed at the attenuation of electropathology and prevention of clinical AF progression. Since various drugs are already on in clinical phase II/III for other indications, it seems worthwhile to test some in clinical AF.
Acknowledgements
This work was supported by the LSH-Impulse grant (40-43100-98-008) and the Dutch Heart Foundation (2013T144, 2013T096 and 2011T046).
Disclosures
None.
References
- 1.Einthoven W. Le télécardiogramme. Arch Int Physiol. 1906;4:132–164. [Google Scholar]
- 2.Wilke Thomas, Groth Antje, Mueller Sabrina, Pfannkuche Matthias, Verheyen Frank, Linder Roland, Maywald Ulf, Bauersachs Rupert, Breithardt Günter. Incidence and prevalence of atrial fibrillation: an analysis based on 8.3 million patients. Europace. 2013 Apr;15 (4):486–93. doi: 10.1093/europace/eus333. [DOI] [PubMed] [Google Scholar]
- 3.Krijthe Bouwe P, Kunst Anton, Benjamin Emelia J, Lip Gregory Y H, Franco Oscar H, Hofman Albert, Witteman Jacqueline C M, Stricker Bruno H, Heeringa Jan. Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur. Heart J. 2013 Sep;34 (35):2746–51. doi: 10.1093/eurheartj/eht280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aizer Anthony, Gaziano J Michael, Cook Nancy R, Manson Joann E, Buring Julie E, Albert Christine M. Relation of vigorous exercise to risk of atrial fibrillation. Am. J. Cardiol. 2009 Jun 1;103 (11):1572–7. doi: 10.1016/j.amjcard.2009.01.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirsh Joel A, Walsh Edward P, Triedman John K. Prevalence of and risk factors for atrial fibrillation and intra-atrial reentrant tachycardia among patients with congenital heart disease. Am. J. Cardiol. 2002 Aug 1;90 (3):338–40. doi: 10.1016/s0002-9149(02)02480-3. [DOI] [PubMed] [Google Scholar]
- 6.Patel Nileshkumar J, Deshmukh Abhishek, Pant Sadip, Singh Vikas, Patel Nilay, Arora Shilpkumar, Shah Neeraj, Chothani Ankit, Savani Ghanshyambhai T, Mehta Kathan, Parikh Valay, Rathod Ankit, Badheka Apurva O, Lafferty James, Kowalski Marcin, Mehta Jawahar L, Mitrani Raul D, Viles-Gonzalez Juan F, Paydak Hakan. Contemporary trends of hospitalization for atrial fibrillation in the United States, 2000 through 2010: implications for healthcare planning. Circulation. 2014 Jun 10;129 (23):2371–9. doi: 10.1161/CIRCULATIONAHA.114.008201. [DOI] [PubMed] [Google Scholar]
- 7.Haissaguerre Michel, Hocini Meleze, Denis Arnaud, Shah Ashok J, Komatsu Yuki, Yamashita Seigo, Daly Matthew, Amraoui Sana, Zellerhoff Stephan, Picat Marie-Quitterie, Quotb Adam, Jesel Laurence, Lim Han, Ploux Sylvain, Bordachar Pierre, Attuel Guillaume, Meillet Valentin, Ritter Philippe, Derval Nicolas, Sacher Frederic, Bernus Olivier, Cochet Hubert, Jais Pierre, Dubois Remi. Driver domains in persistent atrial fibrillation. Circulation. 2014 Aug 12;130 (7):530–8. doi: 10.1161/CIRCULATIONAHA.113.005421. [DOI] [PubMed] [Google Scholar]
- 8.Konrad Torsten, Theis Cathrin, Mollnau Hanke, Sonnenschein Sebastian, Rostock Thomas. Body surface potential mapping for mapping and treatment of persistent atrial fibrillation. Herzschrittmacherther Elektrophysiol. 2014 Dec;25 (4):226–9. doi: 10.1007/s00399-014-0341-7. [DOI] [PubMed] [Google Scholar]
- 9.Guillem Maria S, Climent Andreu M, Millet Jose, Arenal Ángel, Fernández-Avilés Francisco, Jalife José, Atienza Felipe, Berenfeld Omer. Noninvasive localization of maximal frequency sites of atrial fibrillation by body surface potential mapping. Circ Arrhythm Electrophysiol. 2013 Apr;6 (2):294–301. doi: 10.1161/CIRCEP.112.000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodrigo Miguel, Guillem María S, Climent Andreu M, Pedrón-Torrecilla Jorge, Liberos Alejandro, Millet José, Fernández-Avilés Francisco, Atienza Felipe, Berenfeld Omer. Body surface localization of left and right atrial high-frequency rotors in atrial fibrillation patients: a clinical-computational study. Heart Rhythm. 2014 Sep;11 (9):1584–91. doi: 10.1016/j.hrthm.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MOE G K, ABILDSKOV J A. Atrial fibrillation as a self-sustaining arrhythmia independent of focal discharge. Am. Heart J. 1959 Jul;58 (1):59–70. doi: 10.1016/0002-8703(59)90274-1. [DOI] [PubMed] [Google Scholar]
- 12.MA Allessie. Experimental evaluation of Moe’s multiple wavelet hypothesis of atrial fibrillation. Cardiac Electrophysiology and Arrhythmias. 1985;0:265–276. [Google Scholar]
- 13.Allessie M A, Bonke F I, Schopman F J. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The "leading circle" concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ. Res. 1977 Jul;41 (1):9–18. doi: 10.1161/01.res.41.1.9. [DOI] [PubMed] [Google Scholar]
- 14.Allessie MA, Bonke FIM, Lammers WJEP, Rensma PL. The Wavelength of the Atrial Impulse and Re-Entrant Arrhythmias in the Conscious Dog. J Physiol-London. 1985;0:0–0. [Google Scholar]
- 15.Konings K T, Kirchhof C J, Smeets J R, Wellens H J, Penn O C, Allessie M A. High-density mapping of electrically induced atrial fibrillation in humans. Circulation. 1994 Apr;89 (4):1665–80. doi: 10.1161/01.cir.89.4.1665. [DOI] [PubMed] [Google Scholar]
- 16.Mandapati R, Skanes A, Chen J, Berenfeld O, Jalife J. Stable microreentrant sources as a mechanism of atrial fibrillation in the isolated sheep heart. Circulation. 2000 Jan 18;101 (2):194–9. doi: 10.1161/01.cir.101.2.194. [DOI] [PubMed] [Google Scholar]
- 17.Jalife José, Berenfeld Omer, Mansour Moussa. Mother rotors and fibrillatory conduction: a mechanism of atrial fibrillation. Cardiovasc. Res. 2002 May;54 (2):204–16. doi: 10.1016/s0008-6363(02)00223-7. [DOI] [PubMed] [Google Scholar]
- 18.Waldo Albert L. Mechanisms of atrial fibrillation. J. Cardiovasc. Electrophysiol. 2003 Dec;14 (12 Suppl):S267–74. doi: 10.1046/j.1540-8167.2003.90401.x. [DOI] [PubMed] [Google Scholar]
- 19.MOE G K, RHEINBOLDT W C, ABILDSKOV J A. A COMPUTER MODEL OF ATRIAL FIBRILLATION. Am. Heart J. 1964 Feb;67 ():200–20. doi: 10.1016/0002-8703(64)90371-0. [DOI] [PubMed] [Google Scholar]
- 20.Narayan Sanjiv M, Krummen David E, Shivkumar Kalyanam, Clopton Paul, Rappel Wouter-Jan, Miller John M. Treatment of atrial fibrillation by the ablation of localized sources: CONFIRM (Conventional Ablation for Atrial Fibrillation With or Without Focal Impulse and Rotor Modulation) trial. J. Am. Coll. Cardiol. 2012 Aug 14;60 (7):628–36. doi: 10.1016/j.jacc.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis T. Oliver-Sharpey Lectures ON THE NATURE OF FLUTTER AND FIBRILLATION OF THE AURICLE. Br Med J. 1921 Apr 23;1 (3147):590–3. doi: 10.1136/bmj.1.3147.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Groot Natasja M S, Houben Richard P M, Smeets Joep L, Boersma Eric, Schotten Ulrich, Schalij Martin J, Crijns Harry, Allessie Maurits A. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: epicardial breakthrough. Circulation. 2010 Oct 26;122 (17):1674–82. doi: 10.1161/CIRCULATIONAHA.109.910901. [DOI] [PubMed] [Google Scholar]
- 23.Lee Geoffrey, Kumar Saurabh, Teh Andrew, Madry Andrew, Spence Steven, Larobina Marco, Goldblatt John, Brown Robin, Atkinson Victoria, Moten Simon, Morton Joseph B, Sanders Prashanthan, Kistler Peter M, Kalman Jonathan M. Epicardial wave mapping in human long-lasting persistent atrial fibrillation: transient rotational circuits, complex wavefronts, and disorganized activity. Eur. Heart J. 2014 Jan;35 (2):86–97. doi: 10.1093/eurheartj/eht267. [DOI] [PubMed] [Google Scholar]
- 24.Allessie Maurits A, de Groot Natasja M S, Houben Richard P M, Schotten Ulrich, Boersma Eric, Smeets Joep L, Crijns Harry J. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: longitudinal dissociation. Circ Arrhythm Electrophysiol. 2010 Dec;3 (6):606–15. doi: 10.1161/CIRCEP.109.910125. [DOI] [PubMed] [Google Scholar]
- 25.Wijffels M C, Kirchhof C J, Dorland R, Allessie M A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995 Oct 1;92 (7):1954–68. doi: 10.1161/01.cir.92.7.1954. [DOI] [PubMed] [Google Scholar]
- 26.Goette A, Honeycutt C, Langberg J J. Electrical remodeling in atrial fibrillation. Time course and mechanisms. Circulation. 1996 Dec 1;94 (11):2968–74. doi: 10.1161/01.cir.94.11.2968. [DOI] [PubMed] [Google Scholar]
- 27.Ausma J, Dispersyn G D, Duimel H, Thoné F, Ver Donck L, Allessie M A, Borgers M. Changes in ultrastructural calcium distribution in goat atria during atrial fibrillation. J. Mol. Cell. Cardiol. 2000 Mar;32 (3):355–64. doi: 10.1006/jmcc.1999.1090. [DOI] [PubMed] [Google Scholar]
- 28.Brundel B J, Van Gelder I C, Henning R H, Tieleman R G, Tuinenburg A E, Wietses M, Grandjean J G, Van Gilst W H, Crijns H J. Ion channel remodeling is related to intraoperative atrial effective refractory periods in patients with paroxysmal and persistent atrial fibrillation. Circulation. 2001 Feb 6;103 (5):684–90. doi: 10.1161/01.cir.103.5.684. [DOI] [PubMed] [Google Scholar]
- 29.Qi Xiao Yan, Yeh Yung-Hsin, Xiao Ling, Burstein Brett, Maguy Ange, Chartier Denis, Villeneuve Louis R, Brundel Bianca J J M, Dobrev Dobromir, Nattel Stanley. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ. Res. 2008 Oct 10;103 (8):845–54. doi: 10.1161/CIRCRESAHA.108.175463. [DOI] [PubMed] [Google Scholar]
- 30.Schotten Ulrich, Duytschaever Mattias, Ausma Jannie, Eijsbouts Sabine, Neuberger Hans-Ruprecht, Allessie Maurits. Electrical and contractile remodeling during the first days of atrial fibrillation go hand in hand. Circulation. 2003 Mar 18;107 (10):1433–9. doi: 10.1161/01.cir.0000055314.10801.4f. [DOI] [PubMed] [Google Scholar]
- 31.Brundel B J, van Gelder I C, Henning R H, Tuinenburg A E, Deelman L E, Tieleman R G, Grandjean J G, van Gilst W H, Crijns H J. Gene expression of proteins influencing the calcium homeostasis in patients with persistent and paroxysmal atrial fibrillation. Cardiovasc. Res. 1999 May;42 (2):443–54. doi: 10.1016/s0008-6363(99)00045-0. [DOI] [PubMed] [Google Scholar]
- 32.Wang Zhiguo. Role of redox state in modulation of ion channel function by fatty acids and phospholipids. Br. J. Pharmacol. 2003 Jun;139 (4):681–3. doi: 10.1038/sj.bjp.0705307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobrev D, Voigt N. Ion channel remodelling in atrial fibrillation. European Cardiology. 2011;0:97–103. [Google Scholar]
- 34.Anderson Mark E. Calmodulin kinase and L-type calcium channels; a recipe for arrhythmias? Trends Cardiovasc. Med. 2004 May;14 (4):152–61. doi: 10.1016/j.tcm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 35.Christ T, Boknik P, Wöhrl S, Wettwer E, Graf E M, Bosch R F, Knaut M, Schmitz W, Ravens U, Dobrev D. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004 Oct 26;110 (17):2651–7. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- 36.Greiser Maura, Halaszovich Christian R, Frechen Dirk, Boknik Peter, Ravens Ursula, Dobrev Dobromir, Lückhoff Andreas, Schotten Ulrich. Pharmacological evidence for altered src kinase regulation of I (Ca,L) in patients with chronic atrial fibrillation. Naunyn Schmiedebergs Arch. Pharmacol. 2007 Aug;375 (6):383–92. doi: 10.1007/s00210-007-0174-6. [DOI] [PubMed] [Google Scholar]
- 37.Dobrev Dobromir, Nattel Stanley. Calcium handling abnormalities in atrial fibrillation as a target for innovative therapeutics. J. Cardiovasc. Pharmacol. 2008 Oct;52 (4):293–9. doi: 10.1097/FJC.0b013e318171924d. [DOI] [PubMed] [Google Scholar]
- 38.Ke Lei, Meijering Roelien A M, Hoogstra-Berends Femke, Mackovicova Katarina, Vos Michel J, Van Gelder Isabelle C, Henning Robert H, Kampinga Harm H, Brundel Bianca J J M. HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS ONE. 2011;6 (6) doi: 10.1371/journal.pone.0020395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brundel Bianca J J M, Ausma Jannie, van Gelder Isabelle C, Van der Want Johan J L, van Gilst Wiek H, Crijns Harry J G M, Henning Robert H. Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc. Res. 2002 May;54 (2):380–9. doi: 10.1016/s0008-6363(02)00289-4. [DOI] [PubMed] [Google Scholar]
- 40.Sherman A J, Klocke F J, Decker R S, Decker M L, Kozlowski K A, Harris K R, Hedjbeli S, Yaroshenko Y, Nakamura S, Parker M A, Checchia P A, Evans D B. Myofibrillar disruption in hypocontractile myocardium showing perfusion-contraction matches and mismatches. Am. J. Physiol. Heart Circ. Physiol. 2000 Apr;278 (4):H1320–34. doi: 10.1152/ajpheart.2000.278.4.H1320. [DOI] [PubMed] [Google Scholar]
- 41.Bito Virginie, Heinzel Frank R, Weidemann Frank, Dommke Christophe, van der Velden Jolanda, Verbeken Erik, Claus Piet, Bijnens Bart, De Scheerder Ivan, Stienen Ger J M, Sutherland George R, Sipido Karin R. Cellular mechanisms of contractile dysfunction in hibernating myocardium. Circ. Res. 2004 Apr 2;94 (6):794–801. doi: 10.1161/01.RES.0000124934.84048.DF. [DOI] [PubMed] [Google Scholar]
- 42.Todd Derick M, Fynn Simon P, Walden Andrew P, Hobbs W Julian, Arya Sanjay, Garratt Clifford J. Repetitive 4-week periods of atrial electrical remodeling promote stability of atrial fibrillation: time course of a second factor involved in the self-perpetuation of atrial fibrillation. Circulation. 2004 Mar 23;109 (11):1434–9. doi: 10.1161/01.CIR.0000124006.84596.D9. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Deli, Wu Chia-Tung, Qi XiaoYan, Meijering Roelien A M, Hoogstra-Berends Femke, Tadevosyan Artavazd, Cubukcuoglu Deniz Gunseli, Durdu Serkan, Akar Ahmet Ruchan, Sibon Ody C M, Nattel Stanley, Henning Robert H, Brundel Bianca J J M. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of α-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014 Jan 21;129 (3):346–58. doi: 10.1161/CIRCULATIONAHA.113.005300. [DOI] [PubMed] [Google Scholar]
- 44.Brundel Bianca J J M, Henning Robert H, Ke Lei, van Gelder Isabelle C, Crijns Harry J G M, Kampinga Harm H. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J. Mol. Cell. Cardiol. 2006 Sep;41 (3):555–62. doi: 10.1016/j.yjmcc.2006.06.068. [DOI] [PubMed] [Google Scholar]
- 45.Meijering Roelien A M, Zhang Deli, Hoogstra-Berends Femke, Henning Robert H, Brundel Bianca J J M. Loss of proteostatic control as a substrate for atrial fibrillation: a novel target for upstream therapy by heat shock proteins. Front Physiol. 2012;3 () doi: 10.3389/fphys.2012.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brundel Bianca J J M, Shiroshita-Takeshita Akiko, Qi Xiaoyan, Yeh Yung-Hsin, Chartier Denis, van Gelder Isabelle C, Henning Robert H, Kampinga Harm H, Nattel Stanley. Induction of heat shock response protects the heart against atrial fibrillation. Circ. Res. 2006 Dec 8;99 (12):1394–402. doi: 10.1161/01.RES.0000252323.83137.fe. [DOI] [PubMed] [Google Scholar]
- 47.Balch William E, Morimoto Richard I, Dillin Andrew, Kelly Jeffery W. Adapting proteostasis for disease intervention. Science. 2008 Feb 15;319 (5865):916–9. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 48.Powers Evan T, Balch William E. Diversity in the origins of proteostasis networks--a driver for protein function in evolution. Nat. Rev. Mol. Cell Biol. 2013 Apr;14 (4):237–48. doi: 10.1038/nrm3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Xuejun, Robbins Jeffrey. Heart failure and protein quality control. Circ. Res. 2006 Dec 8;99 (12):1315–28. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 50.Galli Alessio. Proteotoxicity and cardiac dysfunction. N. Engl. J. Med. 2013 May 2;368 (18):1754–5. doi: 10.1056/NEJMc1302511. [DOI] [PubMed] [Google Scholar]
- 51.Meijering Roelien A M, Henning Robert H, Brundel Bianca J J M. Reviving the protein quality control system: therapeutic target for cardiac disease in the elderly. Trends Cardiovasc. Med. 2015 Apr;25 (3):243–7. doi: 10.1016/j.tcm.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Deli, Ke Lei, Mackovicova Katarina, Van Der Want Johannes J L, Sibon Ody C M, Tanguay Robert M, Morrow Genevieve, Henning Robert H, Kampinga Harm H, Brundel Bianca J J M. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J. Mol. Cell. Cardiol. 2011 Sep;51 (3):381–9. doi: 10.1016/j.yjmcc.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 53.Ke Lei, Qi Xiao Yan, Dijkhuis Anne-Jan, Chartier Denis, Nattel Stanley, Henning Robert H, Kampinga Harm H, Brundel Bianca J J M. Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J. Mol. Cell. Cardiol. 2008 Nov;45 (5):685–93. doi: 10.1016/j.yjmcc.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 54.Brown Joan Heller, Del Re Dominic P, Sussman Mark A. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ. Res. 2006 Mar 31;98 (6):730–42. doi: 10.1161/01.RES.0000216039.75913.9e. [DOI] [PubMed] [Google Scholar]
- 55.Sah V P, Minamisawa S, Tam S P, Wu T H, Dorn G W, Ross J, Chien K R, Brown J H. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J. Clin. Invest. 1999 Jun;103 (12):1627–34. doi: 10.1172/JCI6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adam Oliver, Frost Gregg, Custodis Florian, Sussman Mark A, Schäfers Hans-Joachim, Böhm Michael, Laufs Ulrich. Role of Rac1 GTPase activation in atrial fibrillation. J. Am. Coll. Cardiol. 2007 Jul 24;50 (4):359–67. doi: 10.1016/j.jacc.2007.03.041. [DOI] [PubMed] [Google Scholar]
- 57.Reil Jan-Christian, Hohl Mathias, Oberhofer Martin, Kazakov Andrey, Kaestner Lars, Mueller Patrick, Adam Oliver, Maack Christoph, Lipp Peter, Mewis Christian, Allessie Maurits, Laufs Ulrich, Böhm Michael, Neuberger Hans-Ruprecht. Cardiac Rac1 overexpression in mice creates a substrate for atrial arrhythmias characterized by structural remodelling. Cardiovasc. Res. 2010 Aug 1;87 (3):485–93. doi: 10.1093/cvr/cvq079. [DOI] [PubMed] [Google Scholar]
- 58.Ogata Takehiro, Ueyama Tomomi, Isodono Koji, Tagawa Masashi, Takehara Naofumi, Kawashima Tsuneaki, Harada Koichiro, Takahashi Tomosaburo, Shioi Tetsuo, Matsubara Hiroaki, Oh Hidemasa. MURC, a muscle-restricted coiled-coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol. Cell. Biol. 2008 May;28 (10):3424–36. doi: 10.1128/MCB.02186-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hubbert Charlotte, Guardiola Amaris, Shao Rong, Kawaguchi Yoshiharu, Ito Akihiro, Nixon Andrew, Yoshida Minoru, Wang Xiao-Fan, Yao Tso-Pang. HDAC6 is a microtubule-associated deacetylase. Nature. 2002 May 23;417 (6887):455–8. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 60.Haggarty Stephen J, Koeller Kathryn M, Wong Jason C, Grozinger Christina M, Schreiber Stuart L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U.S.A. 2003 Apr 15;100 (8):4389–94. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsuyama Akihisa, Shimazu Tadahiro, Sumida Yuko, Saito Akiko, Yoshimatsu Yasuhiro, Seigneurin-Berny Daphné, Osada Hiroyuki, Komatsu Yasuhiko, Nishino Norikazu, Khochbin Saadi, Horinouchi Sueharu, Yoshida Minoru. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002 Dec 16;21 (24):6820–31. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Powers Evan T, Morimoto Richard I, Dillin Andrew, Kelly Jeffery W, Balch William E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009;78 ():959–91. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 63.St Rammos Kyriakos, Koullias George J, Hassan Moustafa O, Argyrakis Nikolaos P, Voucharas Christos G, Scarupa Steven J, Cowte Tomas G. Low preoperative HSP70 atrial myocardial levels correlate significantly with high incidence of postoperative atrial fibrillation after cardiac surgery. Cardiovasc Surg. 2002 Jun;10 (3):228–32. doi: 10.1016/s0967-2109(01)00138-7. [DOI] [PubMed] [Google Scholar]
- 64.Mandal Kaushik, Torsney Evelyn, Poloniecki Jan, Camm A John, Xu Qingbo, Jahangiri Marjan. Association of high intracellular, but not serum, heat shock protein 70 with postoperative atrial fibrillation. Ann. Thorac. Surg. 2005 Mar;79 (3):865–71. doi: 10.1016/j.athoracsur.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 65.Cao Hailong, Xue Lei, Xu Xiaohan, Wu Yanhu, Zhu Jinfu, Chen Liang, Chen Duan, Chen Yijiang. Heat shock proteins in stabilization of spontaneously restored sinus rhythm in permanent atrial fibrillation patients after mitral valve surgery. Cell Stress Chaperones. 2011 Sep;16 (5):517–28. doi: 10.1007/s12192-011-0263-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang Miao, Tan Hao, Cheng Longxian, He Meian, Wei Qingyi, Tanguay Robert M, Wu Tangchun. Expression of heat shock proteins in myocardium of patients with atrial fibrillation. Cell Stress Chaperones. 2007;12 (2):142–50. doi: 10.1379/CSC-253R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Willis Monte S, Patterson Cam. Proteotoxicity and cardiac dysfunction. N. Engl. J. Med. 2013 May 2;368 (18) doi: 10.1056/NEJMc1302511. [DOI] [PubMed] [Google Scholar]
- 68.Liu Jing, Kam Kenneth W L, Borchert Gudrun H, Kravtsov Gennadi M, Ballard Heather J, Wong Tak Ming. Further study on the role of HSP70 on Ca2+ homeostasis in rat ventricular myocytes subjected to simulated ischemia. Am. J. Physiol., Cell Physiol. 2006 Feb;290 (2):C583–91. doi: 10.1152/ajpcell.00145.2005. [DOI] [PubMed] [Google Scholar]
- 69.Kalmar Bernadett, Greensmith Linda. Induction of heat shock proteins for protection against oxidative stress. Adv. Drug Deliv. Rev. 2009 Apr 28;61 (4):310–8. doi: 10.1016/j.addr.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 70.Sugiyama Y, Suzuki A, Kishikawa M, Akutsu R, Hirose T, Waye M M, Tsui S K, Yoshida S, Ohno S. Muscle develops a specific form of small heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. J. Biol. Chem. 2000 Jan 14;275 (2):1095–104. doi: 10.1074/jbc.275.2.1095. [DOI] [PubMed] [Google Scholar]
- 71.Mounier Nicole, Arrigo André-Patrick. Actin cytoskeleton and small heat shock proteins: how do they interact? Cell Stress Chaperones. 2002 Apr;7 (2):167–76. doi: 10.1379/1466-1268(2002)007<0167:acashs>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Golenhofen Nikola, Perng Ming Der, Quinlan Roy A, Drenckhahn Detlev. Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem. Cell Biol. 2004 Nov;122 (5):415–25. doi: 10.1007/s00418-004-0711-z. [DOI] [PubMed] [Google Scholar]
- 73.Salinthone Sonemany, Tyagi Manoj, Gerthoffer William T. Small heat shock proteins in smooth muscle. Pharmacol. Ther. 2008 Jul;119 (1):44–54. doi: 10.1016/j.pharmthera.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Camm A John, Lip Gregory Y H, De Caterina Raffaele, Savelieva Irene, Atar Dan, Hohnloser Stefan H, Hindricks Gerhard, Kirchhof Paulus. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur. Heart J. 2012 Nov;33 (21):2719–47. doi: 10.1093/eurheartj/ehs253. [DOI] [PubMed] [Google Scholar]
- 75.de Paola Angelo A V, Figueiredo Edilberto, Sesso Ricardo, Veloso Henrique H, Nascimento Luiz Olympio T. Effectiveness and costs of chemical versus electrical cardioversion of atrial fibrillation. Int. J. Cardiol. 2003 Apr;88 (2-3):157–66. doi: 10.1016/s0167-5273(02)00380-7. [DOI] [PubMed] [Google Scholar]
- 76.Pisters Ron, Nieuwlaat Robby, Prins Martin H, Le Heuzey Jean-Yves, Maggioni Aldo P, Camm A John, Crijns Harry J G M. Clinical correlates of immediate success and outcome at 1-year follow-up of real-world cardioversion of atrial fibrillation: the Euro Heart Survey. Europace. 2012 May;14 (5):666–74. doi: 10.1093/europace/eur406. [DOI] [PubMed] [Google Scholar]
- 77.LOWN B, AMARASINGHAM R, NEUMAN J. New method for terminating cardiac arrhythmias. Use of synchronized capacitor discharge. JAMA. 1962 Nov 3;182 ():548–55. [PubMed] [Google Scholar]
- 78.Camm A John, Lip Gregory Y H, De Caterina Raffaele, Savelieva Irene, Atar Dan, Hohnloser Stefan H, Hindricks Gerhard, Kirchhof Paulus. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation--developed with the special contribution of the European Heart Rhythm Association. Europace. 2012 Oct;14 (10):1385–413. doi: 10.1093/europace/eus305. [DOI] [PubMed] [Google Scholar]
- 79.Di Biase Luigi, Fahmy Tamer S, Patel Dimpi, Bai Rong, Civello Kenneth, Wazni Oussama M, Kanj Mohamed, Elayi Claude S, Ching Chi Keong, Khan Mohamed, Popova Lucie, Schweikert Robert A, Cummings Jennifer E, Burkhardt J David, Martin David O, Bhargava Mandeep, Dresing Thomas, Saliba Walid, Arruda Mauricio, Natale Andrea. Remote magnetic navigation: human experience in pulmonary vein ablation. J. Am. Coll. Cardiol. 2007 Aug 28;50 (9):868–74. doi: 10.1016/j.jacc.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 80.Lüthje Lars, Vollmann Dirk, Seegers Joachim, Dorenkamp Marc, Sohns Christian, Hasenfuss Gerd, Zabel Markus. Remote magnetic versus manual catheter navigation for circumferential pulmonary vein ablation in patients with atrial fibrillation. Clin Res Cardiol. 2011 Nov;100 (11):1003–11. doi: 10.1007/s00392-011-0333-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saliba Walid, Reddy Vivek Y, Wazni Oussama, Cummings Jennifer E, Burkhardt J David, Haissaguerre Michel, Kautzner Josef, Peichl Petr, Neuzil Petr, Schibgilla Volker, Noelker Georg, Brachmann Johannes, Di Biase Luigi, Barrett Conor, Jais Pierre, Natale Andrea. Atrial fibrillation ablation using a robotic catheter remote control system: initial human experience and long-term follow-up results. J. Am. Coll. Cardiol. 2008 Jun 24;51 (25):2407–11. doi: 10.1016/j.jacc.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 82.Bordignon Stefano, Chun K R Julian, Gunawardene Melanie, Fuernkranz Alexander, Urban Verena, Schulte-Hahn Britta, Nowak Bernd, Schmidt Boris. Comparison of balloon catheter ablation technologies for pulmonary vein isolation: the laser versus cryo study. J. Cardiovasc. Electrophysiol. 2013 Sep;24 (9):987–94. doi: 10.1111/jce.12192. [DOI] [PubMed] [Google Scholar]
- 83.Ganesan Anand N, Shipp Nicholas J, Brooks Anthony G, Kuklik Pawel, Lau Dennis H, Lim Han S, Sullivan Thomas, Roberts-Thomson Kurt C, Sanders Prashanthan. Long-term outcomes of catheter ablation of atrial fibrillation: a systematic review and meta-analysis. J Am Heart Assoc. 2013 Apr;2 (2) doi: 10.1161/JAHA.112.004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bhat Tariq, Baydoun Hassan, Asti Deepak, Rijal Jharendra, Teli Sumaya, Tantray Mohmad, Bhat Hilal, Kowalski Marcin. Major complications of cryoballoon catheter ablation for atrial fibrillation and their management. Expert Rev Cardiovasc Ther. 2014 Sep;12 (9):1111–8. doi: 10.1586/14779072.2014.925802. [DOI] [PubMed] [Google Scholar]
- 85.Yaksh A, Kik C, Knops P, Roos-Hesselink J W, Bogers A J J C, Zijlstra F, Allessie M, de Groot N M S. Atrial fibrillation: to map or not to map? Neth Heart J. 2014 Jun;22 (6):259–66. doi: 10.1007/s12471-013-0481-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nademanee Koonlawee, McKenzie John, Kosar Erol, Schwab Mark, Sunsaneewitayakul Buncha, Vasavakul Thaveekiat, Khunnawat Chotikorn, Ngarmukos Tachapong. A new approach for catheter ablation of atrial fibrillation: mapping of the electrophysiologic substrate. J. Am. Coll. Cardiol. 2004 Jun 2;43 (11):2044–53. doi: 10.1016/j.jacc.2003.12.054. [DOI] [PubMed] [Google Scholar]
- 87.Wu Shao-Hui, Jiang Wei-Feng, Gu Jun, Zhao Liang, Wang Yuan-Long, Liu Yu-Gang, Zhou Li, Gu Jia-Ning, Xu Kai, Liu Xu. Benefits and risks of additional ablation of complex fractionated atrial electrograms for patients with atrial fibrillation: a systematic review and meta-analysis. Int. J. Cardiol. 2013 Oct 25;169 (1):35–43. doi: 10.1016/j.ijcard.2013.08.083. [DOI] [PubMed] [Google Scholar]
- 88.Verma A, Sanders P, Champagne J, Macle L, Nair G, Calkins H, Wilber D. Selective Complex Fractionated Electrogram Targeting For Atrial Fibrillation Study (Select AF): A Multicenter, Randomized Trial. Circulation. 2012;0:0–0. doi: 10.1161/CIRCEP.113.000890. [DOI] [PubMed] [Google Scholar]
- 89.Mikhaylov Evgeny, Kanidieva Anastasia, Sviridova Nina, Abramov Mikhail, Gureev Sergey, Szili-Torok Tamas, Lebedev Dmitry. Outcome of anatomic ganglionated plexi ablation to treat paroxysmal atrial fibrillation: a 3-year follow-up study. Europace. 2011 Mar;13 (3):362–70. doi: 10.1093/europace/euq416. [DOI] [PubMed] [Google Scholar]
- 90.Pokushalov Evgeny, Romanov Alexander, Artyomenko Sergey, Turov Alex, Shugayev Pavel, Shirokova Natalya, Katritsis Demosthenes G. Ganglionated plexi ablation for longstanding persistent atrial fibrillation. Europace. 2010 Mar;12 (3):342–6. doi: 10.1093/europace/euq014. [DOI] [PubMed] [Google Scholar]
- 91.Evelyn Chris R, Wade Susan M, Wang Qin, Wu Mei, Iñiguez-Lluhí Jorge A, Merajver Sofia D, Neubig Richard R. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol. Cancer Ther. 2007 Aug;6 (8):2249–60. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 92.Shang Xun, Marchioni Fillipo, Sipes Nisha, Evelyn Chris R, Jerabek-Willemsen Moran, Duhr Stefan, Seibel William, Wortman Matthew, Zheng Yi. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem. Biol. 2012 Jun 22;19 (6):699–710. doi: 10.1016/j.chembiol.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Inserte Javier, Hernando Victor, Garcia-Dorado David. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2012 Oct 1;96 (1):23–31. doi: 10.1093/cvr/cvs232. [DOI] [PubMed] [Google Scholar]
- 94.Butler Kyle V, Kalin Jay, Brochier Camille, Vistoli Guilio, Langley Brett, Kozikowski Alan P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010 Aug 11;132 (31):10842–6. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.d'Ydewalle Constantin, Krishnan Jyothsna, Chiheb Driss M, Van Damme Philip, Irobi Joy, Kozikowski Alan P, Vanden Berghe Pieter, Timmerman Vincent, Robberecht Wim, Van Den Bosch Ludo. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 2011 Aug;17 (8):968–74. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- 96.Santo Loredana, Hideshima Teru, Kung Andrew L, Tseng Jen-Chieh, Tamang David, Yang Min, Jarpe Matthew, van Duzer John H, Mazitschek Ralph, Ogier Walter C, Cirstea Diana, Rodig Scott, Eda Homare, Scullen Tyler, Canavese Miriam, Bradner James, Anderson Kenneth C, Jones Simon S, Raje Noopur. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012 Mar 15;119 (11):2579–89. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sakabe Masao, Shiroshita-Takeshita Akiko, Maguy Ange, Brundel Bianca J J M, Fujiki Akira, Inoue Hiroshi, Nattel Stanley. Effects of a heat shock protein inducer on the atrial fibrillation substrate caused by acute atrial ischaemia. Cardiovasc. Res. 2008 Apr 1;78 (1):63–70. doi: 10.1093/cvr/cvn019. [DOI] [PubMed] [Google Scholar]
- 98.Murakami M, Oketani K, Fujisaki H, Wakabayashi T, Ohgo T. Antiulcer effect of geranylgeranylacetone, a new acyclic polyisoprenoid on experimentally induced gastric and duodenal ulcers in rats. Arzneimittelforschung. 1981;31 (5):799–804. [PubMed] [Google Scholar]
- 99.Unoshima Masako, Iwasaka Hideo, Eto Junko, Takita-Sonoda Yoshiko, Noguchi Takayuki, Nishizono Akira. Antiviral effects of geranylgeranylacetone: enhancement of MxA expression and phosphorylation of PKR during influenza virus infection. Antimicrob. Agents Chemother. 2003 Sep;47 (9):2914–21. doi: 10.1128/AAC.47.9.2914-2921.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Katsuno Masahisa, Sang Chen, Adachi Hiroaki, Minamiyama Makoto, Waza Masahiro, Tanaka Fumiaki, Doyu Manabu, Sobue Gen. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc. Natl. Acad. Sci. U.S.A. 2005 Nov 15;102 (46):16801–6. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yanaka Akinori, Zhang Songhua, Sato Daisuke, Tauchi Masafumi, Suzuki Hideo, Shibahara Takeshi, Matsui Hirofumi, Nakahara Akira, Hyodo Ichinosuke. Geranylgeranylacetone protects the human gastric mucosa from diclofenac-induced injury via induction of heat shock protein 70. Digestion. 2007;75 (2-3):148–55. doi: 10.1159/000106756. [DOI] [PubMed] [Google Scholar]
- 102.Fujimura Noritaka, Jitsuiki Daisuke, Maruhashi Tatsuya, Mikami Shinsuke, Iwamoto Yumiko, Kajikawa Masato, Chayama Kazuaki, Kihara Yasuki, Noma Kensuke, Goto Chikara, Higashi Yukihito. Geranylgeranylacetone, heat shock protein 90/AMP-activated protein kinase/endothelial nitric oxide synthase/nitric oxide pathway, and endothelial function in humans. Arterioscler. Thromb. Vasc. Biol. 2012 Jan;32 (1):153–60. doi: 10.1161/ATVBAHA.111.237263. [DOI] [PubMed] [Google Scholar]
- 103.Hoogstra-Berends Femke, Meijering Roelien A M, Zhang Deli, Heeres André, Loen Lizette, Seerden Jean-Paul, Kuipers Irma, Kampinga Harm H, Henning Robert H, Brundel Bianca J J M. Heat shock protein-inducing compounds as therapeutics to restore proteostasis in atrial fibrillation. Trends Cardiovasc. Med. 2012 Apr;22 (3):62–8. doi: 10.1016/j.tcm.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 104.van Oosten-Hawle Patricija, Morimoto Richard I. Organismal proteostasis: role of cell-nonautonomous regulation and transcellular chaperone signaling. Genes Dev. 2014 Jul 15;28 (14):1533–43. doi: 10.1101/gad.241125.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Novo Giuseppina, Cappello Francesco, Rizzo Manfredi, Fazio Giovanni, Zambuto Sabrina, Tortorici Enza, Marino Gammazza Antonella, Gammazza Antonella M, Corrao Simona, Zummo Giovanni, De Macario Everly C, Macario Alberto J L, Assennato Pasquale, Novo Salvatore, Li Volti Giovanni, Volti Giovanni L. Hsp60 and heme oxygenase-1 (Hsp32) in acute myocardial infarction. Transl Res. 2011 May;157 (5):285–92. doi: 10.1016/j.trsl.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 106.Giannessi Daniela, Colotti Chiara, Maltinti Maristella, Del Ry Silvia, Prontera Concetta, Turchi Stefano, Labbate Antonio, Neglia Danilo. Circulating heat shock proteins and inflammatory markers in patients with idiopathic left ventricular dysfunction: their relationships with myocardial and microvascular impairment. Cell Stress Chaperones. 2007;12 (3):265–74. doi: 10.1379/CSC-272.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhu J, Quyyumi A A, Rott D, Csako G, Wu H, Halcox J, Epstein S E. Antibodies to human heat-shock protein 60 are associated with the presence and severity of coronary artery disease: evidence for an autoimmune component of atherogenesis. Circulation. 2001 Feb 27;103 (8):1071–5. doi: 10.1161/01.cir.103.8.1071. [DOI] [PubMed] [Google Scholar]
- 108.Metzler B, Schett G, Kleindienst R, van der Zee R, Ottenhoff T, Hajeer A, Bernstein R, Xu Q, Wick G. Epitope specificity of anti-heat shock protein 65/60 serum antibodies in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1997 Mar;17 (3):536–41. doi: 10.1161/01.atv.17.3.536. [DOI] [PubMed] [Google Scholar]
- 109.Burian K, Kis Z, Virok D, Endresz V, Prohaszka Z, Duba J, Berencsi K, Boda K, Horvath L, Romics L, Fust G, Gonczol E. Independent and joint effects of antibodies to human heat-shock protein 60 and Chlamydia pneumoniae infection in the development of coronary atherosclerosis. Circulation. 2001 Mar 20;103 (11):1503–8. doi: 10.1161/01.cir.103.11.1503. [DOI] [PubMed] [Google Scholar]
- 110.Xu Q, Kiechl S, Mayr M, Metzler B, Egger G, Oberhollenzer F, Willeit J, Wick G. Association of serum antibodies to heat-shock protein 65 with carotid atherosclerosis : clinical significance determined in a follow-up study. Circulation. 1999 Sep 14;100 (11):1169–74. doi: 10.1161/01.cir.100.11.1169. [DOI] [PubMed] [Google Scholar]
- 111.Hoppichler F, Lechleitner M, Traweger C, Schett G, Dzien A, Sturm W, Xu Q. Changes of serum antibodies to heat-shock protein 65 in coronary heart disease and acute myocardial infarction. Atherosclerosis. 1996 Oct 25;126 (2):333–8. doi: 10.1016/0021-9150(96)05931-x. [DOI] [PubMed] [Google Scholar]
- 112.Birnie D H, Hood S, Holmes E, Hillis W S. Anti-heat shock protein 65 titres in acute myocardial infarction. Lancet. 1994 Nov 19;344 (8934) doi: 10.1016/s0140-6736(94)90614-9. [DOI] [PubMed] [Google Scholar]
- 113.Lavoie J N, Lambert H, Hickey E, Weber L A, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol. Cell. Biol. 1995 Jan;15 (1):505–16. doi: 10.1128/mcb.15.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vander Heide Richard S. Increased expression of HSP27 protects canine myocytes from simulated ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2002 Mar;282 (3):H935–41. doi: 10.1152/ajpheart.00660.2001. [DOI] [PubMed] [Google Scholar]
- 115.Knowlton A A. The role of heat shock proteins in the heart. J. Mol. Cell. Cardiol. 1995 Jan;27 (1):121–31. doi: 10.1016/s0022-2828(08)80012-0. [DOI] [PubMed] [Google Scholar]
- 116.Pourghadamyari Hossein, Moohebati Mohsen, Parizadeh Seyed Mohammad Reza, Falsoleiman Homa, Dehghani Mashalla, Fazlinezhad Afsoon, Akhlaghi Saeed, Tavallaie Shima, Sahebkar Amirhossein, Paydar Roghayeh, Ghayour-Mobarhan Majid, Ferns Gordon A. Serum antibody titers against heat shock protein 27 are associated with the severity of coronary artery disease. Cell Stress Chaperones. 2011 May;16 (3):309–16. doi: 10.1007/s12192-010-0241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Azarpazhooh Mahmoud Reza, Mobarra Naser, Parizadeh Syyed Mohammad Reza, Tavallaie Shima, Bagheri Mahsa, Rahsepar Amir Ali, Ghayour-Mobarhan Majid, Sahebkar Amirhossein, Ferns Gordon A A. Serum high-sensitivity C-reactive protein and heat shock protein 27 antibody titers in patients with stroke and 6-month prognosis. Angiology. 2010 Aug;61 (6):607–12. doi: 10.1177/0003319709360524. [DOI] [PubMed] [Google Scholar]