

Abstract
A barrier to drug development for nonalcoholic steatohepatitis (NASH) is the absence of translational preclinical human-relevant systems. An in vitro liver model was engineered to incorporate hepatic sinusoidal flow, transport, and lipotoxic stress risk factors (glucose, insulin, free fatty acids) with cocultured primary human hepatocytes, hepatic stellate cells (HSCs), and macrophages. Transcriptomic, lipidomic, and functional endpoints were evaluated and compared with clinical data from NASH patient biopsies. The lipotoxic milieu promoted hepatocyte lipid accumulation (4-fold increase, P < 0.01) and a lipidomics signature similar to NASH biopsies. Hepatocyte glucose output increased with decreased insulin sensitivity. These changes were accompanied by increased inflammatory analyte secretion (e.g., IL-6, IL-8, alanine aminotransferase). Fibrogenic activation markers increased with lipotoxic conditions, including secreted TGF-β (>5-fold increase, P < 0.05), extracellular matrix gene expression, and HSC activation. Significant pathway correlation existed between this in vitro model and human biopsies. Consistent with clinical trial data, 0.5 μM obeticholic acid in this model promoted a healthy lipidomic signature, reduced inflammatory and fibrotic secreted factors, but also increased ApoB secretion, suggesting a potential adverse effect on lipoprotein metabolism. Lipotoxic stress activates similar biological signatures observed in NASH patients in this system, which may be relevant for interrogating novel therapeutic approaches to treat NASH.
An in vitro lipotoxic liver system that exhibits a clinical disease phenotype and therapeutic drug response similar to NASH patients.
Introduction
Nonalcoholic fatty liver disease (NAFLD) encompasses a progressive disease spectrum characterized by excessive accumulation of lipid within the liver and has emerged as the most prevalent liver disease, affecting ~30% of Western populations (1). Development of NAFLD can progress from simple fatty liver (steatosis) to more serious forms of the disease, including nonalcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and cancer. Hepatic lipid accumulation occurs in response to uptake of circulating free fatty acids (FFAs), de novo lipogenesis in the liver, or dietary fats (2) and presents as intracellular hepatocyte lipid droplet formation. Increasing triglyceride deposition with ensuing insulin resistance can lead to NASH, which is characterized by hepatic injury, which drives inflammation, oxidative stress, and apoptosis. Despite the recognition of this disease as a public health crisis affecting up to 75% of type 2 diabetics and 95% of obese individuals (3), there is currently no approved therapy for NAFL or NASH.
While there are currently more than 20 distinct therapeutics in various stages of clinical development (4), the therapeutic landscape includes no clear consensus on target strategy, illustrating a lack of mechanistic clarity with respect to the pivotal drivers of this disease spectrum. There is also limited consensus regarding the utility of animal models, as demonstrated by the array of different models (>25) currently in use. Further, in vitro liver systems typically use supraphysiologic concentrations of insulin/glucose and drug concentrations, and lack key elements of the liver microenvironment, including hemodynamics, transport, and multicellularity (5). The relevance of these models to humans is questionable because they most often essentialize isolated aspects of the human pathophysiology (6).
We previously described an approach that applies liver sinusoidal hemodynamics and interstitial fluid transport parameters to restore a mature, differentiated, in vivo hepatocyte phenotype and function (7, 8). Herein, we adapted this system to model human fatty liver disease with lipotoxic stress. Primary human hepatocytes were cocultured with primary human hepatic stellate cells (HSCs) and macrophages (MΦs) and perfused with media containing higher levels of NASH-associated risk factors (glucose, insulin, and FFA) (9). Activation of liver-resident MΦs (i.e., Kupffer cells) and HSCs significantly contribute to the pathogenesis of NASH by promoting inflammation and fibrosis locally in the liver and systemically via the secretome (10). We demonstrate that lipotoxic stress similar to that seen in NASH patients was recapitulated and we measured its impact on hepatocyte morphology and function. Obeticholic acid, a promising therapeutic for NASH in phase III clinical trials, was used to further validate this system and corroborate clinical findings in vitro. The utilization of this lipotoxic liver system provides a new translational tool for understanding the complex disease progression in NAFLD and a platform to evaluate the impact of new therapies.
Results
Recapitulation of liver tissue architecture.
To mimic the liver microenvironment and multicellularity, primary human hepatocytes were cocultured in a collagen sandwich with primary human MΦs and HSCs, separated by a synthetic membrane (Figure 1A) under conditions of hemodynamics and transport (7, 8, 11). In addition to hepatocytes, MΦs and HSCs have been shown to be critical to the signaling and progression of NAFL/NASH; thus, it was important that these additional cell types be present in the system (1, 12, 13). These 3 cell types communicate through direct cell-cell contact via the porous transwell membrane (11) and/or through secreted factors. Under healthy-milieu conditions (i.e., containing physiological levels of hormones) and lacking lipotoxic risk factors, hepatocytes maintain their polarized morphology as previously described (7, 8). At the conclusion of each experiment, the hepatocytes were separated and analyzed independently from the nonparenchymal cells (NPCs). Representative images of NPCs and hepatocytes exposed to hemodynamics (Figure 1B) demonstrate that CD68 and reelin, markers for MΦs and HSCs, respectively (12, 13), were expressed in our system at the end of the experiment, reflecting the maintenance of the cell types in a differentiated state. E-cadherin, a marker of cell-cell junctions, depicts the retention of polarized morphology by differentiated hepatocytes in our system.
Figure 1. Adaptation of the in vitro human liver system to mimic human nonalcoholic fatty liver under lipotoxic stress.

(A) Liver sinusoidal hemodynamics were applied to the human liver system using a cone-and-plate viscometer incorporated into a transwell multiculture model of nonparenchymal cells (NPCs) (top of transwell) and hepatocytes (bottom of the transwell). Rotation of the cone (orange triangle) imparts shear stress onto the transwell. Medium is continually perfused to recapitulate interstitial flow, as indicated by the inflow and outflow ports. (B) Representative photomicrographs (original magnification, ×20) of NPCs and hepatocytes are shown. NPCs include hepatic stellate cells (reelin+, green) and macrophages (CD68+, red). Hepatocytes are stained for E-cadherin (green). Nuclei stained with DAPI (blue). Scale bars: 100 μm and 50 μm for NPCs and hepatocytes, respectively. (C) Hepatocyte protein-protein interaction network was visualized and colored according to signaling community. (D) The relative directionality of many pathways identified in C were explored by calculating the FDR-scaled fold change for each, revealing key signaling pathways perturbed at the RNA level by lipotoxic stress. *Rotation gene set tests (ROAST), FDR < 5%.
Recapitulation of signaling pathways.
In order to understand the impact of NAFL/NASH risk factors on liver cells, the system was exposed to a lipotoxic milieu composed of elevated glucose (25 mM), insulin (6,900 pM), and FFAs including oleic (65 μM) and palmitic (45 μM) acid, at concentrations derived from the differences between the plasma levels of healthy and NASH patients (14, 15). These factors were present for the duration of the experiment (10 days). To gain a high-level understanding of key differences between hepatocytes in healthy or a lipotoxic milieu, RNA-seq transcriptomic data were evaluated for repeatability and pathway analyses using gene ontology and a protein-protein interaction (PPI) analysis (16). For this study, only hepatocytes were interrogated using transcriptomics, owing to challenges inherent with the mixed-cell population of NPCs. To gain confidence in the response, the study was repeated and reproducibility was evaluated using a gene correlation plot, where the correlation was highly significant (Pearson correlation = 0.90, P value < 2.2 × 10–16, Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/jci.insight.90954DS1). Next, the transcriptomic data were utilized for pathway analysis across multiple gene ontology databases (see Methods). Using this approach, 639 pathways were significantly regulated (complete list in Supplemental Materials), spanning a broad range of metabolic functions. To further distill this transcriptomic data down to their fundamental biological processes, we utilized a PPI analysis. The goal of the PPI network is to identify biological centers of dysregulation induced by the lipotoxic milieu. Figure 1C shows that the data organized into multiple protein communities that represent coordinated changes in activity and are color-coded for visualization. Pathway analysis was performed for each protein community to better understand the biological functions associated with each, which are summarized in Figure 1C. Most communities detected are associated with NAFLD. Community 3, one of the largest regulated groups, represents PI3K/Akt signaling that is important for glucose/insulin signaling and is associated with lipotoxicity in the liver. PKA activation (community 4) and glucose signaling (community 5) are regulated and support the above finding. Furthermore, communities associated with altered lipid metabolism were also detected and include lipid/lipoprotein metabolism (community 1) and cytochrome p450 (CYP) enzymes (community 2). Finally, inflammatory processes were regulated as reflected by the NF-κB pathway (community 6), ER stress (community 7), and mitochondrial activity (community 8). In addition to these lipotoxic-associated communities, basic cell biological pathways (e.g., mitosis, protein modifications) were regulated as seen in communities 10–13.
The relative directionality of these pathways was explored by calculating the average fold change for key pathways identified above, which was visualized as a radar plot relative to the healthy baseline condition (Figure 1D). The average gene expression for each of these pathways (with the exception of cholesterol biosynthesis) was significantly elevated (FDR < 5%). Using these same methods, 2 independent studies that evaluated gene expression changes in NASH patients (17, 18) were analyzed to benchmark the in vitro transcriptomic signature. Table 1 shows pathway analysis results, including directional pathway regulation in patient biopsies and this in vitro lipotoxic model. Strong agreement was observed across all 3 datasets for these key pathways associated with NASH, particularly with the fatty acid synthesis, gluconeogenesis, and inflammatory signaling pathways. Therefore, an unbiased analysis showed that the lipotoxic model activated key signaling pathways associated with NASH and these signaling communities were further investigated using both functional endpoint assays and transcriptomic pathway analysis, as described below.
Table 1. Pathway activation in human biopsies and the in vitro human liver lipotoxic system.

Hepatic steatosis.
In the pathogenesis of NAFLD, lipid accumulates in hepatocytes due to both increased FFA load in the circulation and de novo lipogenesis stemming from elevated glucose and insulin levels (2). To assess lipid accumulation within hepatocytes, neutral lipids were stained with Nile red (Figure 2A). Under healthy conditions, a limited amount of lipid was visible within the cell, while distinct lipid droplets were observed in hepatocytes under lipotoxic stress. The lipid droplet–cytoplasm interface is stabilized by amphiphilic proteins (19) such as adipophilin, which was clearly observed in lipotoxic conditions (Figure 2B). These changes were quantified across multiple hepatocyte cell donors, where a 4-fold increase in steatosis was observed in lipotoxic conditions relative to healthy conditions (Figure 2C). Thus, this system is amendable to modulating lipogenesis via exposure to disease physiological levels of factors.
Figure 2. De novo lipogenesis and cholesterol synthesis are increased in the human liver lipotoxic system.

(A) Representative photomicrographs (original magnification, ×20) of hepatocytes exposed to the healthy or lipotoxic milieu are shown. Hepatocytes are stained for E-cadherin (green), lipid (Nile red+, red), and nuclei (DAPI+, blue). Insets have been magnified to provide greater clarity of lipid droplets. Scale bars: 50 μm. (B) Representative photomicrograph (original magnification, ×100) of hepatocytes exposed to the lipotoxic milieu reveals adipophilin staining (green) around lipid droplets (Nile red+, red). Nuclei stained with DAPI (blue). Scale bar: 100 μm. (C) Nile red staining intensity from hepatocyte images were quantified and represented as fold change relative to healthy controls. n ≥ 11 experiments, 3 donors. ***P < 0.01, Student’s 2-tailed t test. (D and E) Hepatocyte expression of genes of the fatty acid (D) and cholesterol (E) biosynthesis pathways are represented as log2-fold change of lipotoxic vs. healthy milieu (red = upregulation, blue = downregulation). n = 6 experiments, 3 donors.
To further explore lipid accumulation, lipidomics was utilized to evaluate concentrations of specific lipid classes within the cells (Figure 3). Lipotoxic conditions had a profound impact on the metabolite signature. Out of 767 detectable lipid species, 449 were significantly regulated (FDR ≤ 5%). Significant increases in triacylglycerol (TAG), diacylglycerol (DAG), and cholesterol ester (CE) species were mechanistically linked to de novo lipogenesis pathways, including fatty acid and cholesterol synthesis pathways, which were significantly increased in hepatocytes in lipotoxic conditions relative to healthy conditions (Figure 1D and Figure 2, D and E, and pathway visualizations in Supplemental Figures 2 and 3). Comparing our in vitro data with previously published lipidomics performed on NAFL/NASH patient livers (20) revealed significant similarity (Pearson correlation P values < 0.002 compared with NAFL and NASH biopsies) between lipidomic signatures (Figure 3). Therefore, lipid imaging, lipidomics, and transcriptomic pathway analysis show evidence for the promotion of a steatotic phenotype in hepatocytes under lipotoxic stress, a hallmark of human NAFL and NASH.
Figure 3. Hepatic lipid changes in lipotoxic stress and clinical nonalcoholic fatty liver disease.
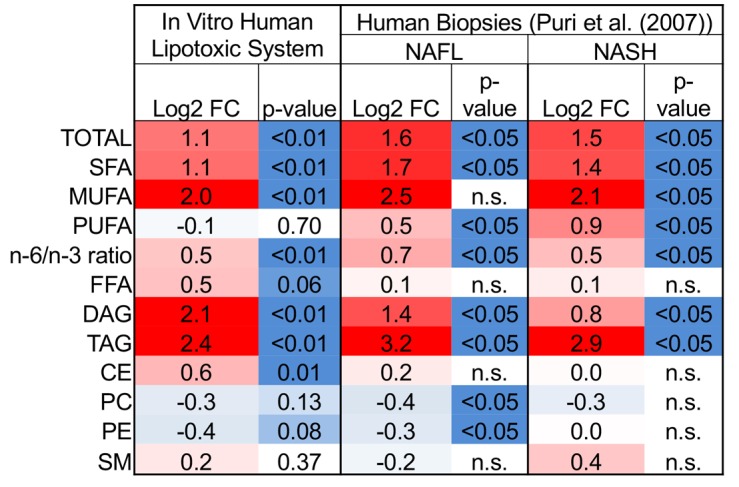
All values are log2-fold changes in lipid concentrations relative to controls. Red indicates upregulated and blue indicates downregulated expression. Abbreviations: FC, fold change; NAFL, nonalcoholic fatty liver; NASH, nonalcoholic steatohepatitis; SFA, saturated fatty acid; MUFA, mono-unsaturated fatty acid; PUFA, polyunsaturated fatty acid; n-6/n-3, omega-6 fatty acids/omega-3 fatty acids; FFA, free fatty acid; DAG, diacylglycerol; TAG, triacylglycerol; CE, cholesterol ester; PC, phosphatidylcholine; PE, phosphatidylethanolamine; SM, sphingomyelin.
Dysregulation of glucose/insulin signaling.
Progression of NAFLD often coincides with elevated systemic glucose levels by increasing gluconeogenesis and decreasing glycolysis, largely by ensuing insulin resistance (21). Thus, we measured glucose regulation in hepatocytes exposed to the lipotoxic milieu and found that these hepatocytes produced 3-fold more glucose than those exposed to a healthy milieu (Figure 4A). This result was corroborated by upregulation of genes in the glycolysis/gluconeogenesis pathway (Figure 4B and Supplemental Figure 4). Insulin normally inhibits gluconeogenesis by inhibiting transcription of phosphoenolpyruvate carboxykinase 1 (PCK1), which is important in catalyzing the first committed step in gluconeogenesis (22). However, PCK1 was upregulated by the lipotoxic milieu (Figure 4B), suggesting that the cells have become unresponsive to insulin. To determine if altered glucose regulation was due to reduced insulin sensitivity, we measured glucose output following an insulin challenge, as insulin should inhibit gluconeogenesis and promote glycolysis (22). Hepatocytes exposed to a lipotoxic milieu were unable to reduce their glucose secretion with an insulin challenge, while the hepatocytes exposed to a healthy milieu exhibited a 4-fold reduction in glucose secretion (Figure 4C). This was further confirmed by measuring phosphorylated Akt, a downstream marker for intact insulin signaling. Akt exhibited a significant reduction in phosphorylation in hepatocytes exposed to the lipotoxic compared with the healthy milieu (12-fold versus 4-fold, respectively, Figure 4D), indicating that insulin signaling was compromised. Thus, these lipotoxic hepatocytes exhibit altered glucose signaling, a well-established risk factor for NAFL and NASH.
Figure 4. Glucose utilization and synthesis is altered in the human liver lipotoxic system.

(A) Baseline levels of secreted glucose were measured from hepatocytes exposed to the healthy or lipotoxic milieu for 10 days and represented as fold change relative to healthy controls. n ≥ 12 experiments, 5 donors. (B) Hepatocyte expression of genes of the glycolysis and gluconeogenesis pathways are represented as log2-fold change of lipotoxic vs. healthy milieu (red = upregulation, blue = downregulation). n = 6 experiments, 3 donors. (C) Hepatocytes exposed to the healthy or lipotoxic milieu for 10 days were serum starved with or without 100 nM insulin for 3 hours and secreted glucose was measured and represented as fold change relative to baseline healthy controls. n ≥ 9 experiments, 5 donors. (D) Hepatocytes exposed to the healthy or lipotoxic milieu for 10 days were serum starved and then stimulated with or without 10 nM insulin for 10 minutes to measure Akt phosphorylation. Levels of phosphorylated Akt induced by insulin are represented as fold change relative to baseline phosphorylated Akt in the absence of insulin challenge. n = 5 experiments, 3 donors. *P < 0.05, **P < 0.01, Student’s 2-tailed t test. NS, not significant.
Increased cell stress and inflammation.
As NAFL progresses to NASH, a distinguishing feature is increased markers of cell stress and subsequent inflammation. While there are no approved biomarkers for NASH, several clinical studies have shown the release of alanine aminotransferase (ALT) and caspase-generated cytokeratin 18 (CK18) fragments to positively correlate with inflammation and the NAFLD activity score (23, 24). In our in vitro system, we observed small, but significant increases in both ALT (1.4-fold, Figure 5A) and CK18 (2.6-fold, Figure 5B) with the lipotoxic milieu compared with the healthy condition on day 10. These conditions corresponded with activation of gene expression associated with apoptotic signaling and oxidative stress (Supplemental Figure 5).
Figure 5. Cellular stress and inflammation are induced in the human liver lipotoxic system.

(A and B) Secreted alanine aminotransferase (ALT) (A) and caspase-cleaved cytokeratin 18 (CK18) (B) were measured in the media effluent from devices at day 10. n ≥ 4 experiments, 4 donors. (C) ATP was measured from hepatocytes exposed to the healthy or lipotoxic milieu for 10 days and represented as fold change relative to healthy controls. n ≥ 13 experiments, 5 donors. *P < 0.05, **P < 0.01, Student’s 2-tailed t test. (D) Secreted analytes were measured in the media effluent at days 5, 7, and 10. n ≥ 4 experiments, 4 donors. Triangles indicate samples that were below the lower limit of quantification. *P < 0.05, **P < 0.01, 2-way ANOVA.
One possible cause of increased apoptotic and oxidative stress gene expression could be increased mitochondrial activity due to elevated FFA load and β-oxidation. Since the end product of increased mitochondrial activity is ATP production, we measured ATP levels at the conclusion of the experiment and observed that lipotoxic conditions promoted a significant 4-fold increase (Figure 5C). Indeed, elevated ATP levels correlated with an upregulation of the β-oxidation gene expression pathway (Supplemental Figure 5). Collectively, these data suggest increased hepatic oxidation and cell stress in response to the lipotoxic milieu.
Increased cell and oxidative stress are key factors in eliciting an inflammatory response in the progression of NAFLD (25). The secretome, which represents contributions from both hepatocytes and NPCs, was interrogated for multiple secreted protein analytes linked to an inflammatory response and the progression of NASH. We compared the secretome from our system with hepatocytes alone or in combination with NPCs when exposed to 65 μM oleic acid and 0.1 ng/ml TNF-α. We observed that the NPC presence significantly impacted the hepatocyte phenotype and composition of the secretome. Supplemental Figure 6 shows that by day 10 NPCs increased the basal concentrations of several proteins (e.g., monocyte chemoattractant protein 1 [MCP1] by 3.6-fold, IL-6 by 26-fold), while also increasing sensitivity to detect treatment-specific differences (e.g., IL-8 and VEGF were 3.6-fold and 2.3-fold more responsive, respectively, to the lipotoxic milieu in the presence of NPCs than without).
To further support the importance of the NPCs to the overall phenotype, Supplemental Figure 7 shows a principle component analysis (PCA) of hepatocyte transcriptomic data from the studies mentioned above that compared multi- versus monocultured hepatocytes. As expected, milieu effects (i.e., healthy vs. lipotoxic) clearly separates the data along principle component 1 (PC1) in both multi- and monocultured cells, showing that NPCs are not required for hepatocytes to respond to the lipotoxic milieu. However, along PC2, multiculture conditions clearly separate from monoculture conditions, creating 4 distinct phenotypes. This shows at a high/unbiased level that the hepatocyte phenotypes are fundamentally different when NPCs are present compared with hepatocytes alone. Thus, NPCs are substantial contributors to the overall phenotype of the tissue and the secretome measurements best represent a composite response of all liver cells in the system, including the NPCs.
In this study, each of the tested analytes showed various temporal responses to the lipotoxic milieu (Figure 5D). Over the tested time course, several proinflammatory cytokines increased, including IL-8, IL-6, and CXCL10, all of which have established chemoattractant properties and are upregulated in NAFL/NASH patients or mouse models (Figure 5D) (26, 27). Studies have found that angiogenic factors, such as VEGF, are increased and contribute to the progression of NASH in animal models (28). In agreement, we found levels of VEGF to be increased at day 5. Plasma levels of YKL40 (also called chitinase-3-like protein 1 [CHI3L1]) are increased in several inflammatory diseases, including rheumatoid arthritis and NASH (29–32). YKL40 increased 6- and 4-fold at days 7 and 10, respectively, in the lipotoxic milieu over the healthy milieu conditions (Figure 5D). Transcriptomic data support an increased inflammatory response triggered by the lipotoxic milieu in hepatocytes, where gene expression changes in inflammation and inflammasome (33) signaling pathways were increased (Supplemental Figure 5). Specifically, Toll-like receptors (e.g., TLR1, TLR2, and TLR4), chemokines (CXCL6, -8, -12, -13, and -16), and STATs were significantly upregulated, all of which have specific roles in the inflammatory response. Collectively, gene expression and protein analyses reveal elevated levels of inflammatory signaling in these liver cells in response to lipotoxic stress, a hallmark feature in the progression from NAFL to NASH.
Modulation of extracellular matrix signaling.
One of the distinct protein communities identified through the unbiased PPI analysis was regulation of extracellular matrix (ECM) signaling proteins (Figure 1C). It is well defined that the NAFL-to-NASH progression includes ECM deposition and can ultimately lead to liver fibrosis (34). Owing to the cell plating within a collagen sandwich, direct measurements of collagen deposition were not possible and thus secreted regulators of ECM deposition were explored. Perhaps the best-studied regulator of fibrosis is TGF-β, which potently regulates gene expression of collagens and matrix-modifying enzymes (34). TGF-β was significantly increased at both tested time points (Figure 6A). Further, there was a high degree of coordinated gene expression activation of collagen-formation genes downstream of this pathway in hepatocytes, further supporting that TGF-β may be an activated upstream regulator in conditions of lipotoxic stress (Figure 6B). In addition to TGF-β, osteopontin (OPN) is a biomarker that has been shown to positively correlate with liver fibrosis and participates in neutrophil recruitment (12). OPN concentrations in the media were higher at each of the tested time points, most significantly at day 5 (Figure 6C). Secreted procollagen 1α1 showed no significant changes at the tested time points between healthy and lipotoxic conditions (Figure 6D).
Figure 6. Evidence for extracellular matrix remodeling in the human liver lipotoxic system.

(A, C, and D) Secreted TGF-β (A), osteopontin (OPN) (C), and procollagen 1α1 (D) were measured in the media effluent from devices at the indicated days. n ≥ 4 experiments, 4 donors. (B) Hepatocyte expression of genes of the collagen formation pathway are represented as log2-fold change of lipotoxic vs. healthy milieu (red = upregulation, blue = downregulation). n = 6 experiments, 3 donors. (E) Representative photomicrographs (original magnification, ×20) of nonparenchymal cells (NPCs) are shown. Macrophages (CD68+, red), nuclei (DAPI+, blue), and hepatic stellate cells (smooth muscle α-actin+ (SMAA+), green). Scale bars: 100 μm. SMAA staining intensity from immunofluorescent NPCs images were quantified and represented as fold change relative to healthy controls. n ≥ 4 experiments, 3 donors. Triangles indicate samples that were below the lower limit of quantification. *P < 0.05, **P < 0.01, Student’s 2-tailed t test.
With the progression of NAFLD, HSCs are well documented to dedifferentiate from quiescent cells to activated myofibroblasts and contribute to the fibrotic response (12). One marker of HSC activation is the presence of smooth muscle α-actin (SMAA), which is accompanied by a change in cell morphology (12). We explored HSC activation via immunofluorescence staining to allow us to simultaneously evaluate cell morphology and quantify levels of SMAA. In healthy milieu conditions, HSCs were more quiescent, displaying a rounder cell morphology (Figure 1B) and lower levels of SMAA (Figure 6E). In contrast, HSCs in the lipotoxic conditions had significantly higher levels of SMAA (3.6-fold) with a more fibroblastic morphology (Figure 6E).
Exploration of obeticholic acid response in the lipotoxic system.
Lastly, having observed recapitulation of pathway (Table 1), lipidomic (Figure 3), and functional (Figures 2 and 4–6) responses compared with human clinical samples, we sought to utilize this system to further validate and better understand the potential impact of a promising new drug for treatment of NASH, obeticholic acid (OCA). OCA is a semisynthetic bile acid analogue that improved histological features of NASH and fibrosis in phase IIb clinical trials (35). OCA was added to the lipotoxic system throughout the duration of the experiment at a concentration of 0.5 μM, which approximates the steady-state blood serum concentration of OCA observed in clinical trials (36). At the conclusion of the experiment, OCA responses were compared with the vehicle control (0.1% DMSO).
On-target effects of farnesoid x receptor (FXR) activation are mediated in part through the secretion of FGF19 primarily by the ileum, but we have previously found that hepatocytes can also secrete FGF19 (Dash et al., unpublished data). When FGF19 was measured in the media, OCA was found to potently induce secretion by 25-fold (Figure 7A). Thus, this evidence confirms on-target impact of FXR signaling due to OCA.
Figure 7. Obeticholic acid restores homeostasis in the human liver lipotoxic system.

Nonparenchymal cells (NPCs) and hepatocytes on device were exposed to the lipotoxic milieu containing 0.5 μM obeticholic acid (OCA) or DMSO vehicle control (Veh) for 10 days. (A) Secreted FGF19 was measured in the media effluent from devices at day 10. n = 3 experiments, 1 donor. (B) Nile red staining intensity from hepatocyte images were quantified and represented as fold change relative to healthy controls. n = 4 experiments, 1 donor. (C) Lipids from hepatocytes from device exposed to the healthy or lipotoxic milieu with 0.5 μM OCA or vehicle control were measured by metabolomics. Scatterplot representation of differentially expressed lipids in these hepatocytes are shown as log2-fold change and colored by response similarity index (RSI). Triacyglycerols (○), cholesterol esters (×), all other lipids (•). n = 4 experiments, 2 donors. (D and E) Secreted analytes were measured in the media effluent from devices at day 10. n ≥ 5 experiments, 3 donors. (F) Secreted apolipoproteins were measured in the media effluent from devices at day 10. n = 4 experiments, 2 donors. Triangles indicate samples that were below the lower limit of quantification. *P < 0.05, **P < 0.01, Student’s 2-tailed t test.
Next, the impact of OCA on NASH-related functional endpoints was assessed. Lipid accumulation was decreased by 25% with OCA treatment relative to the vehicle (Figure 7B), confirming the positive effects seen clinically for the improvement in steatosis. The lipidomic profile within the cells was further explored using the response similarity index (RSI) based on lipidomics data. As previously described (37), correlation plots compare responses of the healthy and OCA treatments relative to the lipotoxic control on the x and y axes, respectively, where the RSI determines the degree to which the 2 conditions alter expression in the same (RSI > 0, purple) or opposite (RSI < 0, green) manner. Figure 7C shows a high degree of similarity between the healthy milieu and OCA response, meaning that OCA treatment in the lipotoxic milieu promotes a change in lipid profile towards that of the healthy controls (see Supplemental Figure 8 for individual plots of lipid species). Most profoundly, TAG species were elevated in lipotoxic conditions (Figure 3) and mostly repressed by OCA (lower left-hand quadrant of Figure 7C). Thus, OCA treatment abrogated aspects of steatosis and lipid signaling.
Clinically, patients treated with OCA showed marked improvement in ALT, lobular inflammation, and fibrosis (35). We found that circulating cell stress and inflammatory factors were reduced at the conclusion of the experiment, including ALT (16% reduced, P = 0.10), MCP1 (66% reduced), IL-6 (48% reduced), and IL-8 (25% reduced) (Figure 7D). Previously mentioned markers associated with NASH, VEGF (60% reduced) and YKL40 (58% reduced), were also suppressed by OCA. In addition to proinflammatory secreted factors, regulators of fibrosis, TGF-β (45% reduced) and OPN (37% reduced), were reduced, but did not reach statistical significance (Figure 7E). However, procollagen 1α1 levels were significantly reduced (59% reduced) with OCA treatment in lipotoxic conditions relative to the vehicle (Figure 7E). These reductions in profibrotic factors align with the reduced fibrosis observed clinically.
Overall, while OCA treatment had several beneficial effects on steatosis and inflammatory endpoints, we did observe that total intracellular levels of cholesterol were increased with OCA treatment (Supplemental Figure 9). Several species of CEs were regulated in the opposite direction (RSI < 0, green) of the healthy response (i.e., increased) as seen in the RSI plot (Figure 7C), with none being significantly regulated in the same direction as the healthy controls (i.e., RSI > 0). Further, several apolipoproteins were measured from the secreted media, where ApoB, primarily found in LDL, was increased 2-fold (Figure 7F). Other apolipoproteins, including ApoCII and ApoE were also increased (Figure 7F).
Discussion
The major focus of this study was to develop an in vitro model of lipotoxic stress that incorporates key physiological aspects of NAFLD and to validate it by comparing it with clinical data. Despite the high prevalence of NAFLD, there are currently no approved therapies, little consensus on disease targets, and few representative investigative models. Thus, there is a strong unmet need for human-relevant systems that are able to recapitulate key elements of this disease for preclinical studies prior to costly clinical trials.
Various chip-based models have attempted to recreate a physiologically relevant liver sinusoidal microenvironment by adopting approaches designed to generate three-dimensional (3D) aggregates or spheroids, micropatterning and bioprinting liver cell types into specific spatial configurations and some introducing fluid flow via microfluidics (38–42). For our lipotoxic system, we chose to use a larger-scale 3D system (7, 8) that utilizes more cells, which enables simultaneous collection of multiple endpoints (e.g., transcriptomics, lipidomics, and imaging) from the same experiment, allowing for interassay comparison within a single experiment. This 3D system design incorporates both perfusion and hemodynamic shear forces while recapitulating elements of sinusoidal spatial architecture. Like hepatocytes in the sinusoid, the hepatocytes in our system are not directly exposed to shear force of fluid movement, an aspect that distinguishes it from various other systems that directly expose the hepatocytes to shear stress and is often outside the physiological range found in the liver sinusoids (40). In addition, we previously reported that our liver system restores an in vivo–like liver phenotype, as evidenced by restoration of a stable polarized morphology, in vivo–like transcriptomic signatures distinct from static cultures, and higher liver-specific biochemical function (e.g., increased albumin and urea production as well as drug and xenobiotic metabolizing enzyme activity) (7, 8). Importantly, hepatocytes in this system exhibit induction and toxicity responses to drugs at levels that match therapeutic concentrations (7, 43). In general, while chip systems are currently used in drug toxicity studies, none comprise all the necessary features and validation required for studying NAFL/NASH, as outlined below.
The lipotoxic system developed in this study preserved and recapitulated key aspects of the lipotoxic state of NAFL/NASH and differentiated itself from other models. First, our system better represents a healthy liver compared with conventional in vitro systems, as we use near-physiological levels of glucose and insulin (generally 10- to 10,000-times lower than other in vitro systems; see ref. 44). This is important to ensure that glucose- and insulin-regulated de novo lipogenesis is not compromised and serves as a meaningful control. Second, our system is human relevant, utilizing human primary cells versus hepatic immortalized cell lines or animal models (5). Third, our multicellular system is composed of primary human hepatocytes, HSCs, and MΦs, which are all key players in the progression of NAFL/NASH (9). Additionally, in other multicellular in vitro systems, e.g., spheroids, 3D bioprinting, liver slices, and organ-on-chips, the signals from individual cell types cannot be separated (5). Fourth, in this system, the liver microenvironment is recapitulated by applying physiologically relevant hemodynamics and biological transport, which is absent in traditional static cultures (including many multicellular systems) (5, 7, 8). Finally, the lipotoxic state is predicated upon FFA concentrations derived from clinical plasma concentrations in NASH patients (14). As we show in this study, the sum of all these differentiators provides phenotypic responses that are more representative of the human NAFL/NAFLD response.
The model demonstrated key clinical aspects of NAFL/NASH not described before in an in vitro NAFL/NASH system. This includes the onset of hepatic injury as evidenced by increased ALT levels. In addition, our ability to examine cell type–specific phenotypic responses in a complex system enabled us to identify potential fibrosis-inducing roles for both hepatocytes and HSCs. While activated HSCs have been widely studied and accepted to be the primary modulators of fibrosis, animal studies suggest a role for both cell types in fibrosis (12, 45) — roles that are difficult to uncouple in an in vivo setting. Finally, our data demonstrate that we have a system that is fluid and amendable to creating various stages along the NAFL/NASH spectrum.
This model best falls into the early stages of NASH progression, supported by the presence of steatosis and inflammation. Elevated levels of ATP and β-oxidation were also observed, consistent with animal studies that found that ATP and β-oxidation were increased in early stages of NASH before falling below normal levels in long-term NASH (46). While there was evidence for early fibrotic signaling, later-stage NASH/fibrosis also includes increased levels of collagen, which were not detected in this study. Therefore, this model system captures many key aspects of early-stage NASH, and preliminary data suggest that additional factors, such as TNF-α, push the phenotype further along the spectrum by enhancing inflammation, apoptosis, and fibrosis (Feaver et al., unpublished data).
To further validate the lipotoxic system for human NASH disease relevance, transcriptomic and metabolomic responses were benchmarked using previously published data from clinical biopsies. Despite the challenges of the samples being reflective of a single time point and inherent differences between material collected from an in vitro system compared with a liver biopsy (e.g., cellular composition, disease duration, isolation techniques), direct comparison with our results showed striking similarities across studies. Specifically, transcriptomic pathway results demonstrated that the in vitro lipotoxic system was equally or more similar to tissue biopsy studies versus comparisons between the 2 independent tissue biopsy studies. Metabolomic signatures also showed striking similarities for the various lipid classes in response to the lipotoxic milieu. To the best of our knowledge no other preclinical system, in vitro or in vivo, has rigorously validated both transcriptomic and metabolomic responses in addition to the functional readouts as were presented in this study. These comparisons provide clinical validation that our in vitro lipotoxic system mimics important signaling aspects of NAFL and early NASH, as outlined in Supplemental Table 1, with the exception of late-stage fibrosis, cirrhosis, and oncogenesis.
Lastly, we challenged the system with OCA, a drug that is currently in phase III clinical trials (4) with published phase II clinical trial data (35). Similar to the results seen in the phase II clinical trial, OCA reduced steatosis, inflammatory cytokines, and fibrotic mediators. Furthermore, we believe that this is the first study to show metabolomics analysis evaluating OCA effects, and we found that OCA reversed the NASH-associated lipidomic signature towards a healthy state. Thus, the reversal of steatosis in OCA-treated patients may occur from the coordinated shift of several specific classes/species of lipids. Defining NAFL/NASH-associated lipid signatures for use in noninvasively benchmarking therapeutic responses will be invaluable (47). The exception to this finding was CEs, which were regulated in an opposite manner to those of the healthy controls for several species (i.e., further increased with OCA). This result may provide additional insight into the most controversial outcome observed with OCA-treated patients in the clinic, which is the increased levels of total cholesterol and LDL (35). We found that hepatic total cholesterol and secreted ApoB were increased, further suggesting dysregulation of this pathway. Outside of clinical trials, to the best of our knowledge this result has never been published before, in vitro or in animal studies. Overall, the response to OCA in the lipotoxic system correlated to outcomes observed in clinical trials that have not been recapitulated in other in vitro systems, validating the system for evaluation of new therapeutic targets in the future. We are currently evaluating head-to-head more than 20 compounds that constitute the bulk of the therapeutic landscape for NAFL/NASH, including on/off-target pharmacodynamic effects and the ability of each to regress the lipotoxic phenotype. These future studies will provide insight regarding the strengths/weaknesses of the compounds currently in development and identify potential novel therapeutic targets for the treatment of NASH.
In conclusion, we demonstrate that lipotoxic stress and therapeutic drug response similar to that seen in NASH patients can be recapitulated in vitro in primary human liver cells. Collectively, we have found that by placing isolated cells into a configuration that better resembles their native microenvironment, the cells begin to regain many aspects of their normal function. This lipotoxic liver system shows responsiveness to in vivo–relevant concentrations of risk factors and drugs, providing a promising new tool for investigating disease mechanisms and interventions. Such systems may prove beneficial in translational preclinical drug discovery in NASH.
Methods
Cell isolation and culture.
Human cryopreserved hepatocytes from various healthy donors were screened and tested to select donors that met the following quality control criteria: (a) postthaw viability greater than 85%, (b) plating efficiency greater than 75%, (c) polarized morphology, and (d) albumin secretion rates greater than 10 mg/million cells/day. The sourcing details and demographics of these donors are included in Supplemental Table 2. Human HSCs were purchased from Triangle Research Labs, maintained in DMEM/F-12 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and used at passages 1–5 for all experiments. MΦs were isolated from human blood. In brief, peripheral mononuclear blood cells were isolated from whole blood (Virginia Blood Services) by density gradient centrifugation. Magnetic isolation of MΦs from this pool was then performed using a Human CD14 Positive Selection Kit (STEMCELL Technologies, 18058). Cells were then incubated with human granulocyte-macrophage colony-stimulating factor (GM-CSF, Peprotech, 300-03) for 10 to 12 days. MΦ phenotype was confirmed by expression of MΦ-specific markers, CD68 and CD163 (BD Pharmingen, 556059 and Abcam, ab174867, respectively). Cells were cryopreserved and stored in liquid nitrogen until ready for experimental plating as described below. No human fetal–derived biomaterial was used in these studies.
Transwell coculture plating conditions and hemodynamic exposure.
On day 1, primary hepatocytes were thawed and plated in a collagen gel sandwich configuration at 220,000 cells/cm2 on the undersurface of the membranes of 75-mm polycarbonate transwells (Corning Inc.) using previously described protocols (7). The cultures were left overnight in maintenance medium (MM) that consisted of DMEM/F-12 supplemented with 10% fetal bovine serum, 50 mg/ml gentamycin, 0.2% ITS (Fisher/MediaTech, MT-25e800CR), and dexamethasone (Sigma-Aldrich, D4902; 1 mM at plating and 250 nM thereafter). The following day, HSCs and MΦs (collectively called NPCs) were plated in the top surface of the transwell in a collagen gel sandwich at 22,000 cell/cm2 and 44,000 cells/cm2, respectively, in MM. NPCs were shielded from direct shear forces with a layer of collagen in the absence of a sinusoidal endothelium. Transwells were then set up within HemoShear devices in a configuration to allow for control of hemodynamics and transport as described previously (see Figure 1A) (7–8, 11). A proprietary hepatocyte flow medium (HFM), modified from MM, was continuously perfused on both sides while shear stress was applied on the top surface based on the calculations described previously (8). Hepatocytes and NPCs were cultured under controlled hemodynamics and terminated on day 10. HFM was supplemented with normal physiological levels of insulin (690 pM) and glucose (5.6 mM) to mimic healthy conditions or high levels of insulin (6,900 pM), glucose (25 mM), FFAs (65 μM sodium oleate [Sigma-Aldrich, O7501] and 45 μM palmitic acid [Cayman, 10006627]) to mimic plasma concentrations of these factors in a NASH patient (14, 15).
Measurement of secreted analytes.
Media effluent was collected from the experimental devices at days 5, 7, and 10. Various human cytokines were measured via ELISA and Magpix kits, according to the manufacturer’s instructions. Kits include: TGF-β1 (R&D Systems, DB100B), OPN (R&D Systems, DOST00), procollagen 1α1 (R&D Systems, DY6220-05), chitinase 3-like protein 1 (YKL40; R&D Systems, DC3L10), M30 CytoDEATH Kit (CK18; diPharma, P10900), ALT (Neo Scientific, HA0395), Magpix Human Cytokine/Chemokine Panel I (VEGF, IL-6, IL-8, CCL2 [MCP1], CXCL10; EMD Millipore, HCYTOMAG-60K), Magpix Apolipoprotein Panel (Apo AI, AII, B, CII, CIII, and E; EMD Millipore, APOMAG-62K).
Metabolic activity assays: glucose output and ATP assay.
For measurement of basal levels of total glucose produced by hepatocytes, NPCs were scraped from the transwell on day 10 and punches of the remaining transwell containing only hepatocytes were collected, washed briefly 3 times in PBS, and incubated at 37°C for 3 hours in glucose-free starvation media (Gibco, A14430-01). Media were then collected and glucose levels in media measured using a Glucose Detection Kit (Abcam, 102517) according to the manufacturer’s instructions. For insulin-mediated glucose response, hepatocytes from the device were washed as above, incubated with media containing high glucose (25 mM) for 1 hour, and then incubated at 37°C for 3 hours in glucose-free starvation media with or without 100 nM insulin. Media were collected and glucose measured as described above.
For measurement of intracellular ATP levels from hepatocytes, these cells were collected as described above and washed with PBS before incubating in 96-well plates. The intracellular ATP levels were measured using the CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega, G7571) according to the manufacturer’s instructions.
Immunofluorescence and microscopy.
Hepatocytes and NPCs plated on transwells and exposed to hemodynamics were fixed in 4% paraformaldehyde for 15 minutes, permeabilized in 0.2% Triton X-100 in PBS for 5 minutes, and incubated with primary antibody for 2 hours at room temperature. After 3 washes in PBS, they were incubated with secondary antibodies for 1 hour at room temperature. Anti-human primary antibodies were against adipophilin (Abcam, ab108323), E-cadherin (Santa Cruz Biotechnology, sc-71009), reelin (Abcam, ab78540), CD68 (BD Pharmingen, 556059), and SMAA (Sigma-Aldrich C6198). Goat anti-mouse or goat anti-rabbit conjugated to Alexa Fluor 488 or 546 (Life Technologies; A11001, A11003, A11008, and A11010) were used as secondary antibodies. Lipids were stained with Nile red (Sigma-Aldrich, 19123) and nuclei were stained with DAPI (Thermo Fisher Scientific, D1306) or TO-PRO-3 (Life Technologies, T3605). Images of immunofluorescently labeled cells were acquired with a Nikon 20× or 100× objective lens on a Nikon C1 confocal microscope with EZ-C1 v3.9 software or a Nikon 20× objective extra-long working distance lens on the ImageXpress Micro XLS Widefield High-Content Analysis System (Molecular Devices). Nile red quantification was performed as total Nile red intensity normalized to nuclei counts and then presented as fold change relative to healthy control.
Cholesterol measurements.
Hepatocytes exposed to hemodynamics were collected and lysed to extract and measure total intracellular cholesterol using the Total Cholesterol Assay Kit (Colorimetric) (Cell Biolabs, STA-384), according to the manufacturer’s instructions.
Metabolomics.
Complex lipids, including TAGs, DAGs, FFAs, CEs, phospholipids, lysophospholipids, sphingomyelins, and ceramides, were measured from 1.2 million hepatocytes using the TrueMass Complex Lipid Panel service through Metabolon.
Differential abundance of individual lipid species was assessed using empirical Bayes variance-moderated linear models with the limma package in R (48). Prior to modeling, lipid abundance was normalized to total protein concentration. Models included experimental treatment and donor as fixed effects, where donor was treated as a nuisance variable. P values for parameter contrasts were adjusted for multiple comparisons using the Benjamini-Hochberg method (49).
Western blot analysis.
Following cessation of experiments at day 10, hepatocytes were collected from transwells and cell pellets lysed in 2× Laemmli sample buffer (Bio-Rad, 161-0737) containing β-mercaptoethanol. Total protein lysates were resolved in a 10% SDS-PAGE gel and transferred to nitrocellulose. For phosphorylated-Akt signaling experiments, hepatocytes were removed from the device at day 10, serum starved at 37°C for 2 hours in media containing low glucose (5.6 mM), and then either left untreated or treated with 10 nM insulin for 10 minutes at 37°C prior to lysing in sample buffer. Blots were probed overnight with primary antibody and 30 minutes at room temperature with secondary antibody in blocking buffer (LI-COR, 927-40000). Primary antibodies used were RPS11 (Bethyl Laboratories, A303-936A-M), phospho-Akt (Ser473) (Cell Signaling Technology, 4060), and Akt (Cell Signaling Technology, 9272). Secondary antibodies (diluted 1:15,000) used were IRDye 680LT donkey anti-mouse (LI-COR, 926-68022), IRDye 800CW donkey anti-mouse (LI-COR, 926-32212), IRDye 680LT donkey anti-rabbit (LI-COR, 926-68023), and IRDye 800CW donkey anti-rabbit (LI-COR, 926-32213). The LI-COR Odyssey infrared imager was used for image acquisition and the LI-COR Odyssey Image Studio software used for densitometry analysis.
Drug treatments.
For drug experiments, hepatocytes and NPCs were exposed to DMSO vehicle control (Sigma-Aldrich, D2650), 0.5 μM OCA (AdipoGen, AG-CR1-3560-M025), or 300 nM atorvastatin (Cayman, 10493) in the lipotoxic milieu with hemodynamics for 10 days.
RNA preparation and RNA deep sequencing.
Hepatocyte cell pellets were collected following cessation of experiments at day 10, and RNA isolated using the Purelink RNA Mini kit (Invitrogen, 12183018A) according to the manufacturer’s instructions. RNA concentration was determined with the Nanodrop and RNA integrity was determined using the Agilent 2100 Bioanalyzer and the RNA 6000 kit (Agilent, 5067-1511) according to the manufacturer’s instructions. Samples with a minimum RNA integrity number of 7.0 were used for further transcriptomics processing. RNA (250 ng per sample) was submitted to Expression Analysis, Inc. for Illumina-based RNA deep sequencing. Approximately 20 million 50-bp paired-end reads were generated per sample. For each treatment, 6 experiments were run using 3 unique hepatocyte donors.
Transcriptomic data analysis.
Following RNA deep sequencing, reads were pseudoaligned to the human transcriptome (GRCh38), and transcripts were quantified using kallisto (50, 51). Transcript abundances were pooled into gene-level estimates using the tximport package in R (52). Three RNA deep sequencing quality control measurements were performed to detect outliers and batch effects associated with the dataset: (a) L1 distance between samples, (b) PCAs, and (c) similarity of density estimates of abundance values (53). The RNA sequencing data has been deposited into an MINSEQE-compliant public database and the accession number is GSE89063.
The gene-level RNA-seq counts were then analyzed to determine differentially expressed genes (DEGs). Only genes that passed our minimum abundance threshold of more than 2 counts per million in at least 3 samples (the minimum number of biological replicates) were used for further analysis. Methods for DEG determination were implemented using the open source R/BioConductor software (http://www.bioconductor.org) and the edgeR package (54). The model for this analysis included treatment and donor as predictor variables, and read count as the response variable. We estimated additional relevant variables using surrogate variable analysis (SVA), which were also included as predictors in the model design (55). A gene was determined to be a DEG by passing an FDR threshold of 10% (49). Select genes of relevant biological processes were chosen to build response heatmaps that depict log2-fold changes in gene expression: blue = downregulation, red = upregulation, white = no change; the intensity of the color reflects the magnitude of change; a white dot indicates a statistically significant change in the expression of the corresponding gene (FDR < 10%). EdgeR-calculated fold changes, raw P values, and FDRs were used as input for downstream pathway analyses with IPA, metacore, SPIA, topGO, and GSEA pathway enrichment algorithms, which included known pathway gene lists from KEGG, Reactome, Biocarta, and Gene Ontology databases (41, 56–62). Pathways found enriched for responses with an FDR of 20% or better were kept for consideration; redundant pathway enrichments were identified and merged based on strongly connected pathway-to-pathway gene overlaps, using the Louvain method for network community detection (63).
For selected pathways, the directionality of the pathway was determined by calculating the average FDR-adjusted fold change for each gene panel/pathway. To do this, for each gene within the specified pathway, the fold change was scaled by (1 – FDR) to provide greater weight to more significant genes. The scaled fold change was then averaged across all genes within the pathway to determine the directionality and magnitude of regulation. The significance of the directional change was determined using rotation gene set tests (ROAST) (64).
PPI networks.
The transcriptomic data were integrated into a global PPI network, composed of high-confidence interactions defined by the STRING database (65). The goal of the PPI network analysis was to identify protein-level centers of dysregulation in response to experimental treatments. Edges in the network were weighted by the joint posterior probability that their incident nodes were differentially expressed (see Calculation of Bayesian posterior probabilities and RSI below). Connections with low weight were filtered, substantially reducing the size of the network. The resulting network of interactions reflects connected portions of the network with the strongest evidence of regulation. The size of any given node is proportional to its eigenvector centrality in the weighted network; thus, node size is an indicator of the relative importance of each node in the context of the treatment response. Communities, which are represented as color in the network figures, are regions of the network comprised of more densely connected nodes, and thus likely represent sets of proteins involved in coordinated biological activities such as signaling pathways. Community partitions were identified using the Louvain algorithm, and the functional themes of the communities were determined by identifying Reactome gene sets with significant overlap via the hypergeometric test (63). The PPI network was spatially organized and viewed using the continuous graphing layout, Force Atlas 2, in GEPHI (66).
Calculation of Bayesian posterior probabilities and RSI.
For tests of differential abundance applied to metabolomic and transcriptomic data sets, we calculated the Bayesian posterior probability of the alternative hypothesis being true for each measured species (i.e., the posterior probability that any given species is differentially abundant). To compute this value, we fit a mixture of 1 uniform distribution and zero or more monotonically decreasing beta distributions to the distribution of P values corresponding to multiple hypothesis tests in a given contrast; this method is described in detail by Allison et al. (67). Then, for a given P value, the probability that it was drawn from a nonuniform component of the mixture was computed using Bayes’ rule. This probability is the posterior probability that the metabolite or transcript species is differentially abundant in the given contrast.
For pairs of contrasts, the posterior probabilities were combined to quantify similarity of treatment response. This quantification, termed the RSI, is based on the composite hypothesis testing procedure described by Erikson et al. (68). The RSI of a given species in 2 contrasts is simply the product of each individual posterior probability signed by the sign of the product of the log2-fold changes. Thus, species demonstrating differential abundance in the same direction will have RSI values approaching 1, species that are differentially abundant in opposite directions will have RSI values approaching –1, and species lacking evidence for differential abundance in one or both contrasts will have RSI values near 0.
Statistics.
Transcriptomic data and pathway analysis were analyzed as stated above. All other data are represented as dot plots that include the mean ± SEM of all data points. The data points represent either raw or normalized data depending on the plot. Various statistical tests were employed including Student’s 2-tailed t test, 1- and 2-way ANOVAs as specified in each figure legend. A Pearson correlation value was used in correlation plots. A P value less than 0.05 was considered significant.
Study approval.
No human or animal studies were performed in this study and thus no approval and/or informed consent was required.
Author contributions
REF was involved in the study conceptualization and design, experiments and procedures, data analysis, statistical analysis, scientific interpretation, drafting of the article, critical revision of the manuscript for important intellectual content, and supervised the study. BKC was involved in the study conceptualization and design, experiments and procedures, data analysis, scientific interpretation, drafting of the article, and critical revision of the manuscript for important intellectual content. MJL and SAH were involved in the study conceptualization and design, data analysis, and statistical analysis. SM was involved in the study conceptualization and design and scientific interpretation. BRB was involved in the study conceptualization and design. RAF, AJS, BRW, and AD were involved in the study conceptualization and design, scientific interpretation, and critical revision of the manuscript for important intellectual content.
Supplementary Material
Acknowledgments
The authors would like to thank Michael B. Simmers, Sandi Walton, Andrew W. Pryor, Diana J. Berry, Nathan Day, Morgan Donovan, and Crystal Passmore for their experimental assistance. This research was supported in part by SBIR grants 2R44DK100136 and 4R44DK104456-02 and HemoShear Therapeutics, LLC.
Footnotes
Conflict of interest: All HemoShear affiliates are employed or were employed by HemoShear, and all authors have stock in the company. Arun J. Sanyal is a consultant to HemoShear.
Reference information:JCI Insight. 2016;1(20):e90954. doi:10.1172/jci.insight.90954.
Contributor Information
Svetlana Marukian, Email: smarukian@gmail.com.
Ajit Dash, Email: dash@hemoshear.com.
References
- 1.Sass DA, Chang P, Chopra KB. Nonalcoholic fatty liver disease: a clinical review. Dig Dis Sci. 2005;50(1):171–180. doi: 10.1007/s10620-005-1267-z. [DOI] [PubMed] [Google Scholar]
- 2.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medina J, Fernández-Salazar LI, García-Buey L, Moreno-Otero R. Approach to the pathogenesis and treatment of nonalcoholic steatohepatitis. Diabetes Care. 2004;27(8):2057–2066. doi: 10.2337/diacare.27.8.2057. [DOI] [PubMed] [Google Scholar]
- 4. U.S. National Institutes of Health . https://clinicaltrials.gov Accessed October 31, 2016.
- 5.Khetani SR, Berger DR, Ballinger KR, Davidson MD, Lin C, Ware BR. Microengineered liver tissues for drug testing. J Lab Autom. 2015;20(3):216–250. doi: 10.1177/2211068214566939. [DOI] [PubMed] [Google Scholar]
- 6.Sanches SC, Ramalho LN, Augusto MJ, da Silva DM, Ramalho FS. Nonalcoholic steatohepatitis: a search for factual animal models. Biomed Res Int. 2015;2015:574832. doi: 10.1155/2015/574832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dash A, et al. Hemodynamic flow improves rat hepatocyte morphology, function, and metabolic activity in vitro. Am J Physiol, Cell Physiol. 2013;304(11):C1053–C1063. doi: 10.1152/ajpcell.00331.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terelius Y, et al. Transcriptional profiling suggests that Nevirapine and Ritonavir cause drug induced liver injury through distinct mechanisms in primary human hepatocytes. Chem Biol Interact. 2016;255:31–44. doi: 10.1016/j.cbi.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia. 2009;13(1):9–19. [PMC free article] [PubMed] [Google Scholar]
- 10.Marra F, Lotersztajn S. Pathophysiology of NASH: perspectives for a targeted treatment. Curr Pharm Des. 2013;19(29):5250–5269. doi: 10.2174/13816128113199990344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hastings NE, Simmers MB, McDonald OG, Wamhoff BR, Blackman BR. Atherosclerosis-prone hemodynamics differentially regulates endothelial and smooth muscle cell phenotypes and promotes pro-inflammatory priming. Am J Physiol, Cell Physiol. 2007;293(6):C1824–C1833. doi: 10.1152/ajpcell.00385.2007. [DOI] [PubMed] [Google Scholar]
- 12.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin Nutr. 2002;21(3):219–223. doi: 10.1054/clnu.2001.0529. [DOI] [PubMed] [Google Scholar]
- 15.Chitturi S, et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35(2):373–379. doi: 10.1053/jhep.2002.30692. [DOI] [PubMed] [Google Scholar]
- 16.Jensen LJ, et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Database issue):D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahrens M, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013;18(2):296–302. doi: 10.1016/j.cmet.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 18.López-Vicario C, et al. Molecular interplay between Δ5/Δ6 desaturases and long-chain fatty acids in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2014;63(2):344–355. doi: 10.1136/gutjnl-2012-303179. [DOI] [PubMed] [Google Scholar]
- 19.Straub BK, et al. Adipophilin/perilipin-2 as a lipid droplet-specific marker for metabolically active cells and diseases associated with metabolic dysregulation. Histopathology. 2013;62(4):617–631. doi: 10.1111/his.12038. [DOI] [PubMed] [Google Scholar]
- 20.Puri P, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46(4):1081–1090. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 21.Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. 2013;5(5):1544–1560. doi: 10.3390/nu5051544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramnanan CJ, Edgerton DS, Cherrington AD. The role of insulin in the regulation of PEPCK and gluconeogenesis in vivo. US Endocrinol. 2009;5(1):34–39. doi: 10.17925/USE.2009.05.1.34. [DOI] [Google Scholar]
- 23.Diab DL, et al. Cytokeratin 18 fragment levels as a noninvasive biomarker for nonalcoholic steatohepatitis in bariatric surgery patients. Clin Gastroenterol Hepatol. 2008;6(11):1249–1254. doi: 10.1016/j.cgh.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44(1):27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 25.Takaki A, Kawai D, Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH) Int J Mol Sci. 2013;14(10):20704–20728. doi: 10.3390/ijms141020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology. 2003;38(2):413–419. doi: 10.1053/jhep.2003.50316. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J Hepatol. 2014;61(6):1365–1375. doi: 10.1016/j.jhep.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Coulon S, et al. Role of vascular endothelial growth factor in the pathophysiology of nonalcoholic steatohepatitis in two rodent models. Hepatology. 2013;57(5):1793–1805. doi: 10.1002/hep.26219. [DOI] [PubMed] [Google Scholar]
- 29.Cho SJ, et al. YKL-40 is a protective biomarker for fatty liver in World Trade Center particulate matter-exposed firefighters. J Mol Biomark Diagn. 2014:5. doi: 10.4172/2155-9929.1000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johansen JS, et al. Serum YKL-40 is increased in patients with hepatic fibrosis. J Hepatol. 2000;32(6):911–920. doi: 10.1016/S0168-8278(00)80095-1. [DOI] [PubMed] [Google Scholar]
- 31.Johansen JS, Kirwan JR, Price PA, Sharif M. Serum YKL-40 concentrations in patients with early rheumatoid arthritis: relation to joint destruction. Scand J Rheumatol. 2001;30(5):297–304. doi: 10.1080/030097401753180381. [DOI] [PubMed] [Google Scholar]
- 32.Kumagai E, et al. Serum YKL-40 as a marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci Rep. 2016;6:35282. doi: 10.1038/srep35282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basaranoglu M, Basaranoglu G, Sentürk H. From fatty liver to fibrosis: a tale of “second hit”. World J Gastroenterol. 2013;19(8):1158–1165. doi: 10.3748/wjg.v19.i8.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neuschwander-Tetri BA, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirschfield GM, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148(4):751–61.e8. doi: 10.1053/j.gastro.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 37.Cole BK, et al. An in vitro cynomolgus vascular surrogate system for preclinical drug assessment and human translation. Arterioscler Thromb Vasc Biol. 2015;35(10):2185–2195. doi: 10.1161/ATVBAHA.115.306245. [DOI] [PubMed] [Google Scholar]
- 38.Bhise NS, et al. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication. 2016;8(1):014101. doi: 10.1088/1758-5090/8/1/014101. [DOI] [PubMed] [Google Scholar]
- 39.Chen Q, Utech S, Chen D, Prodanovic R, Lin JM, Weitz DA. Controlled assembly of heterotypic cells in a core-shell scaffold: organ in a droplet. Lab Chip. 2016;16(8):1346–1349. doi: 10.1039/C6LC00231E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esch MB, et al. Multi-cellular 3D human primary liver cell culture elevates metabolic activity under fluidic flow. Lab Chip. 2015;15(10):2269–2277. doi: 10.1039/C5LC00237K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gori M, Simonelli MC, Giannitelli SM, Businaro L, Trombetta M, Rainer A. Investigating nonalcoholic fatty liver disease in a liver-on-a-chip microfluidic device. PLoS One. 2016;11(7):e0159729. doi: 10.1371/journal.pone.0159729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma X, et al. Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proc Natl Acad Sci U S A. 2016;113(8):2206–2211. doi: 10.1073/pnas.1524510113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dash A, Figler RA, Sanyal AJ, Wamhoff BR. Drug-induced steatohepatitis. Expert Opin Drug Metb Toxicol. doi: 10.1080/17425255.2017.1246534. [published online ahead of print October 19, 2016]. doi: 10.1080/17425255.2017.1246534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.LeCluyse EL, Witek RP, Andersen ME, Powers MJ. Organotypic liver culture models: meeting current challenges in toxicity testing. Crit Rev Toxicol. 2012;42(6):501–548. doi: 10.3109/10408444.2012.682115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gorrell MD. Liver fibrosis: the hepatocyte revisited. Hepatology. 2007;46(5):1659–1661. doi: 10.1002/hep.22003. [DOI] [PubMed] [Google Scholar]
- 46.Serviddio G, et al. Alterations of hepatic ATP homeostasis and respiratory chain during development of non-alcoholic steatohepatitis in a rodent model. Eur J Clin Invest. 2008;38(4):245–252. doi: 10.1111/j.1365-2362.2008.01936.x. [DOI] [PubMed] [Google Scholar]
- 47.Gorden DL, et al. Biomarkers of NAFLD progression: a lipidomics approach to an epidemic. J Lipid Res. 2015;56(3):722–736. doi: 10.1194/jlr.P056002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 50.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 51.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 52.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics--a bioconductor package for quality assessment of microarray data. Bioinformatics. 2009;25(3):415–416. doi: 10.1093/bioinformatics/btn647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3(9):1724–1735. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Croft D, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42(Database issue):D472–D477. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishimura D. Biotech Software and Internet Report. 2004;2(3):117–120. doi: 10.1089/152791601750294344. [Google Scholar]
- 60.Schaefer CF, et al. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37(Database issue):D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tarca AL, et al. A novel signaling pathway impact analysis. Bioinformatics. 2009;25(1):75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blondel V, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech. 2008;P10008(3 Pt 2) doi:10.1088/1742-5468/2008/10/P10008. [Google Scholar]
- 64.Wu D, Lim E, Vaillant F, Asselin-Labat ML, Visvader JE, Smyth GK. ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics. 2010;26(17):2176–2182. doi: 10.1093/bioinformatics/btq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jensen LJ, et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Database issue):D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jacomy M, Venturini T, Heymann S, Bastian M. ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS ONE. 2014;9(6):e98679. doi: 10.1371/journal.pone.0098679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Allison DB, et al. A mixture model approach for the analysis of microarray gene expression data. Comput Stat Data Anal. 2002;39:1–20. doi: 10.1016/S0167-9473(01)00046-9. [DOI] [Google Scholar]
- 68. Erickson S, Kim K, Allison DB. Composite hypothesis testing: an approach built on intersection-union tests and Bayesian posterior probabilities. In: Guerra R, Goldstetin DR, eds. Meta-anlaysis and combining information in genetics and genomics. London: Chapman and Hall/CRC; 2010:83–93. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.