

Abstract
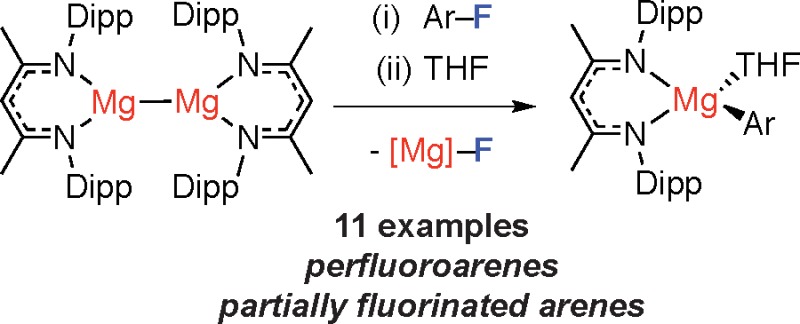
Addition of the carbon–fluorine bond of a series of perfluorinated and polyfluorinated arenes across the Mg–Mg bond of a simple coordination complex proceeds rapidly in solution. The reaction results in the formation of a new carbon–magnesium bond and a new fluorine–magnesium bond and is analogous to Grignard formation in homogeneous solution.
Grignard reagents are ubiquitous in synthesis.1 Widely employed and often taught in practical experiments to undergraduate students, the preparation of Grignard reagents by the simple addition of an alkyl or aryl halide to magnesium metal has stood the test of a century of scientific advances.1 The vast majority of preparations employ substrates containing R–X (X = I, Br, Cl) bonds. The use of finely divided (Rieke) magnesium powder,2 or metal vapor synthesis,3 however, has allowed extension of the reaction scope to a handful of challenging substrates including those containing carbon–fluorine bonds.3 In more recent years, there has been growing interest in the use of main group reagents for the functionalization of strong carbon–fluorine bonds in organic molecules.4 Reactions that transform an unreactive carbon–fluorine bond to a reactive and polarized carbon–element bond (element = boron, aluminum, silicon) are gaining increasing attention.5−8
For example, diborane reagents react with either monofluoroarenes or partially fluorinated arenes in the presence of nickel precatalysts, in combination with either phosphine or N-heterocyclic carbene ligands and additives.6a−6c Similarly, the reaction of bis(pinacolato)diborane with fluoroarenes bearing a 2-pyridyl directing group can be catalyzed by [Rh(1,5-COD)2][BF4] in the presence of 2 equiv KOAc.6d These reactions can be categorized in terms of a formal 1,2-addition of the carbon–fluorine bond across the B–B bond of the diborane reagent (Figure 1, eq 1). In related studies, a number of low-valent main group species have been shown to be capable of the oxidative addition of carbon–fluorine bonds. Silylene, germylene, and related aluminum(I) reagents react with fluoroarenes by addition of the carbon–fluorine bond to the main group center (Figure 1, eqs 2–3).9
Figure 1.
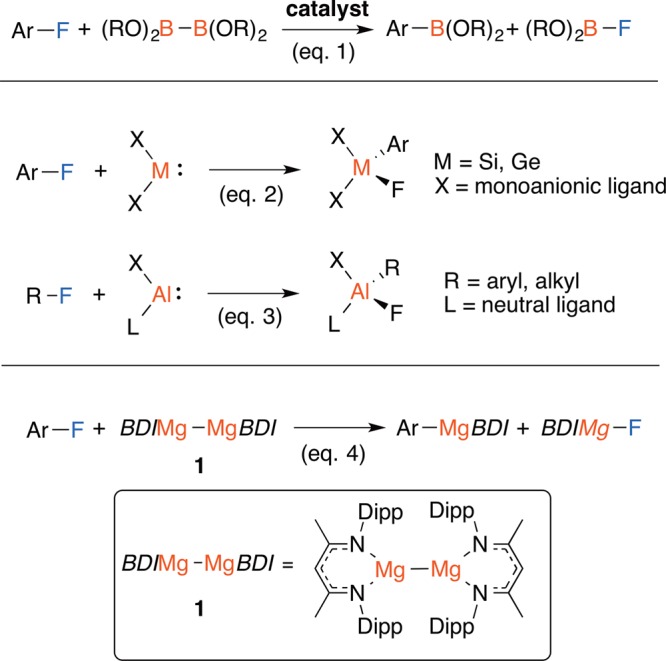
Carbon–fluorine bond borylation, silylation, germylation, stannylation and alumination.
The seminal discovery by Jones, Stasch and Green of 1, a coordination complex containing a Mg–Mg bond (Figure 1, BDI = κ2-{2,6-iPr2C6H3NCMe}2CH),10 offers the possibility of studying the addition of carbon–fluorine bonds across a Mg–Mg bond in homogeneous solution. Despite important work demonstrating that 1 can act as a net 2-electron reductant,11 reactions with alkyl or aryl halides are to the best of our knowledge unreported. Herein, we demonstrate that 1 undergoes facile reactions with fluoroarenes to generate the corresponding magnesium fluoride and aryl reagent (Figure 1, eq 4). The reaction does not require a directing group in the substrate, nor toxic or expensive catalysts.
Upon addition of 10 equiv of C6F6 to a 0.02 M solution of 1 in C6D6 an instant color change from yellow to colorless was observed. Monitoring the reaction by 19F NMR spectroscopy revealed clean formation of new Mg–C6F5 and Mg–F moieties as evidenced by new resonances at δ = −119.6, −156.0, −156.9 ppm and δ = −187.8 ppm, respectively. 1H NMR data at the same time point demonstrated a set of broad resonances consistent with a single ligand environment. The complexity of these observations was resolved by adding a drop of THF at which point the equilibrium mixture collapses to 2:1 mixture of [BDIMg(THF)(C6F5)] and [BDIMg(μ-F)(THF)]2. The same mixture was obtained upon reaction of the THF solvate of 1 with C6F6.
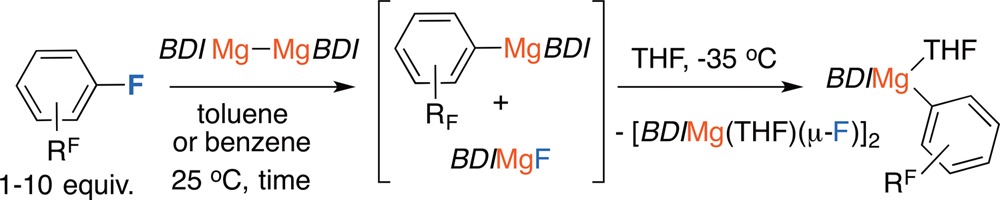
A synthetic procedure was developed using these observations. The scope of this reaction is presented in Table 1 and includes a series of perfluorinated and partially fluorinated arenes. Yields measured by NMR spectroscopy are consistently high (80–95%). The products could be separated by fractional crystallization of the magnesium fluoride at −35 °C and further purification of the magnesium aryl complexes achieved by recrystallization from n-hexane (38–54%). In the case of perfluoroarenes, the reaction occurs at a single carbon–fluorine bond, and the reagent can be forced to the ortho-position by inclusion of a 2-pyridyl directing group. Preparations using partially fluorinated arenes are less selective and although mixtures of regioisomeric products are obtained, in all cases the carbon–fluorine bond that breaks is that with at least one ortho-fluorine substituent.12 This regiochemistry complements that reported by Marder, Radius and co-workers, and C–F borylation using Ni-catalysis proceeds at positions adjacent to an ortho-hydrogen substituent.6d For 2j and 2k, the major products are accompanied by a side-reaction and small amounts of C–H bond cleavage of the acidic proton was observed (∼5% NMR scale; 15–20% preparative scale, see Supporting Information).
Table 1. Scope of the Addition of Fluoroarenes to 1.
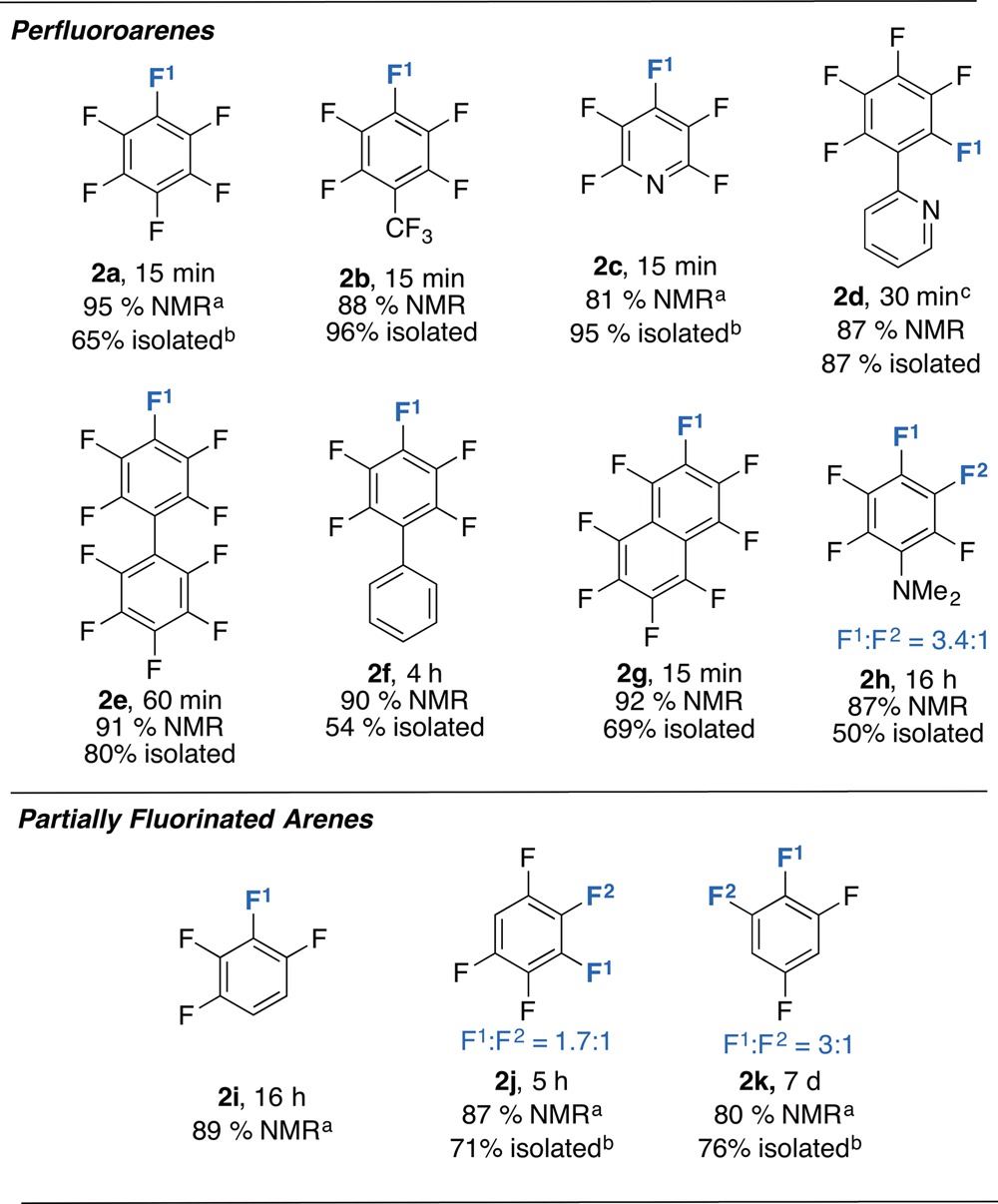
NMR scale reactions use 10 equiv of fluorocarbon.
preparative scale reactions use 1 equiv of fluorocarbon.
For regioisomeric products F1 is the major and F2 the minor product selectivities are consistent across NMR and preparative scale reactions.
Isolated without THF coordinated.
Complexes 2b–d were subject to single-crystal X-ray studies; these are rare examples of crystallographically characterized magnesium fluoroaryl species (Figure 2). The Mg–C bond lengths ranges from 2.1381(15) to 2.176(2) Å. Close contact of a fluorine atom with the magnesium center of 2b is evidenced by the asymmetry in the Mg–F distances to the two ortho-fluorines of 3.2 and 3.4 Å, accompanied by a small canting of the fluoroarene of ∼5° toward the side with the closest Mg–F contact.
Figure 2.

Structures of (a) 2b, (b) 2c and (c) 2d. Selected bond lengths (Å) and angles (deg): 2b, Mg–C 2.176(2); 2c, 2.1719(18); 2d, Mg–N(31) 2.1293(12), Mg–C 2.1381(14), N(31)–Mg(1)–C 79.62(5).
The subtle β-fluorine interaction is reflected in DFT calculations (NBO: 2a–b, ortho-F −0.34 and −0.35, av. F, −0.315) and VT NMR studies in which hindered rotation about the Mg–C bond in 2a is observed below 283 K in toluene-d8 (ΔH‡ = 7.6 kcal mol–1, ΔS‡ = −15.0 cal K–1 mol–1, ΔG‡298 K = 12.1 kcal mol–1). This weak interaction may represent an intermediate along a reaction coordinate to β-fluoride elimination. In line with this finding, thermal stability tests on reaction mixtures show that decomposition of the products occurs slowly at 25 °C or more rapidly at elevated temperatures. For example, heating the reaction mixture of 1 with C6F6 for 50 °C in C6D6 resulted in slow decomposition of the magnesium aryl species over a period of 4 weeks.13 Attempts to expand the scope of carbon–fluorine bond functionalization to substrates with lower fluorine content (e.g 1,2,3-trifluorobenzene) required elevated temperatures and resulted in complex, inseparable, reaction mixtures. No reaction was observed between 1 and either fluorobenzene or α,α,α-trifluorotoluene.
The thermodynamics of carbon–fluorine bond functionalization and THF solvation were evaluated by DFT. Both the carbon–fluorine bond functionalization and the subsequent THF solvation are highly exergonic (Figure 3a). These calculations provide some insight into the complex Schlenk equilibria at play following C–F bond cleavage; the steric demands of the β-diketiminate ligand are such that the magnesium aryl cannot form symmetric dimeric species containing 3-center 2-electron Mg–C–Mg bonds. Instead a number of asymmetric dimers are potentially accessible (Figure 3b).
Figure 3.

Gas-phase calculations using the ωB97X or ωB97XD functional (values in bold) and 6,31G+d,p (C,H,N,O,F)/Lanl2DZ (Mg) basis set. (a) Thermodynamics of C–F bond cleavage and Mg solvation. (b) Relative stability of dimeric species. Gibbs free energies at 298 K; all values in kcal mol–1.
Jones, Stasch and co-workers have demonstrated that 1 reacts with benzophenone, 1,3-cyclohexadiene or tBuNC by single-electron transfer.11 Recent calculations on the addition of CO2 to 1 are consistent with a concerted 2-electron pathway.14 On the basis of preliminary observations, we suggest that carbon–fluorine bond cleavage may not proceed through radical intermediates and the concerted pathway remains most likely.15 Although a crossover experiment between 1 and 3 results in slow formation of the asymmetric species 4 at 25 °C (Figure 4a and 5), this result could be explained by a ligand exchange reaction in which the Mg–Mg core remains intact.
Figure 4.
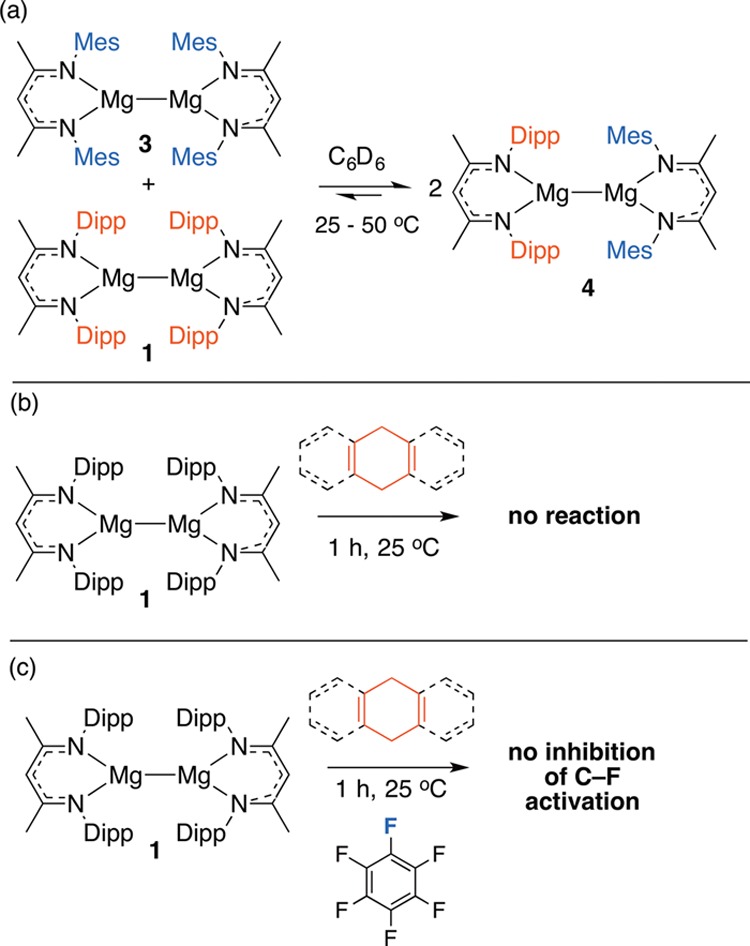
Preliminary experimental support for a concerted pathway for C–F bond cleavage.
Figure 5.
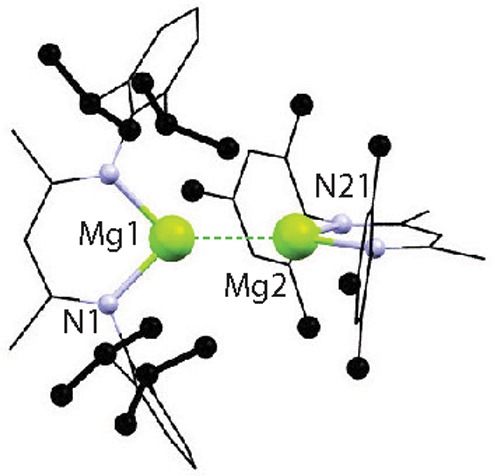
Crystal structure of 4. Selected bond lengths (Å) Mg1–Mg2 2.8700(9), Mg(1)–N(1) 2.0644(12).
To probe production of [BDIMg]•, the reaction of 1 with 9,10-dihydroanthracene or 1,4-cyclohexadiene was conducted and no evidence for hydride abstraction observed under the conditions presented in Table 1 (Figure 4b). Further, the reaction of 1 with C6F6 is not inhibited by addition of either radical trap (Figure 4c). In combination, these experiments suggest that neither [BDIMg]• nor organic radicals are reaction intermediates in carbon–fluorine bond cleavage. In summary, we report the first example of a homogeneous equivalent of Grignard formation. Addition of carbon–fluorine bonds of fluorinated arenes across the Mg–Mg bond of a simple coordination complex proceeds rapidly in solution. We are continuing to study the mechanism and scope of this reaction. We aim to develop further chemical transformations of this new generation of Grignard reagents and are currently investigating reactions with a series of electrophiles.
Acknowledgments
We are grateful to the Royal Society for provision of a University Research Fellowship (MRC) and to the Leverhulme Trust (RPG-2015-248) and ERC (FluoroFix: 677367) for generous funding.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b08104.
The authors declare no competing financial interest.
Supplementary Material
References
- a Garst G. F.; Ungvary F.. Grignard Reagnets: New Developments; Richey H., Ed.; John Wiley and Sons: Chichester, 2000. [Google Scholar]
- a Lee J.-S.; Velarde-Ortiz R.; Guijarro A.; Wurst J. R.; Rieke R. D. J. Org. Chem. 2000, 65, 5428. 10.1021/jo000413i. [DOI] [PubMed] [Google Scholar]; b Rieke R. D.; Hanson M. V. Tetrahedron 1997, 53, 1925. 10.1016/S0040-4020(96)01097-6. [DOI] [Google Scholar]; c Rieke R. D. Science 1989, 246, 1260. 10.1126/science.246.4935.1260. [DOI] [PubMed] [Google Scholar]
- a Tjurina L. A.; Smirnov V. V.; Barkovskii G. B.; Nikolaev E. N.; Esipov S. E.; Beletskaya I. P. Organometallics 2001, 20, 2449. 10.1021/om0100380. [DOI] [Google Scholar]; b Tjurina L. A.; Smirnov V. V.; Potapov D. A.; Nikolaev S. S.; Esipov S. E.; Beletskaya I. P. Organometallics 2004, 23, 1349. 10.1021/om030633x. [DOI] [Google Scholar]
- a Ahrens T.; Kohlmann J.; Ahrens M.; Braun T. Chem. Rev. 2015, 115, 931. 10.1021/cr500257c. [DOI] [PubMed] [Google Scholar]; b Kuehnel M. F.; Lentz D.; Braun T. Angew. Chem., Int. Ed. 2013, 52, 3328. 10.1002/anie.201205260. [DOI] [PubMed] [Google Scholar]; c Amii H.; Uneyama K. Chem. Rev. 2009, 109, 2119. 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]
- a Braun T.; Salomon M. A.; Altenhöner K.; Teltewskoi M.; Hinze S. Angew. Chem., Int. Ed. 2009, 48, 1818. 10.1002/anie.200805041. [DOI] [PubMed] [Google Scholar]; b Teltewskoi M.; Panetier J. A.; Macgregor S. A.; Braun T. Angew. Chem., Int. Ed. 2010, 49, 3947. 10.1002/anie.201001070. [DOI] [PubMed] [Google Scholar]; c Kalläne S. I.; Teltewskoi M.; Braun T.; Braun B. Organometallics 2015, 34, 1156. 10.1021/om500952x. [DOI] [Google Scholar]
- a Guo W.-H.; Min Q.-Q.; Gu J.-W.; Zhang X. Angew. Chem., Int. Ed. 2015, 54, 9075. 10.1002/anie.201500124. [DOI] [PubMed] [Google Scholar]; b Liu X.-W.; Echavarren J.; Zarate C.; Martin R. J. Am. Chem. Soc. 2015, 137, 12470. 10.1021/jacs.5b08103. [DOI] [PubMed] [Google Scholar]; c Niwa T.; Ochiai H.; Watanabe Y.; Hosoya T. J. Am. Chem. Soc. 2015, 137, 14313. 10.1021/jacs.5b10119. [DOI] [PubMed] [Google Scholar]; d Zhou J.; Kuntze-Fechner M. W.; Bertermann R.; Paul U. S. D.; Berthel J. H. J.; Friedrich A.; Du Z.; Marder T. B.; Radius U. J. Am. Chem. Soc. 2016, 138, 5250. 10.1021/jacs.6b02337. [DOI] [PubMed] [Google Scholar]
- Ishii Y.; Chatani N.; Yorimitsu S.; Murai S. Chem. Lett. 1998, 27, 157. 10.1246/cl.1998.157. [DOI] [Google Scholar]
- a Yow S.; Gates S. J.; White A. J. P.; Crimmin M. R. Angew. Chem., Int. Ed. 2012, 51, 12559. 10.1002/anie.201207036. [DOI] [PubMed] [Google Scholar]; b Ekkert O.; Strudley S. D. A.; Rozenfeld A.; White A. J. P.; Crimmin M. R. Organometallics 2014, 33, 7027. 10.1021/om501113j. [DOI] [Google Scholar]
- a Jana A.; Samuel P. P.; Tavcar G.; Roesky H. W.; Schulzke C. J. Am. Chem. Soc. 2010, 132, 10164. 10.1021/ja103988d. [DOI] [PubMed] [Google Scholar]; b Azhakar R.; Roesky H. W.; Wolf H.; Stalke D. Chem. Commun. 2013, 49, 1841. 10.1039/c3cc38669d. [DOI] [PubMed] [Google Scholar]; c Samuel P. P.; Singh A. P.; Sarish S. P.; Matussek J.; Objartel I.; Roesky H. W.; Stalke D. Inorg. Chem. 2013, 52, 1544. 10.1021/ic302344a. [DOI] [PubMed] [Google Scholar]; d Crimmin M. R.; Butler M. J.; White A. J. P. Chem. Commun. 2015, 51, 15994. 10.1039/C5CC07140B. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chu T.; Boyko Y.; Korobkov I.; Nikonov G. I. Organometallics 2015, 34, 5363. 10.1021/acs.organomet.5b00793. [DOI] [Google Scholar]
- a Green S. P.; Jones C.; Stasch A. Science 2007, 318, 1754. 10.1126/science.1150856. [DOI] [PubMed] [Google Scholar]; b Overgaard J.; Jones C.; Stasch A.; Iversen B. B. J. Am. Chem. Soc. 2009, 131, 4208. 10.1021/ja900385u. [DOI] [PubMed] [Google Scholar]; c Stasch A.; Jones C. Dalton. Trans. 2011, 40, 5659. 10.1039/c0dt01831g. [DOI] [PubMed] [Google Scholar]; d Platts J. A.; Overgaard J.; Jones C.; Iversen B. B.; Stasch A. J. Phys. Chem. A 2011, 115, 194. 10.1021/jp109547w. [DOI] [PubMed] [Google Scholar]; e Wu L.-C.; Jones C.; Stasch A.; Platts J. A.; Overgaard J. Eur. J. Inorg. Chem. 2014, 2014, 5536. 10.1002/ejic.201402606. [DOI] [Google Scholar]; f Boutland A. J.; Dange D.; Stasch A.; Maron L.; Jones C. Angew. Chem., Int. Ed. 2016, 55, 9239. 10.1002/anie.201604362. [DOI] [PubMed] [Google Scholar]
- a Bonyhady S. J.; Green S. P.; Jones C.; Nembenna S.; Stasch A. Angew. Chem., Int. Ed. 2009, 48, 2973. 10.1002/anie.200900331. [DOI] [PubMed] [Google Scholar]; b Bonyhady S. J.; Jones C.; Nembenna S.; Stasch A.; Edwards A. J.; McIntyre G. J. Chem. - Eur. J. 2010, 16, 938. 10.1002/chem.200902425. [DOI] [PubMed] [Google Scholar]; c Bonyhady S. J.; Collis D.; Frenking G.; Holzmann N.; Jones C.; Stasch A. Nat. Chem. 2010, 2, 865. 10.1038/nchem.762. [DOI] [PubMed] [Google Scholar]; d Jones C.; Bonyhady S. J.; Holzmann N.; Frenking G.; Stasch A. Inorg. Chem. 2011, 50, 12315. 10.1021/ic200682p. [DOI] [PubMed] [Google Scholar]; e Murphy D. M.; McDyre L. E.; Carter E.; Stasch A.; Jones C. Magn. Reson. Chem. 2011, 49, 159. 10.1002/mrc.2721. [DOI] [PubMed] [Google Scholar]; f Ma M.; Stasch A.; Jones C. Chem. - Eur. J. 2012, 18, 10669. 10.1002/chem.201201030. [DOI] [PubMed] [Google Scholar]; g Lalrempuia R.; Kefalidis C. E.; Bonyhady S. J.; Schwarze B.; Maron L.; Stasch A.; Jones C. J. Am. Chem. Soc. 2015, 137, 8944. 10.1021/jacs.5b06439. [DOI] [PubMed] [Google Scholar]
- Currently, we cannot discriminate a kinetic effect, that the ortho fluorine directs the Mg–Mg reagent to an adjacent C–F, bond from a thermodynamic one, that the weakest C–F bonds are those flanked by fluorine atoms. See for comparison:; a Macgregor S. A.; Mckay D.; Panetier J. A.; Whittlesey M. K. Dalton Trans. 2013, 42, 7386. 10.1039/c3dt32962c. [DOI] [PubMed] [Google Scholar]; For activation of C–H bonds flanked by fluorine atoms, see:; b Selmeczy A. D.; Jones W. D.; Partridge M. G.; Perutz R. N. Organometallics 1994, 13, 522. 10.1021/om00014a025. [DOI] [Google Scholar]; c Evans M. E.; Burke C. L.; Yaibuathes S.; Clot E.; Eisenstein O.; Jones W. D. J. Am. Chem. Soc. 2009, 131, 13464. 10.1021/ja905057w. [DOI] [PubMed] [Google Scholar]; d Clot E.; Eisenstein O.; Jasim N.; Macgregor S. A.; McGrady J. E.; Perutz R. N. Acc. Chem. Res. 2011, 44, 333. 10.1021/ar100136x. [DOI] [PubMed] [Google Scholar]; e Clot E.; Mégret C.; Eisenstein O.; Perutz R. N. J. Am. Chem. Soc. 2009, 131, 7817. 10.1021/ja901640m. [DOI] [PubMed] [Google Scholar]
- The fate of the organic fragment was not clear. No evidence for a benzyne intermediate or [4+2]-cycloaddition products was collected.
- a Lalrempuia R.; Stasch A.; Jones C. Chem. Sci. 2013, 4, 4383. 10.1039/c3sc52242c. [DOI] [Google Scholar]; b Kefalidis C. E.; Stasch A.; Jones C.; Maron L. Chem. Commun. 2014, 50, 12318. 10.1039/C4CC04984E. [DOI] [PubMed] [Google Scholar]
- For experiments relevant to the mechanism of Grignard formation with Mg(0), see:; a Root K. S.; Hill G. L.; Lawrence L. M.; Whitesides G. M. J. Am. Chem. Soc. 1989, 111, 5405. 10.1021/ja00196a053. [DOI] [Google Scholar]; b Ashby E. C.; Oswald J. J. Org. Chem. 1988, 53, 6068. 10.1021/jo00261a016. [DOI] [Google Scholar]; c Walborsky H. M.; Young A. E. J. Am. Chem. Soc. 1964, 86, 3288. 10.1021/ja01070a018. [DOI] [Google Scholar]; d Kruczynski T.; Henke F.; Neumaier M.; Bowen K. H.; Schnöckel H. Chem. Sci. 2016, 7, 1543. 10.1039/C5SC03914B. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.